1. Introduction
Undoubtedly, biofouling can be considered a great drawback limiting the use of membranes in the treatment of domestic or agricultural wastewater. Biofouling causes numerous problems, ranging from the flow declining, with a consequent loss of membrane performance, to the cleaning of the membranes, with consequences on the costs of the wastewater treatment. For these reasons, membranes with increased resistance to biofouling are required; hence, the knowledge of the relationships between the structure and properties, correlated with the antibacterial activity of the molecules, is crucial for the synthesis of novel membrane functionalization.
Because of their well-known antimicrobial activity, since the first half of the 20th century, quaternary ammonium salts (QAS) have been used in many human activities (industrial, agricultural, household, and hospital) [
1,
2]. They are amphiphilic chemicals with a positive charge related to the quaternary ammonium group and have a hydrophobic tail formed by alkyl chains, hence they are also considered surfactants. Several investigations pointed to the crucial role played by the total positive charge of the ammonium salts and their lipophilicity [
3,
4,
5,
6]. Madaan et al. [
7] showed that the antibacterial effect of quaternary pyridinium salts improved by increasing the number of thimethylammonium head-groups [
8]. Such a result was due to the strongest interaction of the ammonium positive charges with the hydrophilic gram-negative cell wall, that is, with the thin layer of lipopolysaccharides (LPS), peptidoglycan [
9]. However, Ye et al. [
10] measured the antimicrobial activity of six QAS tethered polydimethylsiloxanes salts as a function of aliphatic chains length (two QAS-linked polymer groups were tested), in order to explain the effect of the chain length on the antiseptic property of QAS-linked polymers. The authors demonstrated that the biocidal activity of the QAS-linked polymer is reduced if the quaternary ammonium groups are exposed to the suppressive effect of dissolved polyanions. On the contrary, QAS with alkyl chains capable of protecting the quaternary nitrogen showed better antibacterial activity [
10,
11]. Thus, the charge density on the antiseptic surfaces [
12,
13] correlated to the concentration of the quaternary nitrogen and partial charge, as well as the length of the alkyl substituents [
14,
15,
16,
17], affect the antibacterial activity of QAS-linked polymers.
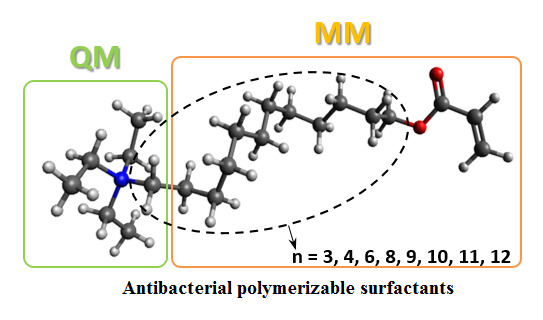
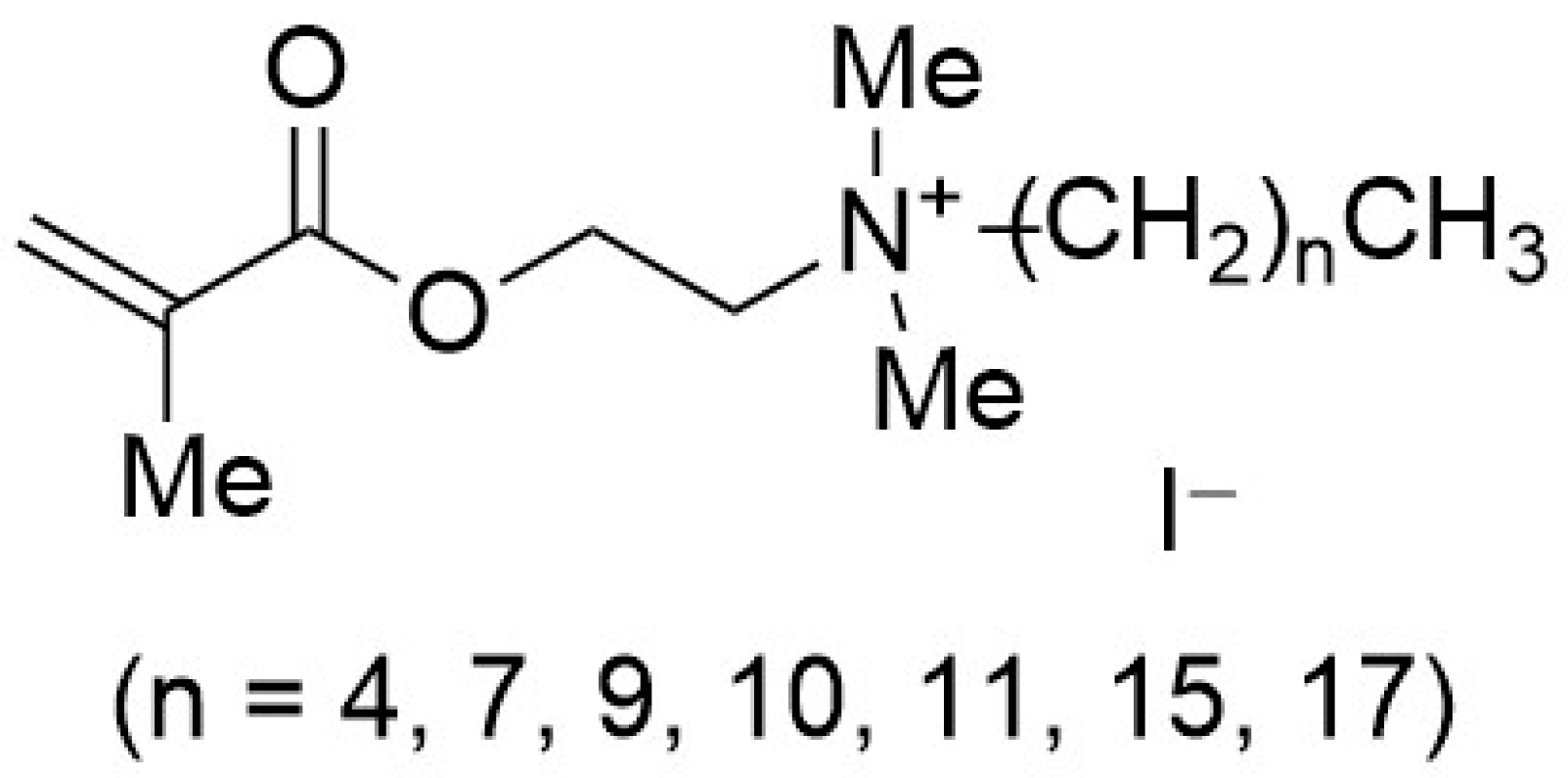
Polymerizable quaternary ammonium salts (PQASs) have found important applications for preventing biofouling through their anchoring on different polymers, resulting in the formation of antiseptic surfaces [
18]. A series of polymerizable surfactants with an alkyl chain of different lengths, as can be seen in
Figure 1, were used as antibacterial agents in methacrylate dental composites.
This study showed that the antibacterial activity against
S. mutans strongly depends on the number of carbon atoms (n) in the main alkyl chain attached to the ammonium group [
18], in agreement with previous works. The 2-dimethyl-2-hexadecyl-1-methacryloxyethyl ammonium iodide (C16) showed the highest antibacterial activity, whereas the 2-dimethyl-2-pentyl-1-methacryloxyethyl ammonium iodide (C5) and 2-dimethyl-2-octyl-1-methacryloxyethyl ammonium (C8) did not show any inhibition of the bacteria.
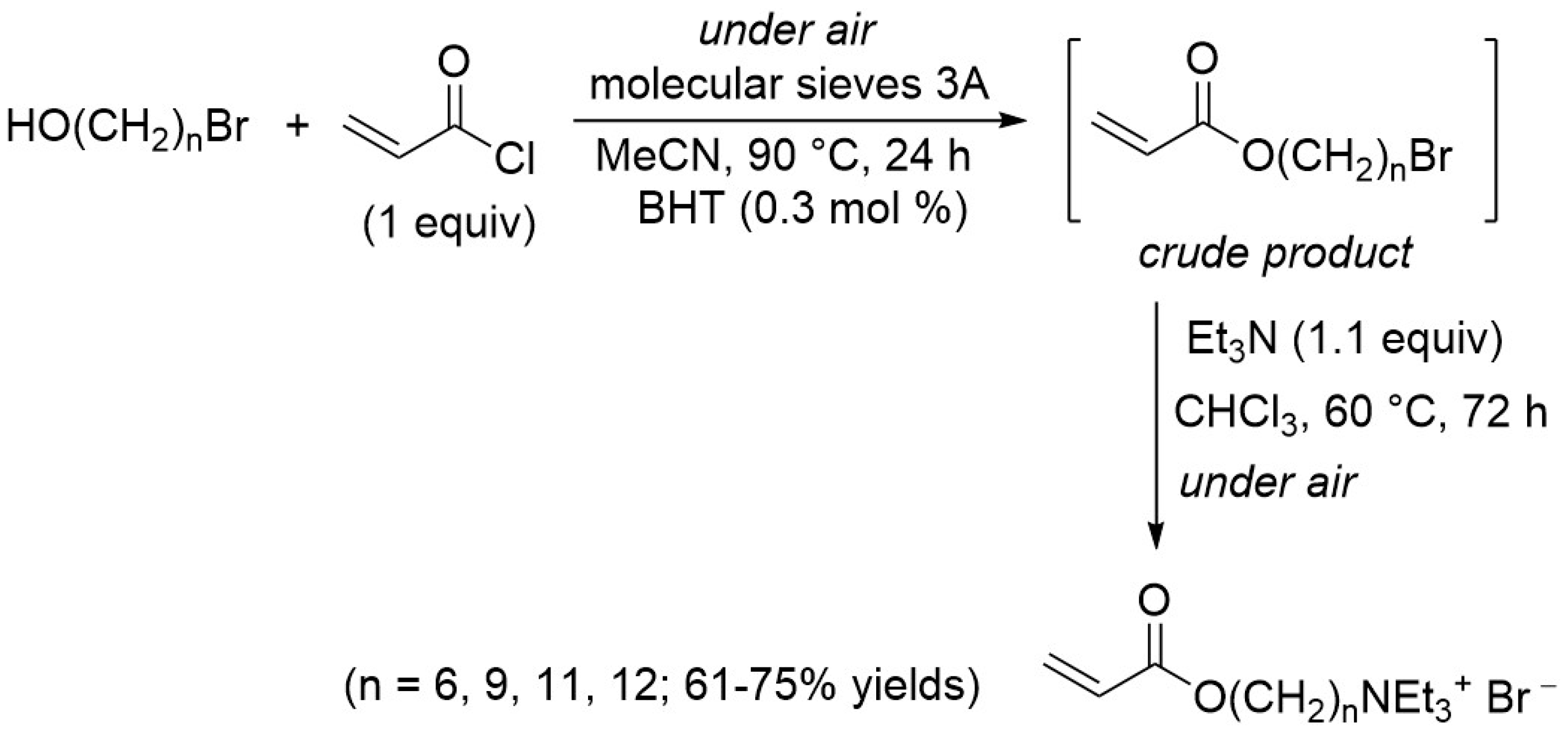
PQASs have also found innovative applications in the membrane framework, so as to prevent biofouling. Recently, some authors of this paper synthesized polymerizable quaternary ammonium salts, acryloxyalkyltriethylammonium bromides (AATEABs) [
19], used as surfactants for the antimicrobial coatings of commercial membranes [
20]. A scheme summarizing the synthesis of these ammonium salts is reported in
Figure 2. With respect to the
N-Alkyl-
N,
N-dimethyl-
N-[(methacryloyloxy)ethyl]ammonium iodides (
Figure 1), the polarizable monomer of the AATEABs is directly bonded to the main aliphatic chain, while three ethyl groups are attached to the quaternary nitrogen. Moreover, the authors found a correlation between the number of carbons in the main aliphatic chain and the antibacterial activity of these compounds [
19].
In the
N-alkyl-
N,
N-dimethyl-
N-[(methacryloyloxy)ethyl]ammonium iodides [
18], the polymerizable group is bonded to a chain with only two carbon atoms, in the AATEABs, this aliphatic chain was stretched to 12 atoms, in order to also obtain good surfactants.
In fact, thanks to its structure, the AATEAB, with n equal to 11 (acryloyloxyundecyltriethylam-monium bromide AUTEAB), has been used as a surfactant for surface modification [
20], with a significant increase of the membrane bio-fouling resistance. Moreover, an analogous of the AUTEAB was also used to study the exchange of bromide ions with antibacterial Keggin polyoxometalates [
21]. The antibacterial activity of the synthetized AATEABs was also measured in previous works [
19], and a correlation between their activity and carbons number of the aliphatic chain was found. In particular, the biocide activity of these surfactants increases along with the carbons number of the chain.
Thus, in this paper, some molecular properties, such as the charge distributions, maximum molecular length, and aspect ratio, correlated to molecular antibacterial activity, were calculated using ab initio-based molecular modeling, with the aim to investigate the correlations between the AATEABs’ structure and target properties. The purpose of the work is to evaluate if the folding of the aliphatic chain, correlated to the carbons in the chain, affects the charges of the head group and the maximum molecular length, through hydrogen bonds or noncovalent interactions between carbonyl and ammonium. Water molecules surrounding the surfactants, as well as hydrophobic interactions are necessary in order to evaluate the folding effect. Indeed, the water molecules could act as a bridge between the carbonyl and ammonium groups, and this bridge could reduce the maximum surfactant length or act as thickness (i.e., shock absorber), avoiding an excessive chain reduction (i.e., winding), mostly for the surfactants with a high number of carbons. Thus, the influence of the water molecules and hydrophobic interaction is not evident. Moreover, it is interesting to evaluate if, for short aliphatic chains, a direct interaction between the carbonyl and the positive ammonium can occur in presence of the water molecules. Although this ab initio modeling was applied to study the structure–property correlations of these particular surfactants, this procedure is quite general, as it does not use any adjustable parameters; thus it can be applied to predict analogous properties of new structures for anti-biofouling functionalization of membranes.
2. Computational Method
According to the computational time necessary for the calculation of the considered properties, three different methodologies, molecular mechanics (mm), quantum/molecular mechanics (qm/mm) and a pure quantum-based approach (DFT), were used in this work. These methodologies were used in sequence, as follows: (i) a conformational analysis was first carried out through an MM approach; (ii) the most stable conformers for each of the surfactants were then used as initial geometries in successive QM/MM optimizations, to consider the interactions with explicit water molecules; and (iii) pure quantum optimizations were performed and the calculation of the target properties was achieved.
The investigated surfactants, at room temperature, show a huge number of conformers that exponentially increase with the aliphatic chain length, thus a huge number of structures should be considered in the quantum calculations. Moreover, for the systems with different local minima, the choice of the initial geometry for the quantum optimizations is important, as it controls the search for the nearest minimum. Thus, a MM based conformational analysis was first performed using the universal force field and Avogadro code [
22], an application of the Open Babel platform [
23]. For each surfactant, the most stable structures were found analyzing a sample of 30 × 10
4 conformers through a weighted rotor search. Then, the thirty most stable conformers were optimized at an MM level, as all the analyzed conformers, using the conjugate gradient algorithm, 10
4 maximum steps, and a threshold of 10
−6 for the energy convergence. The most stable conformers were grouped as a function of their maximum molecular length (
h), so that the structures belonging to a particular group showed a very similar length. The group with the maximum number of conformers was named
pcg (i.e., the most populated conformers group), while the group containing the conformers with smaller
h among the thirty most stable was called
sc. A structure from the
pcg was chosen as the initial geometry for the successive QM/MM optimizations (
pcg initial geometry) and, for each surfactant, another structure was chosen from the
sc group (
sc initial geometry). The ratios between the
pcg and
sc populations, R
popul, were showed in
Table 1. Although the
pcg group is markedly more abundant than
sc, one structure from the latter group was chosen, because the maximum length is an important property for the antibacterial activity of these compounds; thus the shortest conformers, chosen as the initial geometry for the quantum optimizations, are important as lower bounds.
The quantum calculations were performed using NWChem code [
24] in the framework of Density Functional Theory using the Grimme’s DFT-D3 dispersion correction [
25,
26]. The hybrid energy functional Becke–Lee–Yang–Parr (B3LYP) [
27] was employed in addition to linear combinations of Gaussian-type orbitals. Double-ζ orbital bases were used for the carbons, and double-ζ bases with a polarization function were used for the O, N, and H atoms. The coulomb and exchange-correlation potentials were numerically integrated on an adaptive grid with accuracy equal to 10
−5. The threshold for the energy convergence in the self-consistent field procedure was set equal to 10
−6 (a.u.), while the root mean square of the electron density was equal to 2 × 10
−5 (a.u.). The QAS electronic proprieties were calculated with good results using B3LYP and s similar or smaller basis set. Several works, such as [
28,
29,
30,
31,
32], used B3LYP to calculate the partial charges of cations and neutral molecules. In reference [
28], atomic charges were calculated using the hybrid B3LYP, obtaining values of the same quality as the correlated
ab initio methods. Moreover, in reference [
32], the B3LYP and triple-ζ basis set were used to obtain the electrostatic potential around the molecules and partial charges, while in reference [
30], the B3LYP calculations of atomic charges were correlated to a conformational study. Finally, B3LYP with 6-31G (d) basis set was also applied to calculate a set of quantum chemistry properties of eleven quaternary ammonium compounds [
33]. In particular, in this work, DFT-based descriptors were used to derive quantitative structure–activity relationships enabling the correlation of the computed properties with the toxicity of quaternary ammonium compounds on green alga
Chlorella vulgaris with good results [
33]. This investigation showed that the alkyl chain lengths and the most positive net atomic charges on the hydrogen atoms of quaternary group, as well as entropy, are the major descriptors (properties) governing the structure–activity relationship models.
The optimizations of the quantum region of the investigated surfactants and the entire geometries in the successive pure DFT optimizations were performed using the analytical energy gradients and the quasi-Newton algorithm with an approximate energy Hessian. The maximum and root mean square gradient thresholds of 4.5 × 10−3 and 3.0 × 10−3, respectively, were used as the convergence criteria in conjunction with the maximum and root mean square of the nucleus Cartesian displacement vectors, equal to 5.4 × 10−3 and 3.6 × 10−3 (a.u.), respectively. The QAS electronic proprieties were calculated with good results using B3LYP and a similar or smaller basis set.
The interactions between the surfactant and water molecules may change both the solute geometry and the charge distributions, thus the QM/MM optimizations were carried out to take into account this effect. The model in
Figure 3 was used in the QM/MM calculations. In particular, the CH
2N(CH
2CH
3)
3 group was considered as the quantum region, while the rest of the surfactant was considered as the classical part, in addition to the water molecules randomly collocated around the solute (
Figure 3). This model is consistent with the environment encountered by the AATEABs, as some of them are used as surfactants to prepare oil/water polarizable bi-continuous micro-emulsions [
20]. Thus, self-assembled surfactants are not consistent with the oil/water micro-emulsion environment. For the same reason, periodic boundary conditions were not used. The dimensions of the box in
Figure 3, correlated with the water content, depend on the surfactant sizes; however, appropriate dimensions were chosen so as to guarantee a sufficient number of water molecules, in order to investigate the possible water bridge between the carbonyl and ammonium hydrogens. Specifically, for all of the investigated AATEABs, a box with the following dimensions was considered: L
x = 1.5 nm, L
y = 1.5 nm, and L
z = 2.5 nm. The geometry of the quantum region was optimized according the aforementioned procedure, while the classic region was optimized by using an Amber99 force field; the steepest descent algorithm and a threshold for the total energy convergence of 10
−6 were also used.
The structures optimized at the QM/MM level were then used as an input for the final DFT optimizations, but in this case, the effect of the bulk solvent was considered through the quantum continuum COnductor-like Screening MOdel (COSMO) [
34]. While explicit water molecules were considered in the QM/MM calculations, in COSMO, the solvent is considered as continuous, characterized by a specific dielectric constant, with the solute placed in a cavity defined by the van der Walls radii of its atoms. Herein, the surfactants and a few water molecules closer to the hydrogens of the ammonium group were considered as solute, and the relative dielectric constant was set equal to four, in order to simulate the effect of the confined water molecules.
The partial charges were calculated using both the Löwdin and the electro static potential (ESP) methods [
24], in addition to a larger basis set for all of the atoms of the surfactants (i.e., 6-311*). The ESP method evaluates the atomic charges from the fit of the quantum mechanical electrostatic potential of the surfactant on a grid point centered on each atom.
Different empirical formulas are available in the literature in order to calculate the maximum length (
h) as a function of the molecular weights. However, these formulas are not applicable for the conformational conformers that obviously show the same molecular weight. Furthermore, these formulas depend on the specific experimental conditions used, thus they are not general. Herein,
h and the molecular aspect ratios were calculated by a bespoke algorithm previously validated [
35,
36,
37] without resorting any empirical parameters, but only using the QM/MM and full DFT optimized geometries. Briefly, the effective diameter and key molecular sizes are evaluated according to specific dimensions, as follows: the height,
h, and size of the minimum rectangle enclosing the orthogonal projections of all of the atoms perpendicular to the principal molecular axis. The height,
h, is defined as the distance between the two most distant atoms, taking into account their van der Waals radii. The molecular size, d
min, is defined as the smallest size of the rectangle enclosing the orthogonal projections, while the second dimension of this rectangle, d
max, is used to calculate the aspect ratio (i.e., h/d
max). Details on the bespoke algorithm used for the calculations and its validation can be found in reference [
37].
A flow chart is reported in
Scheme 1 to summarize the computational methodology used to investigate the relationships between surfactant’s structure and proprieties correlated to the antibacterial activity.
As shown in
Scheme 1, the computational procedure is quite general and can be applied to predict the analogous properties of functional groups, in order to improve the biofouling resistance of the membrane surfaces.
3. Results and Discussion
In addition to the five synthesized AATEABs, surfactants with
n = 10, 8, and 4 were also studied. After the QM/MM geometry optimizations, the molecules of water formed a cluster around the surfactant, as shown in
Figure 4, with an average distance between the solute atoms, and the nearest water molecules are slightly longer than 3 Å. As a result, the water molecules do not form strong hydrogen bonds that are able to bend the surfactant or act as thickness between these groups with carbonyl or with ammonium; in fact, water molecules were not found between carbonyl and ammonium, as shown in
Figure 4a.
Comparing the optimized structures obtained from initial geometries belonging to the
pcg (
Figure 4a) and
sc (
Figure 4b), respectively, the interaction between the quantum part and the carbonyl of the surfactant is mainly a hydrophobic (dispersion) interaction. It is important to emphasize that this analysis was carried out using the both the QM/MM optimized structures (
Figure 4a,b) and those obtained from the pure DFT calculations, in which all of the surfactants were considered as a quantum system, in addition to the dispersion correction and solvent effect.
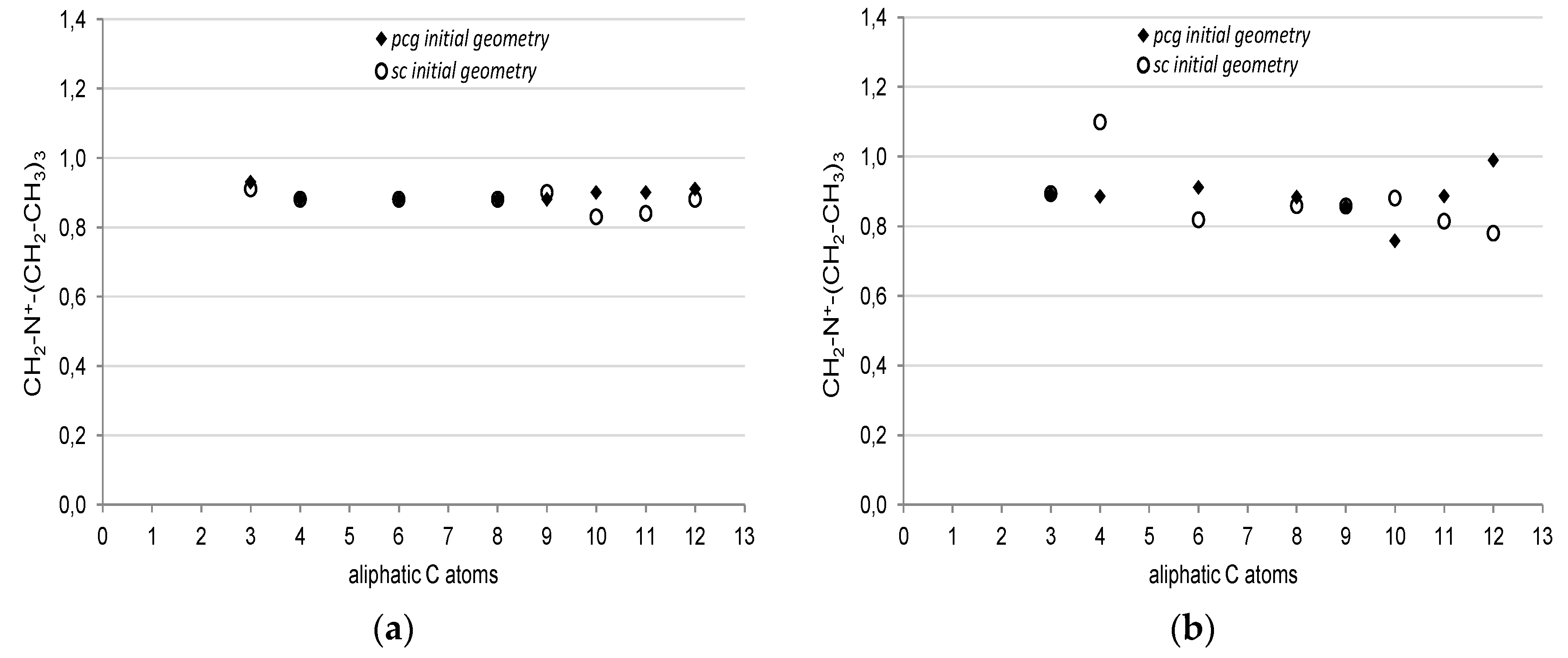
The average charges of the ammonium carbons and hydrogens, as well as the charge of nitrogen are plotted in
Figure 5 as a function of the number of C atoms in the aliphatic chain, while, in
Figure 6, the ammonium net charge, defined as the algebraic sum of the partial charges of its atoms, is reported. Diamonds refer to the values coming out of the optimizations, where the
pcg structures (i.e., initial geometries from the most populated conformers group) were used, while circles refer to the values obtained from the optimized geometries where the shortest isomer (conformer with the shorter h value) for each surfactant was used as the initial geometry.
The average partial charges of C and H do not depend on the number of aliphatic carbons, as well as the Löwdin charge of N, in contrast, the ESP charge of the latter varies in an oscillating way. The conformation of the aliphatic chain affects the ESP charges of C and N, as shown in
Figure 5d,f, conversely, the Löwdin charges do not depend on the conformer structure chosen as the initial geometry.
The ESP net charge of the ammonium group,
Figure 6b, changes with the number of C atoms in an oscillating way around 0.88 a.u., while the Löwdin charges remain almost constant, as can be seen in
Figure 5a. It is worth noting that the ESP charges depend on the conformer chosen as the initial geometry (
Figure 5b). This is physically consistent, because the ESP charges reproduce the quantum electrostatic potential of the entire molecule, thus the conformation affects the potential, and in turn, the ESP charges; conversely, the Löwdin charges depend on the nature of the chemical bonds (i.e., on the electron density). This means that the bonds in the ammonium head do not change with the increase of the aliphatic chain. The surfactant can bend over itself, thus the carbonyl group can interact through an intra-molecular interaction with the ammonium group. The results in
Figure 6b show that this happens for the longer surfactants, and markedly for the C4. In
Figure 7, the optimized geometries of the C4 surfactant obtained from the DFT and COSMO calculations are shown. In particular, a
pcg conformer was used to obtain the structure of
Figure 7a, while the structure of
Figure 7b was obtained using an initial geometry belonging to
sc. A weak hydrogen bonding or electrostatic interaction is present between C=O and a hydrogen of the ammonium group (
Figure 7b), in fact the C=O
...H distance is 2.895 Å. This interaction, absent in the structure obtained starting from a
pcg conformer, can explain the gap between the values of the ammonium charge (
Figure 6b and
Figure 5d) of C4 obtained when
pcg and
sc conformers are used as initial geometries in the quantum optimizations. As pointed out, the antibacterial activity of the PQASs is correlated with the interaction between the ammonium positive charge and the LPS (peptidoglycan) thin layer of the bacteria. Although the number of aliphatic carbons influences the AATEABs antibacterial activity, as measured in reference [
19], this influence is not correlated with an increase in the positive charge on the ammonium group.
The maximum length of the AATEABs as a function of carbons in the main chain was calculated and shown in
Figure 8, while
Figure 9 reports using the structures of the surfactants optimized at a QM/MM level. Similar to previous Figures, the diamonds refer to the
h values obtained from the optimizations, where the
pcg isomer is used as the initial geometry, while circles indicate values derived from optimizations where the shortest isomer is used.
Unlike the partial charges, a different trend was obtained for the maximum length of the surfactants.
Using the
pcg initial geometries,
h increases non-linearly as the number of aliphatic carbons increases, whereas when considering
sc as the initial geometry, the maximum length does not increase. As molecular conformations, water effects, and dispersion interactions were considered in the modeling, the trends in
Figure 8 and
Figure 9 are not trivial; the winding of the main aliphatic chain with the increase of the carbon number was in fact taken into account. The difference between the
h values obtained using the
pcg and
sc initial geometries widened, going from C3 to C12, however, the average value increases with the number of C atoms in the aliphatic chain. The surfactant should show an adequate length so that the positive ammonium group can effectively interact with the bacteria LPS structure; this distance is estimated at 1.5 nm [
38,
39]. Considering this threshold as a reference, the trends shown in
Figure 8 and
Figure 9 are in good agreement with the measured antibacterial activity of the AATEABs [
19]
The suppressive effect due to the solvated polyanions interacting with the ammonium group is other aspect that should be considered; PQASs with alkyl chains capable of protecting the quaternary nitrogen showed a better antibacterial activity [
10,
11]. For the investigated surfactants, this shielding effect can be correlated to the molecular aspect ratio.
A molecule assumes spherical form when its aspect ratio tends to one, whereas it will be elongated when the latter gets closer to two or higher values. Thus, the aspect ratio of the surfactants was evaluated using both QM/MM and DFT optimized structures. In
Table 2, the QM/MM aspect ratios of the syntetized surfactants, shown in
Figure 2, were reported; the DFT calculated values were quite similar. The aspect ratio of C6 was near two when considering the structure obtained from a
pcg initial conformer, while the aspect ratio of C9 was 1.34. For C12, the aspect ratio, evaluated using the structure obtained from a
sc conformer, resulted 1.36, this is a good value for a shielding effect.
However, when using a
pcg conformer as the initial geometry, the aspect ratio was quite high. The maximum length of C12, obtained from a
sc conform optimization (
Figure 8), is bigger than the threshold of 1.5 nm, which is long enough for an antibacterial activity. As result, C9, C11, and C12 (
Figure 4b) show curved structures, with the ammonium group bent towards the polymerizable monomer, while keeping an adequate maximum length. This would make this group less exposed to interactions with the dissolved polyanions, thus the computated aspect ratios were also in good agreement with the measured antibacterial trend of these surfactants [
19]. The differences in the energy between the local minima of all of the surfactants were also calculated, and small differences were obtained. Most of the values fall between 0.7 Kcal/mol and 5 Kcal/mol, except for the C9 structures, which showed an energy difference near 10 Kcal/mol. Considering these energy differences, the thermal distributions [
40] of the thirty low-lying structures at T = 300 K will not provide more insights on the molecular shape and charges distribution than those obtained following the adopted computational procedure.

 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}