Understanding Heteroatom-Mediated Metal–Support Interactions in Functionalized Carbons: A Perspective Review


1
Dipartimento di Chimica, Università degli Studi di Milano, via Golgi 19, 20133 Milano, Italy
2
Institut pour la Maîtrise de l’Énergie—Université d’Antananarivo BP 566, 101 Antananarivo, Madagascar
*
Authors to whom correspondence should be addressed.
Appl. Sci. 2018, 8(7), 1159; https://doi.org/10.3390/app8071159
Submission received: 1 July 2018
/
Revised: 13 July 2018
/
Accepted: 14 July 2018
/
Published: 17 July 2018
(This article belongs to the Section Nanotechnology and Applied Nanosciences)
Abstract
:Featured Application
Surface engineering for catalysis and energy.
Abstract
Carbon-based materials show unique chemicophysical properties, and they have been successfully used in many catalytic processes, including the production of chemicals and energy. The introduction of heteroatoms (N, B, P, S) alters the electronic properties, often increasing the reactivity of the surface of nanocarbons. The functional groups on the carbons have been reported to be effective for anchoring metal nanoparticles. Although the interaction between functional groups and metal has been studied by various characterization techniques, theoretical models, and catalytic results, the role and nature of heteroatoms is still an object of discussion. The aim of this review is to elucidate the metal–heteroatoms interaction, providing an overview of the main experimental and theoretical outcomes about heteroatom-mediated metal–support interactions. Selected studies showing the effect of heteroatom–metal interaction in the liquid-phase alcohol oxidation will be also presented.

1. Introduction
Since their discovery at the end of the 20th century, nanostructured carbon allotropes such as fullerenes [1,2], carbon nanotubes [1,3], carbon nanofibers [4], and graphene [5] have attracted the interest of several researchers from disparate fields. Nowadays, the family of low-dimensional carbon allotropes includes numerous members [6], supporting a wide range of applications from electronics [7] to medicine [8], from optics [9] to environment protection [10], from energy storage [11,12,13,14] to catalysis [15]. While the synthesis strategies as well as the key structural features have already been extensively investigated, the focus has now shifted toward tuning the textural and electronic properties of these materials to optimize their performances in the pursuit of technological progress and manufacturing efficiency. Due to the nanometric sizes, the behavior of low-dimensional materials is dominated by surface phenomena; therefore, the design of carbon nanomaterials with tailored properties is essentially a matter of surface engineering. A common approach in surface engineering is functionalization, aiming to introduce a functional component that is able to confer novel properties to the material [16,17]. In most cases, the surface functionalization results in the incorporation of heteroatoms (commonly boron, nitrogen, oxygen, phosphorus, and sulfur) in the carbon framework [18,19,20,21]. Functional groups can be attached on the surface of carbon materials either by non-covalent or covalent functionalization [22,23]. In the former case, hydrogen bonding, π–cation, π–π stacking, π–anion electrostatic forces, hydrophobic interactions, and van der Waals forces are involved [24,25]. Covalent functionalization implies the formation of stronger interactions, and often it is associated with structural alterations (defects) in the pristine carbon materials [26,27,28,29]. Covalent bonds between carbon atoms and heteroatoms can be created during the synthesis (in situ doping) [30,31] or by post-treatment of carbon materials (post-synthesis doping) [32]. Whatever the strategy used, the functionalization causes drastic changes in the electronic [33,34], mechanical, and thermal [35] properties of carbon materials. For this reason, functionalized carbon materials have been successfully employed in catalysis.
The introduction of heteroatoms generates defects that alter the surface structure and act as anchoring points for metal nanoparticles or active sites. Furthermore, it has been recently reported that the incorporation of heteroatoms (N, B, P, and S) in the framework of ordered mesoporous carbons can affect the d-spacing (and consequently the thickness of framework walls) and the pore texture of the resulting mesostructures, still maintaining well-defined porosity and moderate surface area [36]. In particular, N and S-doping lead to a slight pore size decrease, while B and P-doping produce an increase in the pore size, according to the different incorporation mechanisms, which can proceed, respectively, with the swelling of the carbon framework (N and S incorporation) or with the incorporation of oxygen, acting to etch the carbon (B and P-doping).
In addition, heteroatom-doped carbon nanomaterials possess more pronounced acidic or basic [32] character compared to parent materials. Despite being promising metal-free catalysts [37,38,39], functionalized carbon materials represent suitable support for metal catalysts both in gas-phase and liquid-phase processes [40].
The modified local electron density and the presence of structural defects in the proximity of functionalities contribute to enhance the so-called metal–support interactions. The term strong metal–support interactions (SMSI) was coined by Tauster in 1978 in relation to the altered catalytic activity and chemisorptive properties of group VIII noble metals when supported on reducible oxides [41]. Over the years, the concept of SMSI was also extended to non-oxidic materials to describe the influence exerted by the support on the catalytic performances of metal nanoparticles (NPs) [42,43,44,45,46]. Subsequently, Verykios et al. invoked the dopant-induced metal–support interactions (DIMSI) to rationalize the role of doping cations in mediating metal–support interactions in the case of noble metal crystallites deposited onto doped supports [42,47,48]. In an extreme simplification, the support can be envisaged as a supramolecular ligand strongly interacting with metal NPs. From this point of view, functional groups on the support surface act as preferential interaction sites and as electron-donating and electron-withdrawing substituents.
The influence on the catalytic activity and selectivity can be essentially ascribed to three effects: (i) electronic effects, (ii) geometric effects, and (iii) dispersion effects (Figure 1) [46]. Electronic effects originate from the difference between the Fermi level (EF) of the metal and that of the support, resulting in an electron transfer aimed to equalize the EF at the metal–support interface [49,50,51]. The lattice mismatch between the support and the metal NPs induces changes in the shape of the nanoparticle to optimize the interaction with the support [52]. Consequently, these geometric effects are combined with variations in the exposed crystallographic planes and in the coordination number of surface metal atoms, which affect the catalytic performances. Finally, functional groups can act as anchoring sites, promoting the nucleation of metal NPs, and then, the uniform dispersion on the support [53,54,55,56]. However, electronic, geometric, and dispersion effects are interrelated, and it is not possible to clearly discriminate each single contribution.
Although the existence of metal–support interactions in carbon-supported metal catalysts has been confirmed by various characterization techniques, theoretical models, and catalytic results, their nature and the role of heteroatoms is still an object of discussion. The purpose of this review is to contribute to this ongoing debate by providing an overview of the main experimental and theoretical outcomes about heteroatom-mediated metal–support interactions. We begin by describing the main theoretical models proposed to account for strong metal–support interactions. Following this, the advances of characterization techniques in probing metal–support interactions are explored. We conclude with a collection of examples reporting the role of heteroatoms in enhancing metal–support interactions in catalysis.
2. Theoretical Studies on Heteroatom-Mediated Metal–Support Interactions
2.1. Basic Concepts for Understanding Heteroatom Mediated Metal–Support Interactions
The deposition of metal nanoparticles or clusters onto the surface of carbon support generates an interface accompanied by a local electrostatic potential perturbation. The entity of this perturbation reflects the strength of the interactions between metal and support atoms at the interface, and it is strictly related to the electronic structures of these two actors [51]. The electronic structure of solids is usually described in terms of electronic bands. The nature and width of the electronic bands can be evaluated considering the density of states (DOS), which are defined as the number of allowed electron states per volume at a given energy. Two parameters related to DOS are of paramount importance for understanding metal–support interactions: the Fermi level and the work function. The Fermi level (EF) is an energy state having a 50% probability of being filled, and it coincides with the electrochemical potential of electrons in the solid. The position of the Fermi level with respect to the D-band is an indicator of the D unoccupied density. The work function, Φ, is the minimum energy that must be furnished to the metal system to extract an electron from the Fermi level, EF, and transport it at infinite distance, when the metal surface is electrically neutral. According to the metal–semiconductor boundary-layer theory, when two phases are contacted, the thermodynamic equilibrium is attained. The equilibrium condition requires that the electrochemical potential and then the Fermi energy level of the electrons of the two solids is the same at the interface. The equalization of Fermi levels implies charge transfers across the interface from one material to the other, whose direction can be predicted from the analysis of the electronic structures. In cases where the work function of the electrons of the metal is larger than in the support, electrons will migrate from the support to the metal, until the Fermi level at the interface is equilibrated, and vice versa. This flow of charge sets up a partial positive charge on the support in the proximity of the interface, and an induced negative charge on the surface of the metal. Moreover, in the presence of the charge at the interface, the rehybridization of electron orbitals occurs, leading to changes in the D-band population and in the density of states at the Fermi level. These phenomena are at the basis of the metal–support interactions of the electronic type.
The introduction of heteroatoms in the support framework causes electron relocalization and local variations in the density of states of the support (LDOS, local density of states), thus producing an increase or decrease of the work function. This modification unavoidably influences the charge transfer, the hybridization between the electron states of the metal and those of the support, and definitively the metal–support interactions (Figure 2).
Theoretical simulations are a valuable technique to explore the phenomena occurring at metal–support interface. Binding energy, atomic distances, and relaxed geometries are useful descriptors can be derived from density functional theory (DFT). Molecular dynamics simulations based on a generic force field have been implemented to obtain information on the mobility of adsorbed metal species. Molecular orbital analysis provides a useful indication of the electron density and charge distribution.
2.2. Metal Single-Atom Adsorption on Doped Carbon Surfaces
Among the several functionalizations, the substitution of carbon atoms with nitrogen or boron has received great attention. In the case of substitutional B doping, a carbon atom is replaced by an atom with one less electron; consequently, sharp and localized states below the Fermi level (i.e., in the valence band) are generated. The electronic structure modifications induced by nitrogen doping are more complex, and they depend on the possible bonding configurations for N in graphitic networks, as detailed below.
Acharya et al. [57] provided a deep insight into the adsorption of Pt and Ru atoms onto isolated B and N-doped graphene surfaces. C18, C16B2, and C16N2 were selected as the carbon structural models for the adsorption of metal species respectively on pristine, B-doped and N-doped graphene. In addition, the co-presence of boron and nitrogen (co-doping) with different connections (heteroatom–carbon or heteroatom–heteroatom bonds) was also investigated. According to DFT calculations, the incorporation of boron atoms in the graphene lattice produces a lowering of the Fermi energy (from −2.25 eV to −3.43 eV) and a consequent increase in the work function from 4.31 eV to 5.43 eV. Looking at the projected density of states (PDOS) for the p-orbital (Figure 3), a lower Fermi energy implies an augmented number of states (bonding and antibonding states) close to the Fermi energy and the appearance of more antibonding empty states above the Fermi level, which can help in minimizing the antibonding repulsions during the adsorption of metal species on the surface. In other words, a strong hybridization between the platinum d-orbitals and boron p-orbitals occurs. The essential consequence is the strengthening of the interaction between the B-doped surface and metal atoms.
Computed values for binding energy of both Pt and Ru atoms confirmed this hypothesis, since the highest values are associated with the adsorption on boron-modified surfaces. In the presence of nitrogen atoms, the increased Fermi energy (−1.27 eV) and the lower work function (3.37 eV) lead to the presence of fewer antibonding empty states close to the Fermi energy, and thus to a weakening of the interaction between metal and the nitrogen atoms. However, although the hybridization between the nitrogen p-orbital and platinum d-orbital is very weak, the presence of nitrogen atoms can result in an enhancement of the interaction with Pt compared to undoped surfaces. The promotion effect of nitrogen can be ascribed to the activation of the neighboring carbon atoms, whose reciprocal π–π interactions are weakened by the lower electron delocalization induced by the close proximity of nitrogen atoms with large electron affinity [58]. An analogous effect can be envisaged in defective carbon surfaces, where the presence of curvature, vacancies, or edges produces a disruption of the delocalized electronic structure of the graphene surface. Consequently, the actual interaction energy is often the balance between electronic effects deriving from both doping and local geometric strain. As an example, comparing the Pt adsorption energy on doped single-walled carbon nanotubes B(N)-SWCNT(6,6) and B(N)-SWCNT(8,0), in the former system, a more moderate enhancement was unveiled compared to the SWCNT(8,0), which was probably due to the reduced curvature in SWCNT(6,6) [59].
In the co-doped surfaces, the effect of nitrogen and boron doping are mixed; therefore, the interaction of Pt and Ru results stronger close to boron atoms and weaker close to nitrogen atoms.
However, it is worth demonstrating that the coordination of nitrogen species in the carbon framework can determine the interaction of metal atoms with nitrogen-doped supports. Indeed, when a nitrogen atom replaces a carbon atom of graphitic plane, it will be bonded with three carbon neighbors, thus generating a so-called graphitic nitrogen: a sp2 nitrogen species. Nevertheless, graphitic nitrogen is not the unique species that can be created in an N-doped surface. The substitution of a carbon atom with nitrogen can occur according to almost three primary structural models differing in their nitrogen bonding configuration (Figure 4): pyridine-like (Npyr), pyrrole-like (NH), and graphitic (Ngr) [21]. Pyridinic nitrogen refers to sp2 two-fold coordinated nitrogen atoms contributing one p electron to the π-system. It could be incorporated as a vacancy (Nv) or as atom on an edge site (Ne). Nitrogen atom in pyrrole-like configuration is an sp3 coordinated (usually in a five-membered ring) species sharing two p electrons in the π system. Doping with pyridine-like, pyrrole-like, and graphitic nitrogen species impacts differently on the electronic band structure of graphitic carbon materials, having unavoidable implications for the interactions with adsorbates. The adsorption of single atoms of Pt-group metals on graphitic surfaces containing nitrogen species with different coordination has been modeled by DFT. The results from theoretical modeling unanimously identify the following ascending order of the binding energies of noble metals: N(graphitic) > N(pyrrolic) > N(pyridinic) [60,61,62].
Bulushev et al. [63] modeled the adsorption of a Pd atom on four different nitrogen forms: graphitic (Ngr), pyrrolic (NH), pyridinic at the monovacancy boundary (Nv), and pyridinic at the edge (Ne) nitrogen atoms, probing different adsorption sites in the graphene fragment.
The obtained relaxed structures and binding energies revealed that the Pd atom preferentially interacts with pyridinic nitrogen. A similar behavior was observed with other Pt-group metals (Pt, Ru). In general, all Pt-group metal atoms strongly interact with Npyr sites, although the interaction with the edge Npyr sites is significantly weaker than the one with the vacancy (mono or di) Npyr atoms. This evidence can be rationalized considering the number and the stability of the bonds generated upon the interaction between metal and vacancy Npyr atoms. In particular, the highest affinity for divacancy Npyr atoms can be related to the formation of four stable bonds among the central metal atom and the adjacent heteroatom and carbon atoms. The strong interaction between metal atoms and monovacancy Npyr atoms is associated with the breaking of a C–C bond in the pentagon ring, the formation of three new bonds, and the concomitant creation of three more stable six-member ring systems. Differently, just two and one new bonds are formed when metal atoms are adsorbed on Npyr atoms on the armchair or zigzag graphene edge, respectively. As expected, the interaction with graphitic nitrogen is not energetically favorable. Pd and Pt atoms do not bind to this site, whilst they prefer to migrate to the neighboring carbon sites and locate above the center of the C–C bond, forming a bridge with two carbon atoms. Ru is the only one that is able to interact directly with the Ngr nitrogen, forming a bond with this atom and two neighboring carbon atoms. The sequence for the binding energy of metal atoms with a particular site was Ru > Pt > Pd, which is in agreement with Acharya’s results [57]. Conflicting conclusions were deduced regarding the influence of the delocalized π-electron system of the graphene surface on the electron density redistribution in the case of Pt and Pd atoms. The comparison of the electron density surfaces for N-doped graphene fragments before and after the interaction with the Pd atom suggests that the interaction with Pd induces a more positive charging of N atoms compared to the initial states. Otherwise, looking at natural bond orbital (NBO) analysis, the interaction of the Pt atom with Npyr sites imparts a partial positive charge to the metal center (except for the multiple interactions with two Npyr atoms at the zigzag edge). Conversely, electron density is transferred from the support to Pt, when the latter is bonded close to Ngr.
It is obvious that also the electronic structure of the metal plays a central role. According to Hammer and Nørskov [64], the interaction of the transition metals tends to increase from right to left, while it decreases as one progresses down (from top to bottom) a group on the periodic table. The number of D electrons, and then, the position of D-band center related to the Fermi energy, play a crucial role.
This appears more evident when the study is extended to other transition metal atoms. An et al. [59] used density functional theory (DFT) calculations to investigate the adsorption of 11 transition metal atoms (i.e., Au, Pt, Ru, Pd, Ag, Fe, Co, Ni, Cu, W, and Ti) on the sidewalls of boron and nitrogen-doped single-wall carbon nanotubes (B-SWCNTs and N-SWCNTs). In general, the highest binding energies were obtained for Sc (3d14s2), Ti (3d24s2), and Fe(3d64s2), which was likely due to their capability to easily lose one or two electrons for reaching the full or half-filled steady states [65]. Again, an enhanced binding energy was observed for boron-doped surfaces, whereas N-doping produces a slighter increase in the interaction energies. In addition, a quenching to zero of the spin-magnetic moment (μ) of the individual metal atoms was predicted for almost all of the species (except Fe and W) upon their bonding on doped carbon nanotubes [59]. Mulliken population analysis [65] indicated the occurrence of charge transfer ranging from 1.32 e (for Sc) to 0.40 e (for Cu and Pt).
Ab initio molecular dynamics (AIMD) simulations allowed investigating the short-time mobility of Pt and Ru clusters on pristine and boron-doped carbon over a range of different temperatures for a very short period. The evaluation of the mean squared displacement (MSD) provided useful details on the stability of metal atoms at increasing temperature. As expected, metal atoms tend to migrate easier on an undoped surface compared to the boron-doped one. However, the differences in MSD over the two surfaces became even less evident as the temperature increases (up to 873 K).
In any case, AIMD operates in a restricted simulation time and size of the system, which are not sufficient to obtain results with general validity. Classical molecular dynamics simulations could guarantee a longer observation time. However, a proper analytical potential should be selected to adequately take into account the metal–support interactions.
Besides tuning the interaction with the adsorbed metal species, the doping can also address the insertion of oxygen functionalities. The adsorption and the stability of epoxide and hydroxyl groups on undoped and B-doped graphene surfaces has been considered [57]. Epoxide functionalities are created under the adsorption of oxygen atoms on the bridge site connecting the boron and carbon atoms. The preferential site for hydroxyl group adsorption was the boron on-top site. Furthermore, the B-doping positively affects the adsorption of oxygen functionalities. A shift of the Fermi level to lower energy and an intensification of the bonding and antibonding states close to the Fermi level occur, contributing therefore to an increase of the binding energies compared to a pure graphene surface. The functionalization with oxygen-containing groups does not have an univocal effect on the metal adsorption properties: the adsorption energies of Pt or Ru atoms rise in the presence of O-groups for pure graphene, whereas a detrimental effect is observed when epoxide and hydroxyl functionalities are incorporated in B-doped graphene. According to the Bader analysis, the metal atoms assume a positive charge, while the functional groups have a net negative charge.
2.3. From Single Metal Atoms to Metal Clusters
The single atom systems described above represent just ideal situations, which can be used as a model for the interpretation and the prediction of the more realistic metal clusters or metal nanoparticles. In multi-atom systems, additional interactions can be established between adsorbed atoms that are topologically close (adatom–adatom interactions). A general relationship approximating the binding energy (BE) of metal nanoparticles or clusters with carbon surfaces has been proposed by Efremenko [66], who proposed a linear dependency described by the equation: BE = an + b, where (n) is the number of metal atoms in the cluster, and (a) is an index of surface unsaturation. The intercept (b) accounts for the interactions between the first-layer metal atoms and the carbon surface, and it is strictly related to the electron and structural properties of the metal–support interface. The metal–support interactions between carbon and metal NPs have been disclosed by density functional theory (DFT) supported by experimental investigation. Acharya et al. examined the adsorption of several different metal clusters (Pt6, Pt10, Pt32, Pt2 Ru4, and Pt4Ru6) on pristine, boron (B), nitrogen (N), and B,N-co-doped graphene surfaces [57]. The trend in binding energies reproduced the predicted one for the monoatomic counterparts (Pt and Ru atoms): the greatest interaction involved metal clusters and boron-doped graphene, while a weak interaction was produced on pristine graphene. Monitoring the minimum distance between metal clusters and graphene as well as the bond lengths in the metal clusters and the graphene models before and after adsorption provided precious details on atom displacement and surface rearrangement induced by the interaction between metal atoms and carbon atoms at the surface. In general, if the interaction is stronger, the minimum distance will be smaller. Calculated cluster–surface distances reflected the binding energy trend and therefore the interaction order. The higher stability of metal clusters when adsorbed on boron-doped surfaces compared to pristine graphene was confirmed also by AIMD simulations, which predicted lower MSDs for Pt and Ru clusters on B-doped graphene compared to the ones immobilized on pristine graphene. Moreover, as expected, the diffusion of clusters depended on the size of the clusters: the larger the cluster, the lower the diffusion.
Concerning N-doping, analogously to single atoms, metal clusters interacted preferentially with pyridinic nitrogen species [67]. The strong covalent chemical adsorption between modified surfaces and metal clusters affected also the density of states of the metal, carbon, and heteroatoms, and the charge distribution. As an instance, the total density of states (TDOS) of Pd in Pd@PdO clusters is deeply altered after adsorption on N-doped graphene, and simultaneously also, the TDOS of C and N atoms are changed after interacting with Pd@PdO shell clusters [68]. Consequently, the electron density was also affected by the adsorption. Electron density difference (EDD) diagrams for Pd@PdO–N-graphene revealed an electron density enhancement around an N atom associated with an electron density depletion around a C atom, indicating that C–N is weakened due to the interaction with Pd atoms. In addition, according to the Mulliken population analysis, an electron transfer process occurred from Pd cluster to support. However, these variations are not observed when Pd@PdO clusters are immobilized onto pristine graphene. This highlights the central role of heteroatoms in activating the carbon atom of the support and mediating the interaction with the metal clusters. QUAMBO (QUAsi-atomic Minimal Basis set Orbitals) charge analysis can be alternatively used to gain theoretical evidence for charge transfer [69]. Rao et al. applied periodic DFT and QUAMBO charge analysis to explore the interactions at the metal–support interface in the case of Pd nanoclusters varied in size and deposited onto carbon nanofibers with different graphitization degrees and O-functionality content. As expected, DFT calculations evidenced a significantly stronger interaction between Pd clusters and the carbon support, increasing the functionalization degree and decreasing the graphitization degree. QUAMBO analysis revealed the occurrence of charge transfer from the metal to the support, independently of particle size, for the metal atoms close to the interface, but these effects rapidly dissipated with distance from the surface. Combining these results with the information deduced from characterization experiments, the authors concluded that the Pdδ+ contributions are consistent with a partial charge localized and distributed over the first one to two atomic layers at the palladium–carbon interface, while Pd atoms in the second layer are negatively charged through a compensation effect.
Transition metal-based systems are not the unique ones; their strong interactions with the support have been investigated. The confinement of Li2S and polysulfides (Li2Sx) on the conductive carbon host framework is an attractive challenge to be faced for the development of Li–S batteries with superior performance [70]. The Li–S battery is an emerging rechargeable device with high specific energy, whose commercialization is currently limited by some drawbacks, such as the so-called shuttle effect. The latter is related to the loss of active mass, and it can be minimized by controlling the phase evolution of S at the interface and the interactions between the conductive host framework (typically carbon-based materials) and S-containing guests. From this point of view, the introduction of N-containing functional groups in the carbon hosts is expected to improve the interaction and stabilize the S guests. To predict and rationalize the effect of N-doping on the conductive carbon host and in general in the Li-S cell, host–guest interaction has been simulated by performing DFT calculations devoted to describing the interaction between three surfaces (pristine carbon nanotubes, CNT, Npyr-doped CNT, and Ngr-doped CNT) and several S-containing guest models (S atom and Li2Sx (1< x <4) clusters with high polarity and positive charged terminal Li atoms). Relaxed geometries and binding energy values revealed a comparable trend with respect to transition metals. Also in this case, S-containing species interacted more strongly with N-CNTs than pure CNT. The most stable configuration for the S-species interacting with Npyr consisted of a direct bond between highly positive charged Li atoms and the electron-rich Npyr center. In the case of Ngr, S-species are adsorbed on the neighboring carbon atoms, and the nitrogen center acts only as promoter, activating carbon atoms. These promising results demonstrated that N-doping assures the anchoring and the permanent trapping of S-species, and it strengthens the host–guest interactions, thus reducing the loss of active mass. In addition, N atoms can promote electron transfer in the electrocatalytic processes.
To generalize and summarize, the role of heteroatoms in mediating metal–support interaction is the result of the interplay of different electronic effects. It includes several contributions:
- (a)
- the match between the electronic structure of the metal and that of the support
- (b)
- the number of bonds that are created with the surface atoms (carbon and heteroatoms)
- (c)
- breaking of the long-range electronic delocalization
- (d)
- local surface deformation, corrugation, and defect introduction
- (e)
- electron density redistribution and charge transfer
- (f)
- the presence of atoms near the adsorption sites that are available to saturate the eventually dangling bonds.
2.4. Predicting the Effect of Heteroatom-Mediated SMSI on the Catalytic Performances
The catalytic consequences of the electronic modification induced by doping can be essentially ascribed to two factors: (i) modifications of the metal adsorption properties because of electronic structure changes; (ii) changes in particle shape; and (iii) increased metal dispersion.
2.4.1. Effects on Molecule Adsorption and Metal Cluster Shape
The adsorption of reactant on the metal surface is the first step of a catalytic cycle, occurring on supported transition metal nanoparticles. The description of the formation of a chemical bond between a surface and a molecule can be performed in terms of a d-band model. According to this model, the bond formation at a transition metal surface is the result of the interaction between the valence states of the adsorbate molecule and the S and D states of the transition metal surface. The interaction of valence states with the S states of the metal produces a shift and broadening of the adsorbate. These renormalized valence states interact with the metal D states, thus generating separate bonding and antibonding states. The strength of the bond is determined by the relative occupancy of these states. The filling of antibonding states results in a bond weakening, which is a function of the energy of the antibonding states relative to the Fermi level. The higher the antibonding states are in energy relative to the Fermi level, the lower their filling degree, and the stronger the bond. Since D antibonding states are always above the D states, the evaluation of the D state energy (the center of the D band) relative to the Fermi level can give a rough idea of the bond strength.
Consequently, the variation in the position of the metal D band induced by heteroatom mediated metal–support interaction will affect the strength of the adsorbate bonding.
Kim et al. [71] compared the adsorption of H2 on transition metal (TM) supported on boron and pyridine-like nitrogen-doped graphenes. The binding energies were similar for adsorption on TM/N-graphene and TM/B-graphene with values slightly smaller than the one on TM/graphene. The number of maximal H2 adsorption is lower for TM/N-graphene compared to B-doped and pristine graphene. These trends can be explained considering that upon H2 adsorption, electrons are transferred from metal D orbitals to the antibonding state of H2 (backdonation). As described in the previous section, the interaction between transition metals and doped surfaces is stronger than the interaction with undoped surfaces. Consequently, when metals are supported on N or B-containing graphene, their back-donation ability is reduced, and then the H2-metal binding is weakened.
The adsorption and decomposition of formic acid have been studied on transition metal (Pt, Pd, Ru) NPs supported on nitrogen-doped graphene. The interaction with a couple of edged pyridinic nitrogen atoms was the most energetically favorable, and promoted the formic acid decomposition to CO2 and H2 (except in the case of Ru). The easy decomposition of formic acid is favored by the proximity of a formed adsorbed hydrogen atom and a hydrogen atom of the carboxyl fragment, which are directed toward each other. This is an ideal configuration for the formation of hydrogen and CO2 molecules [72].
The position of the D-band is a fundamental parameter to describe the adsorption of molecules on metal surfaces, since it can be correlated with the adsorbate binding energy. However, this is not always true. Kim et al. [73] demonstrated that a direct correlation between the D-band center and the adsorption energy of H2 and CO is not applicable in the case of Pt nanoparticles supported on nitrogen-doped graphene. Otherwise, in these cases, an affordable correlation has been disclosed between the D-band center and the difference in the averaged adsorption energies between H2 and CO, ΔEad = Ead(CO) − Ead(H2). Authors also reported the weakened adsorption strength on Pt NPs supported on doped graphene. This difference in adsorption energy was more pronounced for CO adsorption than H2, and it is a consequence of the strong metal–support interactions. The latter was also responsible for the bond rearrangement and change in the cluster morphology to maximize the contact with heteroatoms present on the support surfaces (Figure 5). In any case, the impact of metal–support interactions decreases as the particles become larger [57].
2.4.2. SMSI and Metal Dispersion
The interactions between the metal phase and the support at the interface can also determine the dispersion of metal clusters onto the support surface, and finally the particle sizes. In fact, the nucleation and the growth of metal clusters are governed by a delicate balance between metal cohesion and metal support interactions: if the former prevails over the latter, clustering and agglomeration will preferentially occur. Conversely, when the metal binding energy exceeds the cohesive energy of the bulk metal, metal aggregation is prevented, and a high metal dispersion is achieved. This is the case of some transition metals (Sc, Ti, and V) deposited on nitrogen-doped graphene [65]. When pyridine-like functionalities are introduced on the surface of graphene, highly localized acceptor-like states close to the Fermi level are created, leading to a stronger interaction between graphene and metal atoms, and thus favoring a high metal dispersion. The DFT method has been implemented to determine how far the nucleation and growth of Pt NPs onto a carbon matrix (described by a 4 × 4 supercell of graphene) are affected by the presence of nitrogen (pyridinic, pyrrolic, graphitic nitrogen) and and/or sulfur (thiophenic defects) [74]. For this purpose, the interaction energy of Pt on doped graphene surfaces was evaluated in terms of adsorption energy. In all of the cases, a higher interaction energy was obtained for nitrogen or sulfur singularly doped surfaces compared to pristine graphene, and in particular, the following interaction order was observed: N pyrrolic > N graphitic > thiophenic > N pyridinic > graphene. Surprisingly, a weaker interaction and a lower stabilization of Pt nuclei were predicted on a surface co-doped with nitrogen (pyrrolic defects) and sulfur (thiophenic defects). In particular, probing the interaction energy for N and S co-doped surfaces with different dopant location, it appears evident that the interaction and the Pt stabilization decrease by increasing the closeness of the two hetero-defects (nitrogen and sulfur). It seems like sulfur acts as preferential anchoring site for Pt, which is probably due to the electron transfer to the Pt atom.
Metal phase can be deposited on the support according to several strategies. Ning et al. focused on the influence of a catalyst synthesis strategy on the metal–support interaction of Pt on nitrogen-doped carbon nanotubes (Pt/NCNTs), comparing ethylene glycol (EG) reduction, sodium borohydride reduction, and impregnation H2 reduction [75].
In the case of the EG reduction method, neutral Pt NPs preferentially interacted with graphitic nitrogen, whereas in sodium borohydride reduction and impregnation H2 reduction, the adsorption of charged metal ion precursors is favored on pyridinic nitrogen.
3. Probing the Metal–Support Interactions at the Interface
Theoretical simulations can help predict and understand metal–support interactions, and in many cases, they are aimed at supporting experimental evidence deriving from characterization and catalytic tests.
Gaining a deeper insight on metal–support interactions and on the role of surface doping requires a huge amount of information to be collected. The combination of different complementary analytical techniques that are able to probe the metal–support interface then represents the most suitable approach. Considering the nature and the effects of SMSI, the characterization process should provide details on the electronic phenomena and structural rearrangements occurring at the metal–support interface.
Electron microscopies are the ideal tools to give a direct illustration of the morphology and structure of the metal–support interface and describe the metal dispersion on the support.
Among spectroscopic techniques, X-ray photoelectron spectroscopy (XPS) and X-ray absorption spectroscopy (XAS) are the most used ones. The former elucidates the electronic properties of the atoms exposed at the interface. XAS is usually divided in two categories: X-ray absorption near-edge structure (XANES or NEXAFS, including metal K-edge, metal L-edge, and ligand K-edge) and extended X-ray absorption fine structure (EXAFS). Precious evidence on the local structure, the individual environment of atoms, binding geometry, binding distances, and coordination number can be derived from the analysis of XANES and EXAFS spectra.
Infrared spectroscopy as well as thermal techniques (thermal programmed desorption, TPD or thermogravimetric analysis, TGA) can be used to monitor the adsorption of probe molecules onto the metal surfaces, thus attaining indirect information on the electronic structure of metal phases and on eventual changes induced by SMSI. Similarly, electrochemical characterization techniques (e.g., voltammetry, chronoamperometry) allow investigating the electrical behavior of supported metal species, and then deducing the effect resulting from the interactions with the support.
Far from claiming to give an exhaustive overview of the current scenario in the field of material characterization, herein we report selected examples. The latter point out the potential, the applicability, and the limitations of the main characterization techniques that are usually employed for the study of the metal–support interface.
3.1. X-ray Photoelectron Spectroscopy (XPS)
XPS is a powerful tool that is crucial for detecting the surface layer element composition, chemical state, and electronic structure of metal and carbon materials. Successively, it has been widely used to study the electronic structure change and the doping effect in supported metal nanostructures on doped carbon materials. Significant shifts in binding energy values are symptomatic of electronic structure alterations resulting from metal–support interactions. However, a core level binding energy shift can be ascribed to three different factors (changes in the local electronic structure, changes in the relaxation process, and cluster charging), and it is often difficult to discriminate among them.
In some cases, the interaction can produce a new electronic state that is detectable as an additional peak (or a peak component in the case of complex signals) in the XPS spectrum.
Among transition metals, noble metals (Pd, Pt, Au) are the most studied systems, due to their large use as catalysts in the form of supported metal nanoparticles.
The strong interaction between Pd species and nitrogen functionalities embedded in carbon-based support has been reported in several papers. In most cases, an increase in the Pd2+/Pd0 ratio and a shift to the higher energy of the Pd 3d signal have been observed and ascribed to a change in the electronic structure or in the coordination environment [76]. The simultaneous analysis of N 1s signals allowed unraveling a prevalent interaction of Pd species with pyridinic and pyrrolic nitrogen [68]. This interaction leads to a charge transfer between the metal and the carbon support, resulting in the stabilization of electron-depleted palladium atoms (Pdδ+), whose signal has been detected in XPS spectra [67,69]. In some cases, a very strong bond between Pd2+ precursor ions and pyridinic nitrogen is created before and during the synthesis, resulting in the low reducibility of palladium species, and thus in the stabilization of isolated Pd2+ ions in the final catalyst [63].
Arrigo et al. [60,77] proposed a convincing rationalization of the Pd 3d XPS signal of Pd NPs supported on nitrogen-doped carbon nanotubes. Four components were separated under the broad and complex Pd 3d signal (Figure 6). The signal at lower binding energy (Pd1, 335 eV) was attributed to bulk-like metallic palladium, and was associated with large Pd particles formed under drastic annealing conditions. The Pd2 signal (335.55−335.7 eV) accounted for metallic Pd nanoparticles electronically interacting with the carbon support. These species were predominant in the samples after calcination and reduction. The Pd3 signal (336.9 eV) was related to partially positive charged Pd atoms located at the interface, while the Pd4 component (337.8 eV) is generated by divalent Pd species, namely atomically dispersed Pd. The latter were stabilized by pyridinic nitrogen and possessed low reducibility. After annealing at 300 °C, Pd3 and Pd4 species evolved toward the formation of Pd2 species.
The interpretation of XPS spectra is not trivial, and often, opposing results can appear in the literature. Specifically, in the case of Pd–N interactions, the presence of reduced Pd species [78] or negatively charged Pd has been reported because of an increased electron density by the charge transferred from N to the Pd species. The fact that these species have been revealed when prepared by the pyrolysis of chitosan and melamine or dicyandiamide [79] and Ketjen black carbon [80] suggests a possible role of the synthetic routes in addressing metal–support interactions.
Similar results were obtained in the case of Pt–N and Au–N [81] interactions. The Pt 4f spectra usually present three transitions corresponding to the different oxidation states of Pt (Pt0, Pt2+, and Pt4+). After deposition on nitrogen-doped supports, the oxidation state of the Pt and the ratio between these three components is altered, even if a univocal trend cannot be derived. In most cases, an increase in the Pt0/Ptδ+ ratio was exposed [80,82]. In addition, the BE of the Pt is affected by the electronic interaction with nitrogen, undergoing a shift to higher energy values. Although this shift has been reported to be proportional to the number and proximity of nitrogen atoms [83], concomitant size and extra atomic relaxation effects cannot be ruled out. A shift toward higher values was produced also in N 1s BE in species interacting with Pt. In addition, the appearance of new components in the N 1s spectra was assigned alternatively to chemisorbed nitrogen oxide (405.2 eV) [84] deriving from the conversion of graphitic nitrogen, or to a nitride (398.1 eV) resulting from the presence of a metal–nitrogen bond.
The effect of heteroatom-mediated electron transfer at the metal–support interface has been also reported for other transition metals (Ru [85], Fe [49,86], Co [86,87]) and for ZnCo2O4 perovskite quantum dots [88].
In any case, the main limitations of XPS are the minimal detectable concentrations and the difficult signal interpretation and deconvolution.
3.2. X-ray Absorption Spectroscopy (XAS)
XAS measurements describe intermolecular interactions, probing the valence and local structures of investigated elements. The carbon and nitrogen K-edge and metal L-edge near-edge X-ray absorption fine structure (NEXAFS) spectra together with the extended X-ray absorption fine structure (EXAFS) data provide a detailed picture of chemical and electronic structure speciation at the metal–support interface.
Concerning carbon K-edge spectra, the introduction of nitrogen in graphene sheet has been reported to produce a shift in the C–C σ* peak (286.5 eV), a shift to higher energy and a broadening of the C–C π* peak (292.8 eV), and the presence of an additional sharp peak in the region between the π* and σ* resonance (289.1 eV), which is related to the introduction of structural defects. Moreover, other peaks can appear due to π* transitions deriving from the introduction of functionalities (e.g., amine or imide groups) [89].
Nitrogen K-edge spectra is usually dominated by π*-transitions at 398.5 eV, 400 eV, and 401.4 eV, which can be credited to pyridinic, amino, and pyrrolic/graphitic type N-groups. The immobilization of metal on the surface on N-doped surfaces results in the decrease of the π* resonance intensity in N K-edge NEXAFS spectra (Figure 7). This evidence can be explained by invoking the Stöhr model: a metal–support π back-bonding interaction involves the N1σ–π* transition. Thus, a σ donor is formed by the lone pair orbital of the pyridine-like N, which is stabilized by the back-bonding of metal dπ–pπ orbitals with the N π* antibonding orbitals. Due to the enhanced bond strength, the metal dπ–pπ contributes in a greater extent to the π* orbital, thus producing the decrease of π* resonance intensity. In addition, a shoulder to the lower energy side of the π* resonance appears, indicating a charge localization on the N atom and a charge transfer between donor/acceptor sites [60].
In some cases, a new peak at ~398.9 eV has been observed after Pd deposition, which is ascribable to the nitrogen species interacting with Pd. [63].
Also, the K-edge and L-edge NEXAFS spectra of the metal are affected by the interaction with the heteroatoms incorporated in the support, inducing changes in electron occupancy in the valence orbital and ligand field environments. As an instance, monitoring Co K-edge XANES provided information on the oxidation state of Co and allowed unraveling strong covalent interactions between Co3O4 and N-graphene, resulting in a partial reduction of Co3+ [90]. A shift toward higher energy in K-edge of the X-ray absorption near-edge structure spectra (XANES) for Pd@PdO on nitrogen-doped graphene revealed a change of the metal’s electronic structure and strong interface interaction between the Pd@PdO particle and nitrogen-doped graphene [80].
In many cases, evidence of XANES is corroborated by EXAFS data, reporting lower metal–metal coordination numbers compared with bulk materials (i.e., high metal dispersion in the form of small nanoparticles), shorter metal–heteroatom and longer metal–metal distances, which are indicative of the strong character of metal–support interaction and demonstrate that these interactions are mediated by the heteroatom present in the support surface [80].
3.3. Transmission Electron Microscopies and Other Imaging Techniques
Transmission electron microscopy is a powerful technique that is able to disclose the structural and morphological features of the metal–support interface at the atomic scale. TEM images combined with energy-dispersive X-ray spectroscopy (EDS) and electron energy loss spectroscopy (EELS) allowed appreciating the metal dispersion on support and individuating the presence of defects, unsaturation, heteroatoms, and functional groups. Therefore, TEM represents the ideal tool to unravel the effects of heteroatom-mediated metal–support interactions [91].
In fact, the comparison of TEM images of metal nanoparticles supported on undoped and doped surfaces evidenced the fundamental role of homogeneously distributed heteroatoms in providing nucleation and anchoring sites and promoting a higher metal dispersion. High metal dispersion was detected for different metal species (Au [92], Pd [93], Pt [94,95], Co [87], MoS2 [96]) on several doped carbon surfaces (N and O-doped carbon nanotubes, N-doped mesoporous carbon, N-doped carbon shells, carbon nanosphere, polyaniline (PANI) fibers). The presence of nitrogen has been demonstrated to assure excellent metal dispersion, even at very high metal loading [93].
Electron micrographs (Figure 8) unveiled the drastic difference between the intimate contact at the interface of Pd particles on defective carbon surfaces, including heteroatom and topological defects, and the undamaged grain boundary of Pd NP supported on an undoped graphitic surface [69].
Besides TEM, other imaging techniques have been successfully used to characterize supported metal nanoparticles. For instance, atomic force microscopy (AFM) revealed that Au nucleation occurs selectively at step edges in a freshly cleaned highly oriented pyrolytic graphite (HOPG) surface, whereas a homogeneous dispersion of Au nanoparticles in the middle of terraces has been attained on HNO3-treated surfaces due to the introduction of O functionalities and defects acting as preferential anchoring sites [81].
Scanning transmission X-ray microscopy (STXM) uses a nanoscaled focused soft X-ray beam to provide microscopic measurements with high spatial resolution. It has been successfully applied to obtain the spatial mapping of reduced and stoichiometric Co3O4 on nitrogen-doped graphene, demonstrating the nitrogen and oxygen sites are favored sites for the immobilization of Co3O4 nanocrystals [90].
3.4. Other Techniques: Electrochemical and Adsorption Properties Characterization
The electronic promotional effects originating from metal–support interactions can result in an enhanced electrocatalytic activity of the supported metal species. A useful parameter that can be used as indicator of the electrocatalytic activity is the electrochemical surface area (ECSA). ECSA can be calculated from the charges of H2 adsorption/desorption peaks in cyclic voltammetry (CV) curves, according to the equation:
where QH is the amount of charge for the electrodesorption of hydrogen atoms on the metal surface, M is the mass of metal, and Qref is the metal surface density [97].
Pt-based nanoparticles dispersed on several N-doped carbon surfaces have been demonstrated to possess a higher ECSA compared to the Pt nanoparticles supported on undoped carbon surfaces. This effect could be associated with the enhanced adsorption of protons and electrolyte ions in the electrical double layer, due to the reinforcement in π bonding and higher wettability at the electrode/electrolyte interface associated with nitrogen species.
While the electrocatalytic activity is determined by ECSA, the catalytic activity is correlated to the adsorption properties of metal surfaces. CO and H2 are classical probe molecules that are usually adsorbed on metal surfaces to determine their binding properties. High-pressure infrared (HP-FTIR) spectroscopy, as well as attenuated total reflection (ATR) spectroscopy, have been used to monitor the adsorption of probe gases on the surface of metal supported on doped surfaces.
4. Effect of Heteroatoms–Metal Interaction in the Liquid Phase Oxidation of Benzyl Alcohol
In the last decades, the liquid phase oxidation of alcohol using noble metals (Pd, Au, Pd, Ru) deposited on carbonaceous supports has been the subject of several studies [98,99,100]. Many reports demonstrated that the presence of heteroatoms on the carbon surface can modify the reactivity of the metal. In this section, we will limit the discussion to some examples of the effect of metal–heteroatom interaction in the liquid phase alcohol oxidation. Our group was among the first to show that Au, Pd, and AuPd immobilized on nitrogen functionalized carbon nanofibers exhibited better performance than the same metals supported on pristine carbon in the benzyl alcohol (entry 1–4) [101,102]. The enhanced performances of N-containing materials were ascribed to the increased metal–support interaction, limiting the leaching and the coarsening of metal NPs [101,103]. In successive studies, we demonstrated that the amount and the nature of the nitrogen groups have influenced both activity and selectivity [52,104]. Pd deposited on pyridine-containing carbon nanofibers CNFs (entry 5) resulted more efficient than pyrrole/pyridine-containing ones (entry 6–7). The presence of pyridine resulted in a higher surface basicity compared to pyrrole/pyridine, facilitating the H-abstraction, which is proposed to be the rate-determining step in alcohol oxidation [100]. Moreover, a strong metal–N interaction prevented the agglomeration and sintering of small Pd particles. The metal–support interaction was also verified by the enhanced metal wetting observed. TEM studies showed that small particles better wet the carbon surface, resulting in oval-like particles rather than the round particle shape observed in colloids [52]. Ravat et al. demonstrated that B-doped carbon structures are better support that pristine carbon for Pd nanoparticles [105]. The presence of B induced a strong Pd–B interaction, increasing the activity and the durability of the catalyst during the benzyl alcohol oxidation. The effect of carbon dopant on the activity of non-noble metal was also reported. Sun et al. demonstrated the highest activity of cobalt nanoparticles deposited on nitrogen-doped graphitic carbon compared to Co on bare carbon in the veratryl alcohol oxidation [106]. The presence of N showed a positive synergistic effect on Co, influencing the electronic properties of the metal.
Su et al. were among the first to clearly demonstrate that the role of nitrogen on the enhancing activity of cobalt in benzyl alcohol oxidation is due to an electron transfer at the metal/nitrogen-doped carbon interface [107]. Co/N-Carbon catalysts with different nitrogen amounts, in the form of pyridinic and graphitic N (Figure 9b) ([email protected], Co@NC-2, Co@NC-4) were prepared using different amounts of C3N4 as the nitrogen source. The catalytic results clearly showed that the activity increases by increasing the nitrogen amount (entry 10–12, Table 1). By a combination of X-ray photoelectron spectroscopy (XPS) and extended X-Ray absorption fine structure (EXAFS) studies, they have proved that the presence of nitrogen can modify the electron density of Co particles, boosting the oxidative properties of the metal (Figure 9f). XPS showed that by increasing the nitrogen amount, the electron density on Co decreases (Figure 9b). As cobalt is in direct contact with N (Figure 9a), there is a flow of electrons (Mott–Schottky effect) at the metal/nitrogen interface from Co to N (Figure 9d) that depends on the nitrogen amount. Nitrogen-containing carbons with a higher flat band potential will accept electrons from cobalt nanoparticles till the Fermi level reaches an equilibrium.
5. Conclusions
The interaction between metal nanoparticles and heteroatoms (N, B, P, S) present on the carbon surface has been a matter of debate in recent years. The functional groups on the carbons have been reported to be (1) effective for anchoring metal nanoparticles, and (2) alter the electronic properties. Consequently, the reactivity of the metal nanoparticles is often improved. A combination of theoretical studies, characterization by spectroscopy (i.e., XPS and EXAFS), microscopy (i.e., TEM), and catalytic experiments has been utilized to understand the nature of metal–heteroatom interaction. Although theoretical models and predictions provide useful tools to rationalize experimental evidence and trends, a huge gap still separates the most common models from the actual systems studied under realistic conditions. Computational approaches suffer from several limitations in terms of time, power, cost, and knowledge. Concerning the rational study of metal–support interactions, great efforts should be made to develop new models. These models should consider some important features, such as the role of long-range dispersion forces at the interface or the reciprocal influence of defects, heteroatoms, and impurity or molecules adsorbed in the proximity of the interface.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ebert, L.B. Science of fullerenes and carbon nanotubes. Carbon 1997, 35, 437–438. [Google Scholar] [CrossRef]
- Hirsch, A. The chemistry of fullerenes: An overview. Angew. Chem. Int. Ed. 1993, 32, 1138–1141. [Google Scholar] [CrossRef]
- Guldi, D.M.; Martìn, N. Carbon Nanotubes and Related Structures: Production and Formation; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- De Jong, K.P.; Geus, J.W.; Jong, K. De Carbon Nanofibers: Catalytic Synthesis and Applications. Catal. Rev. 2000, 42, 481–510. [Google Scholar] [CrossRef]
- Geim, A.K. Graphene: Status and Prospects. Prospects 2009, 324, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, V.; Perman, J.A.; Tucek, J.; Zboril, R. Broad Family of Carbon Nanoallotropes: Classification, Chemistry, and Applications of Fullerenes, Carbon Dots, Nanotubes, Graphene, Nanodiamonds, and Combined Superstructures. Chem. Rev. 2015, 115, 4744–4822. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yan, J.; Fan, Z. Carbon materials for high volumetric performance supercapacitors: Design, progress, challenges and opportunities. Energy Environ. Sci. 2016, 9, 729–762. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, L.; Yan, M.; Yu, J. Carbon nanostructures in biology and medicine. J. Mater. Chem. B 2017, 5, 6437–6450. [Google Scholar] [CrossRef]
- Zieleniewska, A.; Lodermeyer, F.; Roth, A.; Guldi, D.M. Fullerenes-how 25 years of charge transfer chemistry have shaped our understanding of (interfacial) interactions. Chem. Soc. Rev. 2018, 47, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.H.-O.; Ralph, S.F. Carbon Nanotube Membranes: Synthesis, Properties, and Future Filtration Applications. Nanomaterials 2017, 7, 99. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Dou, Y.; Zhao, D.; Fulvio, P.F.; Mayes, R.T.; Dai, S. Carbon materials for chemical capacitive energy storage. Adv. Mater. 2011, 23, 4828–4850. [Google Scholar] [CrossRef] [PubMed]
- Qiang, Z.; Chen, Y.M.; Xia, Y.; Liang, W.; Zhu, Y.; Vogt, B.D. Ultra-long cycle life, low-cost room temperature sodium-sulfur batteries enabled by highly doped (N, S) nanoporous carbons. Nano Energy 2017, 32, 59–66. [Google Scholar] [CrossRef]
- Chen, Y.M.; Liang, W.; Li, S.; Zou, F.; Bhaway, S.M.; Qiang, Z.; Gao, M.; Vogt, B.D.; Zhu, Y. A nitrogen doped carbonized metal-organic framework for high stability room temperature sodium-sulfur batteries. J. Mater. Chem. A 2016, 4, 12471–12478. [Google Scholar] [CrossRef]
- Liu, X.; Zou, F.; Liu, K.; Qiang, Z.; Taubert, C.J.; Ustriyana, P.; Vogt, B.D.; Zhu, Y. A binary metal organic framework derived hierarchical hollow Ni3S2/Co9S8/N-doped carbon composite with superior sodium storage performance. J. Mater. Chem. A 2017, 5, 11781–11787. [Google Scholar] [CrossRef]
- Serp, B.; Machado, B. Nanostructured Carbon Materials for Catalysis; RSC Catalysis Series No. 23; Royal Society of Chemistry: London, UK, 2015. [Google Scholar]
- Thakur, V.K.; Thakur, M.K. Chemical Functionalization of Carbon Nanomaterials Chemistry and Applications; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Hirsch, A. Functionalization of single-walled carbon nanotubes. Angew. Chem. Int. Ed. 2002, 41, 1853–1859. [Google Scholar] [CrossRef]
- Wei, D.; Liu, Y.; Wang, Y.; Zhang, H.; Huang, L.; Yu, G. Synthesis of N-Doped Graphene by Chemical Vapor Deposition and Its Electrical Properties. Nano Lett. 2009, 9, 1752–1758. [Google Scholar] [CrossRef] [PubMed]
- Duclaux, L. Review of the doping of carbon nanotubes (multiwalled and single-walled). Carbon 2002, 40, 1751–1764. [Google Scholar] [CrossRef]
- Park, Y.; Yoo, J.; Lim, B.; Kwon, W.; Rhee, S.-W. Improving the functionality of carbon nanodots: Doping and surface functionalization. J. Mater. Chem. A 2016, 4, 11582–11603. [Google Scholar] [CrossRef]
- Ewels, C.P.; Glerup, M. Nitrogen Doping in Carbon Nanotubes. J. Nanosci. Nanotechnol. 2005, 5, 1345–1363. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Kahn, M.G.C.; Wong, S.S. Rational chemical strategies for carbon nanotube functionalization. Chemistry 2003, 9, 1898–1908. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, F.V.; Cividanes, L.D.S.; Brito, F.S.; Menezes, B.R.C.D.; Franceschi, W.; Simonetti, E.A.N.; Thim, G.P. Functionalizing Graphene and Carbon Nanotubes: A Review; Springer: New York, NY, USA, 2016. [Google Scholar]
- Georgakilas, V.; Tiwari, J.N.; Kemp, K.C.; Perman, J.A.; Bourlinos, A.B.; Kim, K.S.; Zboril, R. Noncovalent Functionalization of Graphene and Graphene Oxide for Energy Materials, Biosensing, Catalytic, and Biomedical Applications. Chem. Rev. 2016, 116, 5464–5519. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.L.; Stoddart, J.F. Noncovalent functionalization of single-walled carbon nanotubes. Acc. Chem. Res. 2009, 42, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Dyke, C.A.; Tour, J.M. Covalent functionalization of single-walled carbon nanotubes for materials applications. J. Phys. Chem. A 2004, 108, 11151–11159. [Google Scholar] [CrossRef]
- Georgakilas, V.; Kordatos, K.; Prato, M.; Guldi, D.M.; Holzinger, M.; Hirsch, A. Organic functionalization of carbon nanotubes. J. Am. Chem. Soc. 2002, 124, 760–761. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Alemany, L.B.; Margrave, J.L.; Khabashesku, V.N. Sidewall Carboxylic Acid Functionalization of Single-Walled Carbon Nanotubes Sidewall Carboxylic Acid Functionalization of Single-Walled Carbon Nanotubes. J. Am. Chem. Soc. 2003, 15174–15182. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.; Wang, Z.; Fierke, M.A. Functionalization of porous carbon materials with designed pore architecture. Adv. Mater. 2009, 21, 265–293. [Google Scholar] [CrossRef]
- Campisi, S.; Marzorati, S.; Spontoni, P.; Chan-Thaw, C.E.; Longhi, M.; Villa, A.; Prati, L. Tailored N-Containing Carbons as Catalyst Supports in Alcohol Oxidation. Materials 2016, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Nxumalo, E.N.; Coville, N.J. Nitrogen doped carbon nanotubes from organometallic compounds: A review. Materials 2010, 3, 2141–2171. [Google Scholar] [CrossRef]
- Prati, L.; Chan-Thaw, C.E.; Campisi, S.; Villa, A. N-Modified Carbon-Based Materials: Nanoscience for Catalysis. Chem. Rec. 2016, 16, 2187–2197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldonado, S.; Morin, S.; Stevenson, K.J. Structure, composition, and chemical reactivity of carbon nanotubes by selective nitrogen doping. Carbon 2006, 44, 1429–1437. [Google Scholar] [CrossRef]
- Strano, M.S.; Dyke, C.A.; Usrey, M.L.; Barone, P.W.; Allen, M.J.; Shan, H.; Kittrell, C.; Hauge, R.H.; Tour, J.M.; Smalley, R.E. Electronic structure control of single-walled carbon nanotube functionalization. Science 2003, 301, 1519–1522. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Santos, C.; Martínez-Hernández, A.L.; Fisher, F.T.; Ruoff, R.S.; Castaño, V.M. Improvement of Thermal and Mechanical Properties of Carbon Nanotube Composites through Chemical Functionalization. Chem. Mater. 2003, 15, 4470–4475. [Google Scholar] [CrossRef]
- Qiang, Z.; Xia, Y.; Xia, X.; Vogt, B.D. Generalized synthesis of a family of highly heteroatom-doped ordered mesoporous carbons. Chem. Mater. 2017, 29, 10178–10186. [Google Scholar] [CrossRef]
- Lee, W.J.; Maiti, U.N.; Lee, J.M.; Lim, J.; Han, T.H.; Kim, S.O. Nitrogen-doped carbon nanotubes and graphene composite structures for energy and catalytic applications. Chem. Commun. 2014, 50, 6818–6830. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wei, H.; Lu, Y.; Wei, S.; Wujcik, E.; Guo, Z. Multifunctional Carbon Nanostructures for Advanced Energy Storage Applications. Nanomaterials 2015, 5, 755–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Q.; Tong, X.; Zhang, G.; Qiao, J.; Gong, Q.; Sun, S. Nitrogen-Doped Carbon Nanotube and Graphene Materials for Oxygen Reduction Reactions. Catalysts 2015, 5, 1574–1602. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Mayoral, E.; Calvino-Casilda, V.; Soriano, E. Metal-supported carbon-based materials: Opportunities and challenges in the synthesis of valuable products. Catal. Sci. Technol. 2016, 6, 1265–1291. [Google Scholar] [CrossRef]
- Tauster, S.J. Strong metal-support interactions. Acc. Chem. Res. 1987, 20, 389–394. [Google Scholar] [CrossRef]
- Nicole, J.; Comninellis, C.; Tsiplakides, D.; Pliangos, C.; Verykios, X.E.; Vayenas, C.G. Electrochemical promotion and metal-support interactions. J. Catal. 2001, 204, 23–34. [Google Scholar] [CrossRef]
- Ryndin, Y.A.; Hicks, R.F.; Bell, A.T.; Yermakov, Y.I. Effects of metal-support interactions on the synthesis of methanol over palladium. J. Catal. 1981, 70, 287–297. [Google Scholar] [CrossRef] [Green Version]
- Román-Martínez, M.C.; Cazorla-Amorós, D.; Linares-Solano, A.; De Lecea, C.S.M.; Yamashita, H.; Anpo, M. Metal-support interaction in Pt/C catalysts. Influence of the support surface chemistry and the metal precursor. Carbon 1995, 33, 3–13. [Google Scholar] [CrossRef]
- Fujiwara, K.; Okuyama, K.; Pratsinis, S.E. Metal-support interactions in catalysts for environmental remediation. Environ. Sci. Nano 2017, 4, 2076–2092. [Google Scholar] [CrossRef]
- Joyner, W.; Pendry, J.B.; Tennison, R.; Joyner, R.W.; Pendry, J.B.; Saldin, D.K.; Tennison, S.R. Metal-support interactions in heterogeneous catalysis. Surf. Sci. 1984, 138, 84–94. [Google Scholar] [CrossRef]
- Akubuiro, E.C.; Verykios, X.E. Effects of dopants on performance of metal crystallites. Further characterization of doped supports and catalysts. J. Catal. 1988, 113, 106–119. [Google Scholar] [CrossRef]
- Matsubu, J.C.; Zhang, S.; DeRita, L.; Marinkovic, N.S.; Chen, J.G.; Graham, G.W.; Pan, X.; Christopher, P. Adsorbate-mediated strong metal-support interactions in oxide-supported Rh catalysts. Nat. Chem. 2017, 9, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Xia, W. Interactions between metal species and nitrogen-functionalized carbon nanotubes. Catal. Sci. Technol. 2016, 6, 630–644. [Google Scholar] [CrossRef]
- Hu, P.; Huang, Z.; Amghouz, Z.; Makkee, M.; Xu, F.; Kapteijn, F.; Dikhtiarenko, A.; Chen, Y.; Gu, X.; Tang, X. Electronic metal-support interactions in single-atom catalysts. Angew. Chem. Int. Ed. 2014, 53, 3418–3421. [Google Scholar] [CrossRef] [PubMed]
- Vayenas, C.G.; Bebelis, S.; Pliangos, C.; Brosda, S.; Tsiplakides, D. Electrochemical Activation of Catalysis: Promotion, Electrochemical Promotion, and Metal-Support Interactions; Springer Science & Business Media: New York, NY, USA, 2001. [Google Scholar]
- Arrigo, R.; Wrabetz, S.; Schuster, M.E.; Wang, D.; Villa, A.; Rosenthal, D.; Girsgdies, F.; Weinberg, G.; Prati, L.; Schlögl, R.; et al. Tailoring the morphology of Pd nanoparticles on CNTs by nitrogen and oxygen functionalization. Phys. Chem. Chem. Phys. 2012, 14, 10523–10532. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Eitan, A.; Schadler, L.S.; Ajayan, P.M.; Siegel, R.W.; Grobert, N.; Mayne, M.; Reyes-Reyes, M.; Terrones, H.; Terrones, M. Selective attachment of gold nanoparticles to nitrogen-doped carbon nanotubes. Nano Lett. 2003, 3, 275–277. [Google Scholar] [CrossRef]
- Lepró, X.; Terrés, E.; Vega-Cantú, Y.; Rodríguez-Macías, F.J.; Muramatsu, H.; Kim, Y.A.; Hayahsi, T.; Endo, M.; Torres, R.M.; Terrones, M. Efficient anchorage of Pt clusters on N-doped carbon nanotubes and their catalytic activity. Chem. Phys. Lett. 2008, 463, 124–129. [Google Scholar] [CrossRef]
- Kim, B.; Sigmund, W.M. Functionalized multiwall carbon nanotube/gold nanoparticle composites. Langmuir 2004, 20, 8239–8242. [Google Scholar] [CrossRef] [PubMed]
- Zamudio, A.; Elías, A.L.; Rodríguez-Manzo, J.A.; López-Urías, F.; Rodríguez-Gattorno, G.; Lupo, F.; Rühle, M.; Smith, D.J.; Terrones, H.; Díaz, D.; et al. Efficient anchoring of silver nanoparticles on n-doped carbon nanotubes. Small 2006, 2, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Acharya, C.K.; Sullivan, D.I.; Turner, C.H. Characterizing the interaction of Pt and PtRu clusters with boron-doped, nitrogen-doped, and activated carbon: Density functional theory calculations and parameterization. J. Phys. Chem. C 2008, 112, 13607–13622. [Google Scholar] [CrossRef]
- Li, Y.H.; Hung, T.H.; Chen, C.W. A first-principles study of nitrogen- and boron-assisted platinum adsorption on carbon nanotubes. Carbon 2009, 47, 850–855. [Google Scholar] [CrossRef]
- An, W.; Turner, C. Chemisorption of transition-metal atoms on boron-and nitrogen-doped carbon nanotubes: Energetics and geometric and electronic structures. J. Phys. Chem. C 2009, 113, 7069–7078. [Google Scholar] [CrossRef]
- Arrigo, R.; Schuster, M.E.; Xie, Z.; Yi, Y.; Wowsnick, G.; Sun, L.L.; Hermann, K.E.; Friedrich, M.; Kast, P.; Hävecker, M.; et al. Nature of the N-Pd interaction in nitrogen-doped carbon nanotube catalysts. ACS Catal. 2015, 5, 2740–2753. [Google Scholar] [CrossRef]
- Kabir, S.; Artyushkova, K.; Kiefer, B.; Atanassov, P. Computational and experimental evidence for a new TM–N 3/C moiety family in non-PGM electrocatalysts. Phys. Chem. Chem. Phys. 2015, 17, 17785–17789. [Google Scholar] [CrossRef] [PubMed]
- Artyushkova, K.; Kiefer, B.; Halevi, B.; Knop-Gericke, A.; Schlogl, R.; Atanassov, P. Density functional theory calculations of XPS binding energy shift for nitrogen-containing graphene-like structures. Chem. Commun. 2013, 49, 2539–2541. [Google Scholar] [CrossRef] [PubMed]
- Bulushev, D.A.; Zacharska, M.; Shlyakhova, E.V.; Chuvilin, A.L.; Guo, Y.; Beloshapkin, S.; Okotrub, A.V.; Bulusheva, L.G. Single Isolated Pd2+ Cations Supported on N-Doped Carbon as Active Sites for Hydrogen Production from Formic Acid Decomposition. ACS Catal. 2016, 6, 681–691. [Google Scholar] [CrossRef]
- Hammer, B.; Nørskov, J.K. Theoretical surface science and catalysis—Calculations and concepts. In Advances in Catalysis; Academic Press: Cambridge, MA, USA, 2000; Volume 45, pp. 71–129. [Google Scholar]
- Feng, H.; Ma, J.; Hu, Z. Nitrogen-doped carbon nanotubes functionalized by transition metal atoms: A density functional study. J. Mater. Chem. 2010, 20, 1702–1708. [Google Scholar] [CrossRef]
- Efremenko, I.; Sheintuch, M. Carbon-supported palladium catalysts. Molecular orbital study. J. Catal. 2003, 214, 53–67. [Google Scholar] [CrossRef]
- Tang, D.; Sun, X.; Zhao, D.; Zhu, J.; Zhang, W.; Xu, X.; Zhao, Z. Nitrogen-Doped Carbon Xerogels Supporting Palladium Nanoparticles for Selective Hydrogenation Reactions: The Role of Pyridine Nitrogen Species. ChemCatChem 2018, 10, 1291–1299. [Google Scholar] [CrossRef]
- Jiang, B.; Song, S.; Wang, J.; Xie, Y.; Chu, W.; Li, H.; Xu, H.; Tian, C.; Fu, H. Nitrogen-doped graphene supported Pd@PdO core-shell clusters for C-C coupling reactions. Nano Res. 2014, 7, 1280–1290. [Google Scholar] [CrossRef]
- Rao, R.G.; Blume, R.; Hansen, T.W.; Fuentes, E.; Dreyer, K.; Moldovan, S.; Ersen, O.; Hibbitts, D.D.; Chabal, Y.J.; Schlögl, R.; et al. Interfacial charge distributions in carbon-supported palladium catalysts. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.J.; Hou, T.Z.; Zhang, Q.; Huang, J.Q.; Cheng, X.B.; Guo, M.Q.; Yuan, Z.; He, L.Y.; Wei, F. Strongly coupled interfaces between a heterogeneous carbon host and a sulfur-containing guest for highly stable lithium-sulfur batteries: Mechanistic insight into capacity degradation. Adv. Mater. Interfaces 2014, 1, 1–10. [Google Scholar] [CrossRef]
- Kim, G.; Jhi, S.H.; Park, N. Effective metal dispersion in pyridinelike nitrogen doped graphenes for hydrogen storage. Appl. Phys. Lett. 2008, 92, 2006–2009. [Google Scholar] [CrossRef]
- Bulushev, D.A.; Zacharska, M.; Lisitsyn, A.S.; Podyacheva, O.Y.; Hage, F.S.; Ramasse, Q.M.; Bangert, U.; Bulusheva, L.G. Single Atoms of Pt-Group Metals Stabilized by N-Doped Carbon Nanofibers for Efficient Hydrogen Production from Formic Acid. ACS Catal. 2016, 6, 3442–3451. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.; Jhi, S.H. Carbon monoxide-tolerant platinum nanoparticle catalysts on defect-engineered graphene. ACS Nano 2011, 5, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Perazzolo, V.; Brandiele, R.; Durante, C.; Zerbetto, M.; Causin, V.; Rizzi, G.A.; Cerri, I.; Granozzi, G.; Gennaro, A. Density Functional Theory (DFT) and Experimental Evidences of Metal-Support Interaction in Platinum Nanoparticles Supported on Nitrogen- and Sulfur-Doped Mesoporous Carbons: Synthesis, Activity, and Stability. ACS Catal. 2018, 8, 1122–1137. [Google Scholar] [CrossRef]
- Ning, X.; Li, Y.; Dong, B.; Wang, H.; Yu, H.; Peng, F.; Yang, Y. Electron transfer dependent catalysis of Pt on N-doped carbon nanotubes: Effects of synthesis method on metal-support interaction. J. Catal. 2017, 348, 100–109. [Google Scholar] [CrossRef]
- Li, Z.; Yang, X.; Tsumori, N.; Liu, Z.; Himeda, Y.; Autrey, T.; Xu, Q.; Li, Z.; Yang, X.; Tsumori, N.; et al. Tandem Nitrogen Functionalization of Porous Carbon: Toward Immobilizing Highly Active Palladium Nanoclusters for Dehydrogenation of Formic Acid Tandem Nitrogen Functionalization of Porous Carbon: Toward Immobilizing Highly Active Palladium Nanoclusters for Dehydrogenation of Formic Acid. ACS Catal. 2017, 7, 2720–2724. [Google Scholar]
- Arrigo, R.; Schuster, M.E.; Abate, S.; Wrabetz, S.; Amakawa, K.; Teschner, D.; Freni, M.; Centi, G.; Perathoner, S.; Hävecker, M. Dynamics of Pd on nanocarbon in the direct synthesis of H2O2. ChemSusChem 2014, 7, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Chew, L.M.; Kostka, A.; Muhler, M.; Xia, W. The structural and electronic promoting effect of nitrogen-doped carbon nanotubes on supported Pd nanoparticles for selective olefin hydrogenation. Catal. Sci. Technol. 2013, 3, 1964–1971. [Google Scholar] [CrossRef]
- Bi, Q.Y.; Lin, J.D.; Liu, Y.M.; He, H.Y.; Huang, F.Q.; Cao, Y. Dehydrogenation of Formic Acid at Room Temperature: Boosting Palladium Nanoparticle Efficiency by Coupling with Pyridinic-Nitrogen-Doped Carbon. Angew. Chem. Int. Ed. 2016, 55, 11849–11853. [Google Scholar] [CrossRef] [PubMed]
- Jeon, M.; Jung, D.; Lee, K.; Hee, S.; Han, J.; Woo, S.; Chul, S.; Park, H.S. Electronically modified Pd catalysts supported on N-doped carbon for the dehydrogenation of formic acid. Int. J. Hydrogen Energy 2016, 41, 15453–15461. [Google Scholar] [CrossRef]
- Burgess, R.; Buono, C.; Davies, P.R.; Davies, R.J.; Legge, T.; Lai, A.; Lewis, R.; Morgan, D.J.; Robinson, N.; Willock, D.J. The functionalisation of graphite surfaces with nitric acid: Identification of functional groups and their effects on gold deposition. J. Catal. 2015, 323, 10–18. [Google Scholar] [CrossRef]
- Zhang, M.; Shi, J.; Sun, Y.; Ning, W.; Hou, Z. Selective oxidation of glycerol over nitrogen-doped carbon nanotubes supported platinum catalyst in base-free solution. Catal. Commun. 2015, 70, 72–76. [Google Scholar] [CrossRef]
- Perini, L.; Durante, C.; Favaro, M.; Perazzolo, V.; Agnoli, S.; Schneider, O.; Granozzi, G.; Gennaro, A. Metal-Support Interaction in Platinum and Palladium Nanoparticles Loaded on Nitrogen-Doped Mesoporous Carbon for Oxygen Reduction Reaction. ACS Appl. Mater. Interfaces 2015, 7, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, J.; Liu, H.; Banis, M.N.; Li, R.; Sun, X.; Sham, T.-K.; Ye, S.; Knights, S. Nitrogen Doping Effects on Carbon Nanotubes and the Origin of the Enhanced Electrocatalytic Activity of Supported Pt for Proton-Exchange Membrane Fuel Cells. J. Phys. Chem. C 2011, 115, 3769–3776. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Swaidan, R.; Li, D.; Li, N. Enhanced methanol electro-oxidation activity of PtRu catalysts supported on heteroatom-doped carbon. Electrochim. Acta 2008, 53, 7622–7629. [Google Scholar] [CrossRef]
- Vinayan, B.P.; Ramaprabhu, S. Platinum-TM (TM = Fe, Co) alloy nanoparticles dispersed nitrogen doped (reduced graphene oxide-multiwalled carbon nanotube) hybrid structure cathode electrocatalysts for high performance PEMFC applications. Nanoscale 2013, 5, 5109–5118. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Moyo, M.; Rayner, M.K.; Jewell, L.L.; Billing, D.G.; Coville, N.J. Autoreduction and Catalytic Performance of a Cobalt Fischer-Tropsch Synthesis Catalyst Supported on Nitrogen-Doped Carbon Spheres. ChemCatChem 2010, 2, 514–518. [Google Scholar] [CrossRef]
- Liu, Z.Q.; Cheng, H.; Li, N.; Ma, T.Y.; Su, Y.Z. ZnCo2O4 Quantum Dots Anchored on Nitrogen-Doped Carbon Nanotubes as Reversible Oxygen Reduction/Evolution Electrocatalysts. Adv. Mater. 2016, 28, 3777–3784. [Google Scholar] [CrossRef] [PubMed]
- Melke, J.; Peter, B.; Habereder, A.; Ziegler, J.; Fasel, C.; Nefedov, A.; Sezen, H.; Wöll, C.; Ehrenberg, H.; Roth, C. Metal-Support Interactions of Platinum Nanoparticles Decorated N-Doped Carbon Nanofibers for the Oxygen Reduction Reaction. ACS Appl. Mater. Interfaces 2016, 8, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhou, J.; Hu, Y.; Regier, T. Chemical interaction and imaging of single Co3O4/graphene sheets studied by scanning transmission X-ray microscopy and X-ray absorption spectroscopy. Energy Environ. Sci. 2013, 6, 926–934. [Google Scholar] [CrossRef]
- Zhang, B.; Su, D.S. Probing the Metal-Support Interaction in Carbon-Supported Catalysts by using Electron Microscopy. ChemCatChem 2015, 7, 3639–3645. [Google Scholar] [CrossRef]
- Suarez-Martinez, I.; Bittencourt, C.; Ke, X.; Felten, A.; Pireaux, J.J.; Ghijsen, J.; Drube, W.; Van Tendeloo, G.; Ewels, C.P. Probing the interaction between gold nanoparticles and oxygen functionalized carbon nanotubes. Carbon 2009, 47, 1549–1554. [Google Scholar] [CrossRef]
- Yoon, H.; Ko, S.; Jang, J. Nitrogen-doped magnetic carbon nanoparticles as catalyst supports for efficient recovery and recycling. Chem. Commun. 2007, 1468–1470. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.; Yang, F.; Rasouli, S.; Qiu, Y.; Li, Z.F.; Uzunoglu, A.; Sun, C.J.; Liu, Y.; Ferreira, P.; Li, W.; et al. Understanding Pt Nanoparticle Anchoring on Graphene Supports through Surface Functionalization. ACS Catal. 2016, 6, 2642–2653. [Google Scholar] [CrossRef]
- He, D.; Jiang, Y.; Lv, H.; Pan, M.; Mu, S. Nitrogen-doped reduced graphene oxide supports for noble metal catalysts with greatly enhanced activity and stability. Appl. Catal. B Environ. 2013, 132–133, 379–388. [Google Scholar] [CrossRef]
- Yu, X.-Y.; Hu, H.; Wang, Y.; Chen, H.; Lou, X.W.D. Ultrathin MoS2 Nanosheets Supported on N-doped Carbon Nanoboxes with Enhanced Lithium Storage and Electrocatalytic Properties. Angew. Chem. Int. Ed. 2015, 54, 7395–7398. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Dai, C.; Wang, D.; Li, D.; Li, N. Nitrogen-doped magnetic onion-like carbon as support for Pt particles in a hybrid cathode catalyst for fuel cells. J. Mater. Chem. 2010, 20, 3059–3068. [Google Scholar] [CrossRef]
- Besson, M.; Gallezot, P. Selective oxidation of alcohols and aldehydes on metal catalysts. Catal. Today 2000, 57, 127–141. [Google Scholar] [CrossRef]
- Mallat, T.; Baiker, A. Oxidation of alcohols with molecular oxygen on solid catalysts. Chem. Rev. 2004, 104, 3037–3058. [Google Scholar] [CrossRef] [PubMed]
- Dimitratos, N.; Lopez-Sanchez, J.A.; Hutchings, G.J. Selective liquid phase oxidation with supported metal nanoparticles. Chem. Sci. 2012, 3, 20–44. [Google Scholar] [CrossRef]
- Villa, A.; Wang, D.; Spontoni, P.; Arrigo, R.; Su, D.; Prati, L. Nitrogen functionalized carbon nanostructures supported Pd and Au–Pd NPs as catalyst for alcohols oxidation. Catal. Today 2010, 157, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Prati, L.; Villa, A.; Chan-Thaw, C.E.; Arrigo, R.; Wang, D.; Su, D.S. Gold catalyzed liquid phase oxidation of alcohol: The issue of selectivity. Faraday Discuss. 2011, 152, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Shang, J.-H.; Chen, Y.; Wang, Y.; Li, Y.-X. Palladium nanoparticles supported on mesoporous carbon nitride for efficiently selective oxidation of benzyl alcohol with molecular oxygen. Appl. Catal. A Gen. 2017, 542, 380–388. [Google Scholar] [CrossRef]
- Chan-Thaw, C.E.; Villa, A.; Veith, G.M.; Prati, L. Identifying the Role of N-Heteroatom Location in the Activity of Metal Catalysts for Alcohol Oxidation. ChemCatChem 2015, 7, 1338–1346. [Google Scholar] [CrossRef]
- Ravat, V.; Nongwe, I.; Coville, N.J. Palladium-Supported Boron-Doped Hollow Carbon Spheres as Catalysts for the Solvent-free Aerobic Oxidation of Alcohols. ChemCatChem 2012, 4, 1930–1934. [Google Scholar] [CrossRef]
- Sun, Y.; Ma, H.; Luo, Y.; Zhang, S.; Gao, J.; Xu, J. Activation of Molecular Oxygen Using Durable Cobalt Encapsulated with Nitrogen-Doped Graphitic Carbon Shells for Aerobic Oxidation of Lignin-Derived Alcohols. Chem. Eur. J. 2018, 24, 4653–4661. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Zhang, K.-X.; Zhang, B.; Wang, H.-H.; Yu, Q.-Y.; Li, X.-H.; Antonietti, M.; Chen, J.-S. Activating Cobalt Nanoparticles via the Mott-Schottky Effect in Nitrogen-Rich Carbon Shells for Base-Free Aerobic Oxidation of Alcohols to Esters. J. Am. Chem. Soc. 2017, 139, 811–818. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Strong metal–support interaction: electronic, geometric, and dispersion effects.
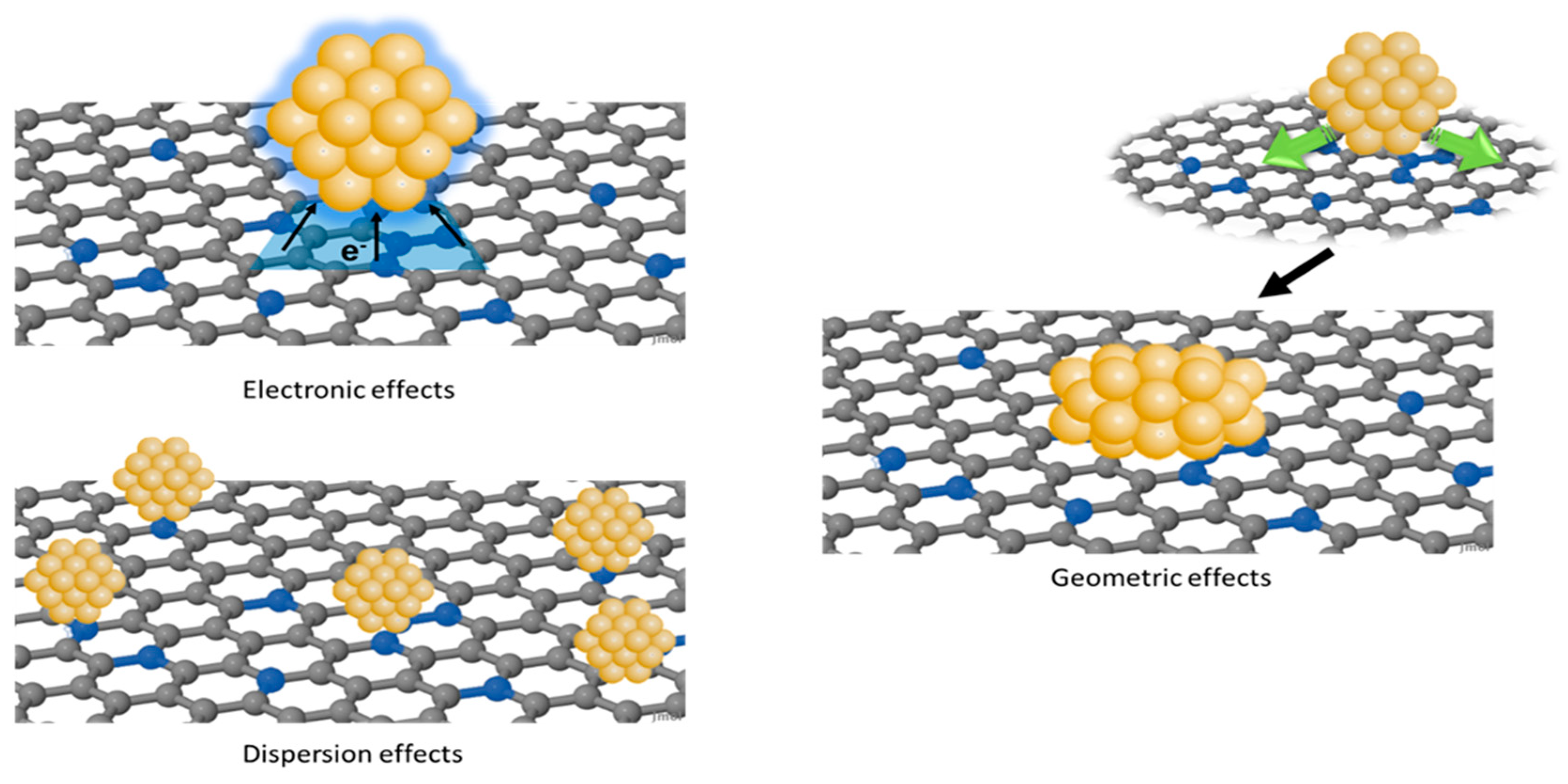
Figure 2.
Effect of the introduction of a heteroatom on metal nanoparticles and the carbon support.

Figure 3.
Projected density of states (PDOS) of different doped and undoped graphene models. The Fermi energy is adjusted to zero. Reprinted with permission from reference [57]. Copyright 2008 American Chemical Society.
Figure 3.
Projected density of states (PDOS) of different doped and undoped graphene models. The Fermi energy is adjusted to zero. Reprinted with permission from reference [57]. Copyright 2008 American Chemical Society.

Figure 4.
Bonding configurations for N in graphitic networks. Reprinted with permission from reference [18]. Copyright 2009 American Chemical Society.
Figure 4.
Bonding configurations for N in graphitic networks. Reprinted with permission from reference [18]. Copyright 2009 American Chemical Society.
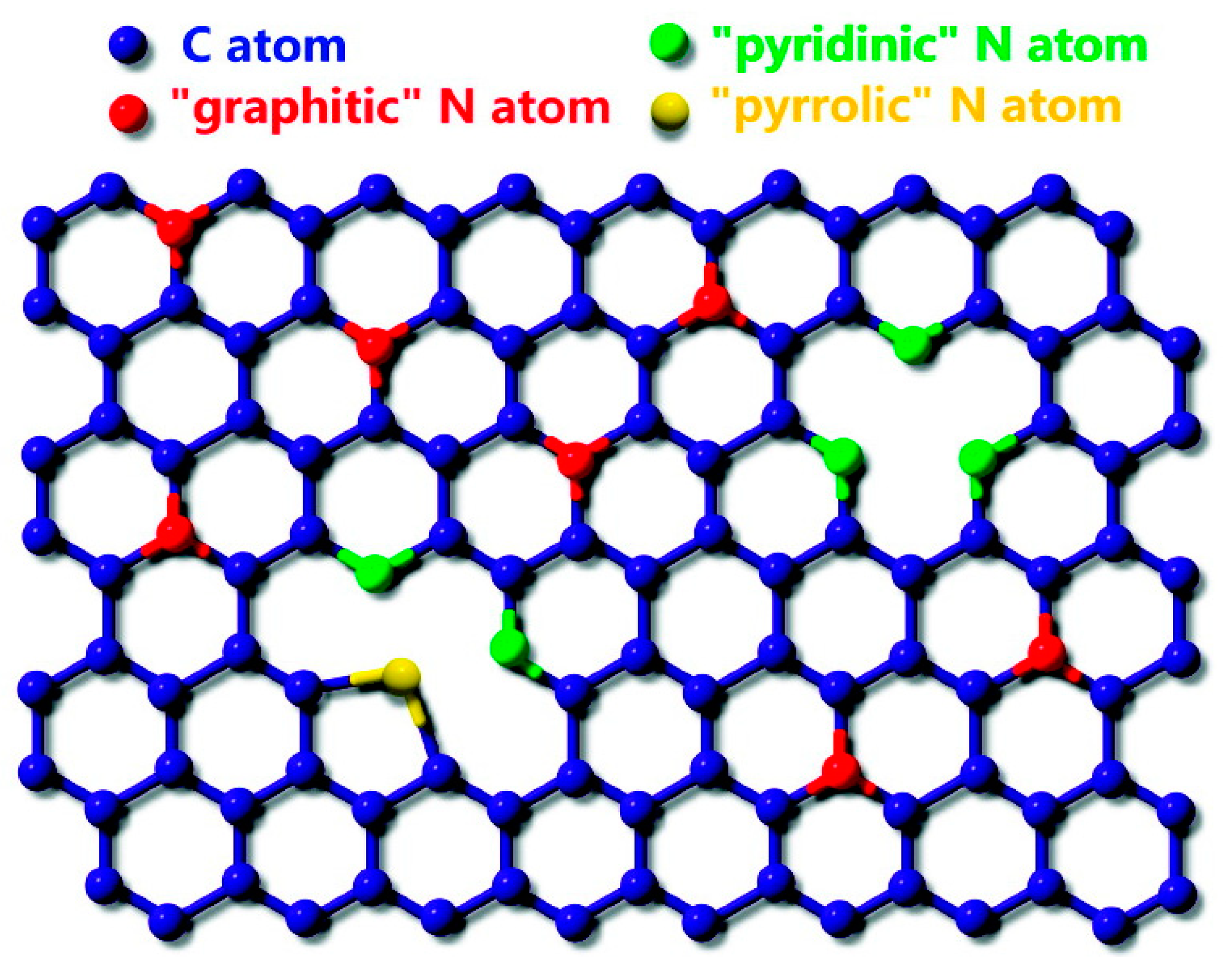
Figure 5.
Structural rearrangement of Pt clusters supported on nitrogen-doped graphene. Reprinted with permission from reference [73]. Copyright 2011 American Chemical Society.
Figure 5.
Structural rearrangement of Pt clusters supported on nitrogen-doped graphene. Reprinted with permission from reference [73]. Copyright 2011 American Chemical Society.
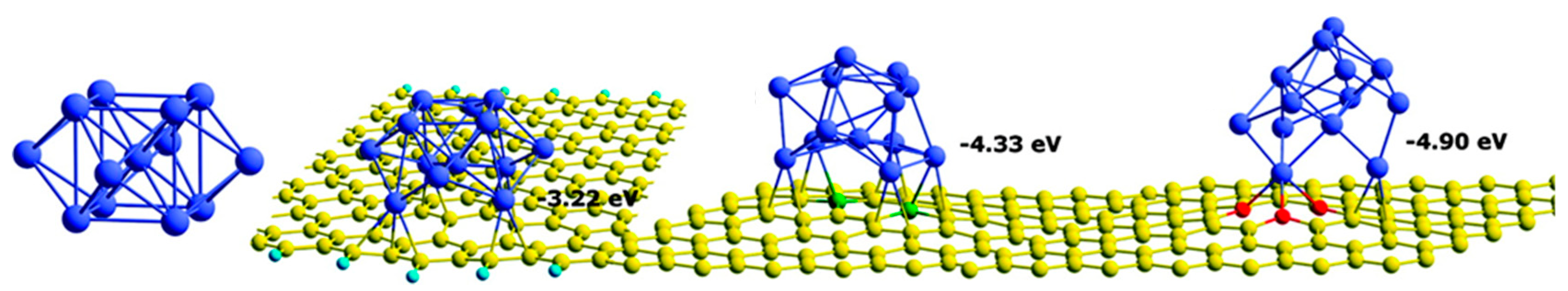
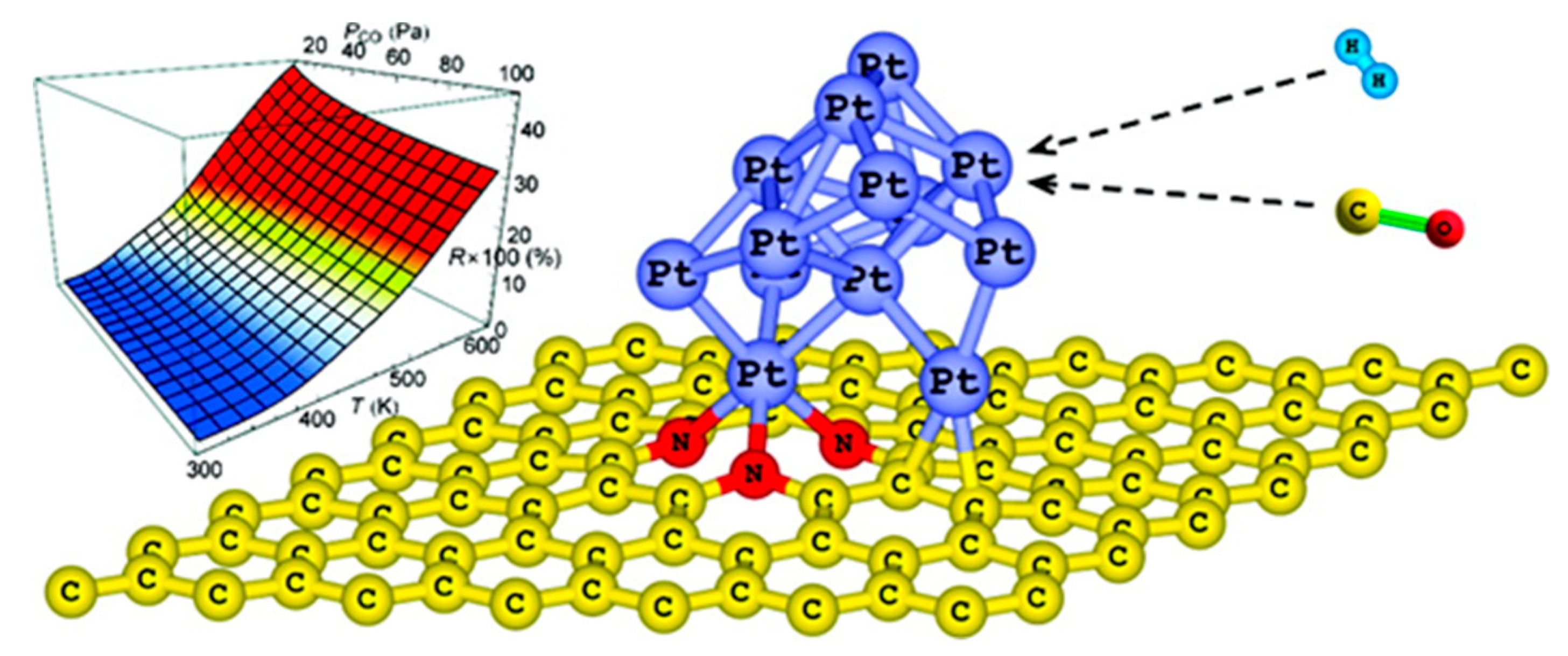
Figure 6.
Components in Pd 3d core level spectra at 600 eV Kinetic Energy for the reduced Pd on the different nitrogen-doped carbon nanotubes (NCNT) samples. Reprinted with permission from reference [60]. Copyright 2015 American Chemical Society.
Figure 6.
Components in Pd 3d core level spectra at 600 eV Kinetic Energy for the reduced Pd on the different nitrogen-doped carbon nanotubes (NCNT) samples. Reprinted with permission from reference [60]. Copyright 2015 American Chemical Society.
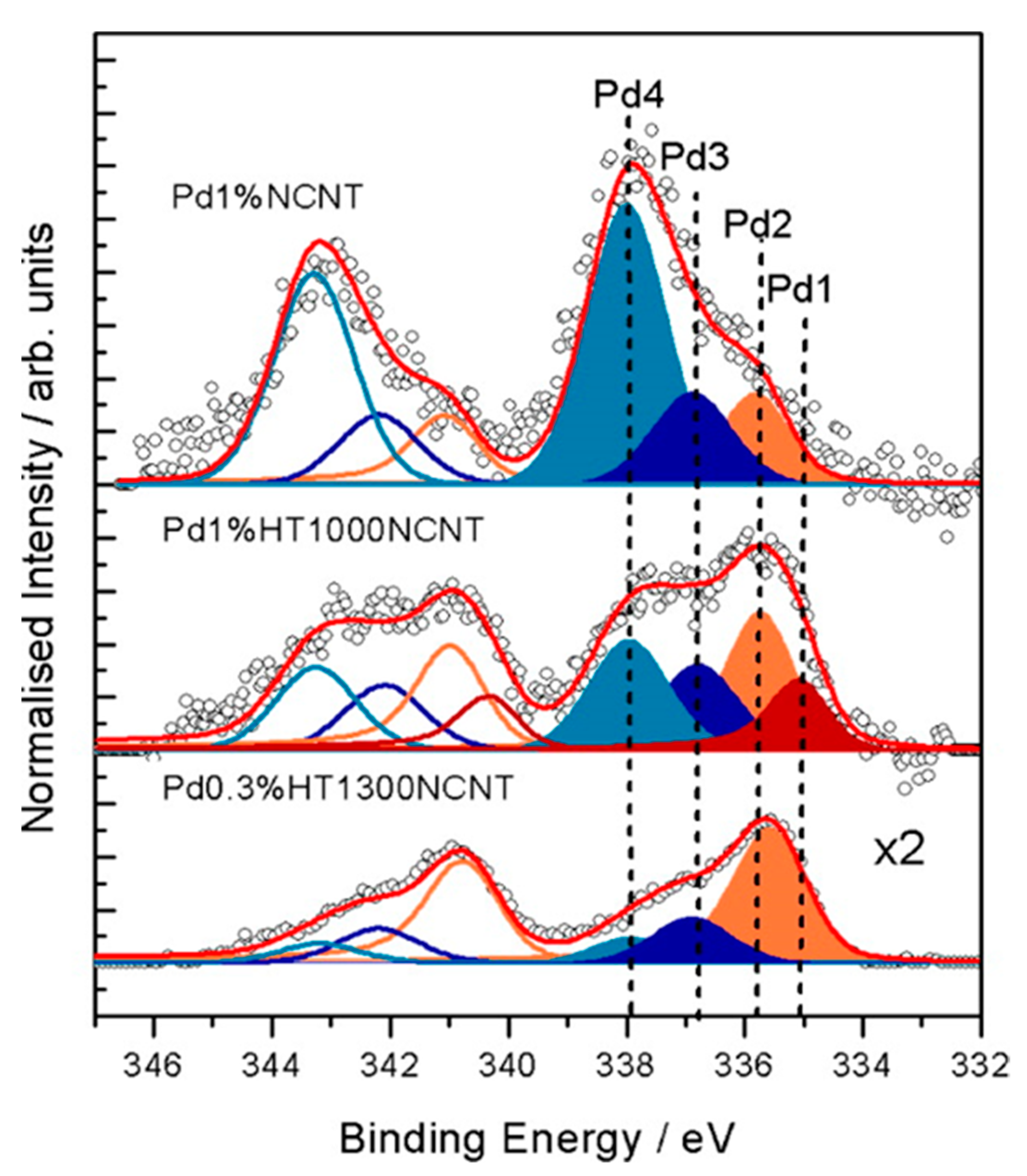
Figure 7.
Near-edge X-ray absorption fine structure (NEXAFS) Nitrogen K-edge spectra for the N-doped surfaces. Reprinted with permission from reference [63]. Copyright 2016 American Chemical Society.
Figure 7.
Near-edge X-ray absorption fine structure (NEXAFS) Nitrogen K-edge spectra for the N-doped surfaces. Reprinted with permission from reference [63]. Copyright 2016 American Chemical Society.

Figure 8.
Aberration-corrected TEM images for the (a) Pd/PS (where PS are pyrolytically stripped carbon nanofibers containing defects and O-functionalities) and (b) Pd/HHT (where HHT are carbon nanofiber heat-treated to high temperatures, thus possessing graphitic surface) catalysts provide an atomic resolution view of the Pd nanoparticles (blue) and their interface with the carbon support (red). The insets show the fast Fourier transform results and the particle size distribution. The scale bars in the micrographs (a,b) represent 5 nm. Reproduced with permission from reference [69]. Copyright 2017.
Figure 8.
Aberration-corrected TEM images for the (a) Pd/PS (where PS are pyrolytically stripped carbon nanofibers containing defects and O-functionalities) and (b) Pd/HHT (where HHT are carbon nanofiber heat-treated to high temperatures, thus possessing graphitic surface) catalysts provide an atomic resolution view of the Pd nanoparticles (blue) and their interface with the carbon support (red). The insets show the fast Fourier transform results and the particle size distribution. The scale bars in the micrographs (a,b) represent 5 nm. Reproduced with permission from reference [69]. Copyright 2017.
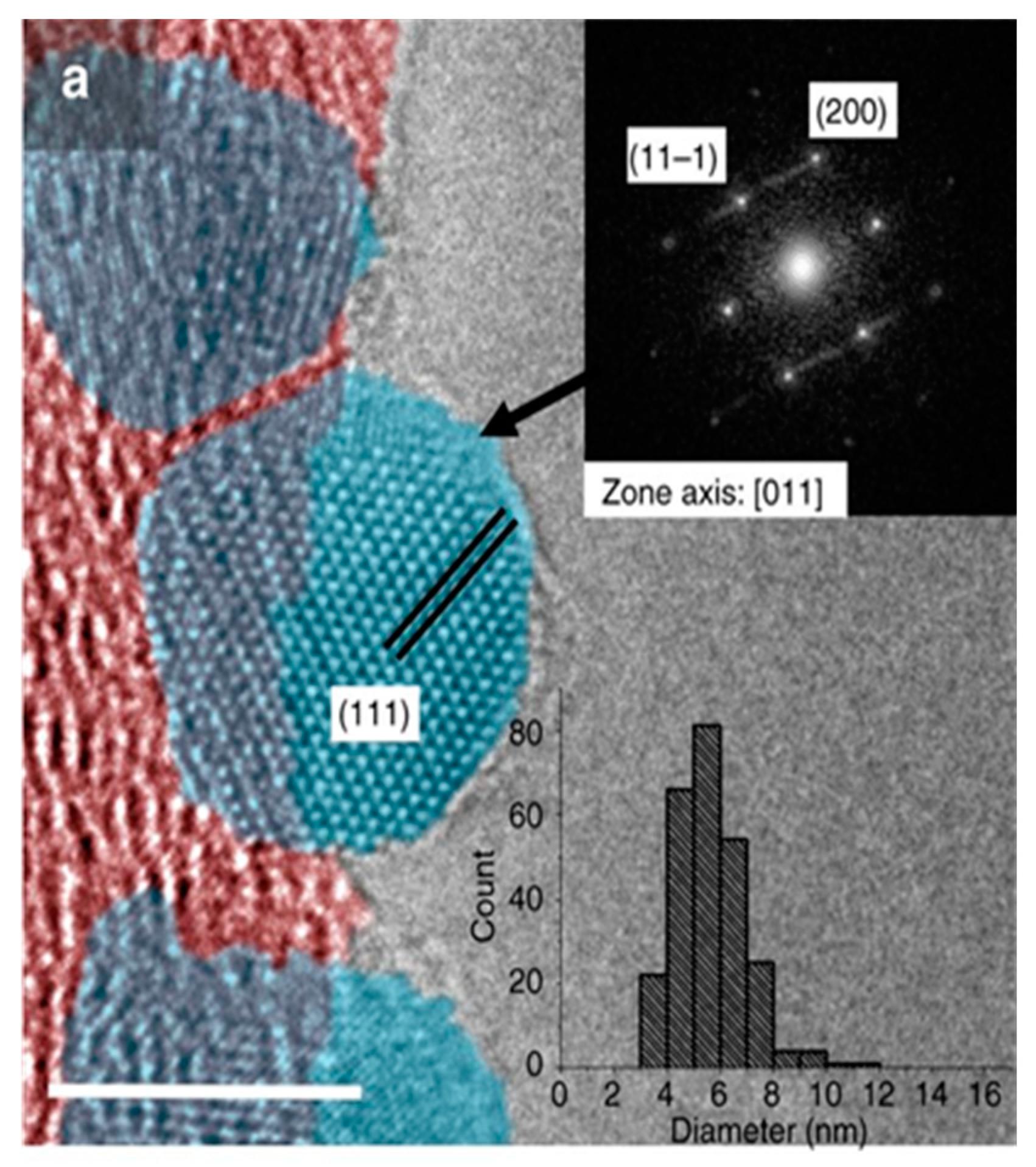

Figure 9.
Mott–Schottky effect on the catalytic performance of the Co@NC-x catalysts. (a) Nitrogen (green) and cobalt (red) elemental-mapping images and the corresponding line profile of Co@NC-4 sample; (b) N 1s and (c) Co 2p X-ray photoelectron spectroscopy (XPS) spectra of Co@NC-x; (d) Schematic illustration of Mott–Schottky-type contact of Co@NC; (e) The N/C mole ratio of Co@NC-x samples as determined by elemental analysis and the corresponding schematic structures; (f) Turnover Frequency (TOF) values for methyl benzoate (MB) production via aerobic esterification of benzyl alcohol and methanol over Co@NC-x. Reprinted with permission from reference [107]. Copyright 2017 American Chemical Society.
Figure 9.
Mott–Schottky effect on the catalytic performance of the Co@NC-x catalysts. (a) Nitrogen (green) and cobalt (red) elemental-mapping images and the corresponding line profile of Co@NC-4 sample; (b) N 1s and (c) Co 2p X-ray photoelectron spectroscopy (XPS) spectra of Co@NC-x; (d) Schematic illustration of Mott–Schottky-type contact of Co@NC; (e) The N/C mole ratio of Co@NC-x samples as determined by elemental analysis and the corresponding schematic structures; (f) Turnover Frequency (TOF) values for methyl benzoate (MB) production via aerobic esterification of benzyl alcohol and methanol over Co@NC-x. Reprinted with permission from reference [107]. Copyright 2017 American Chemical Society.
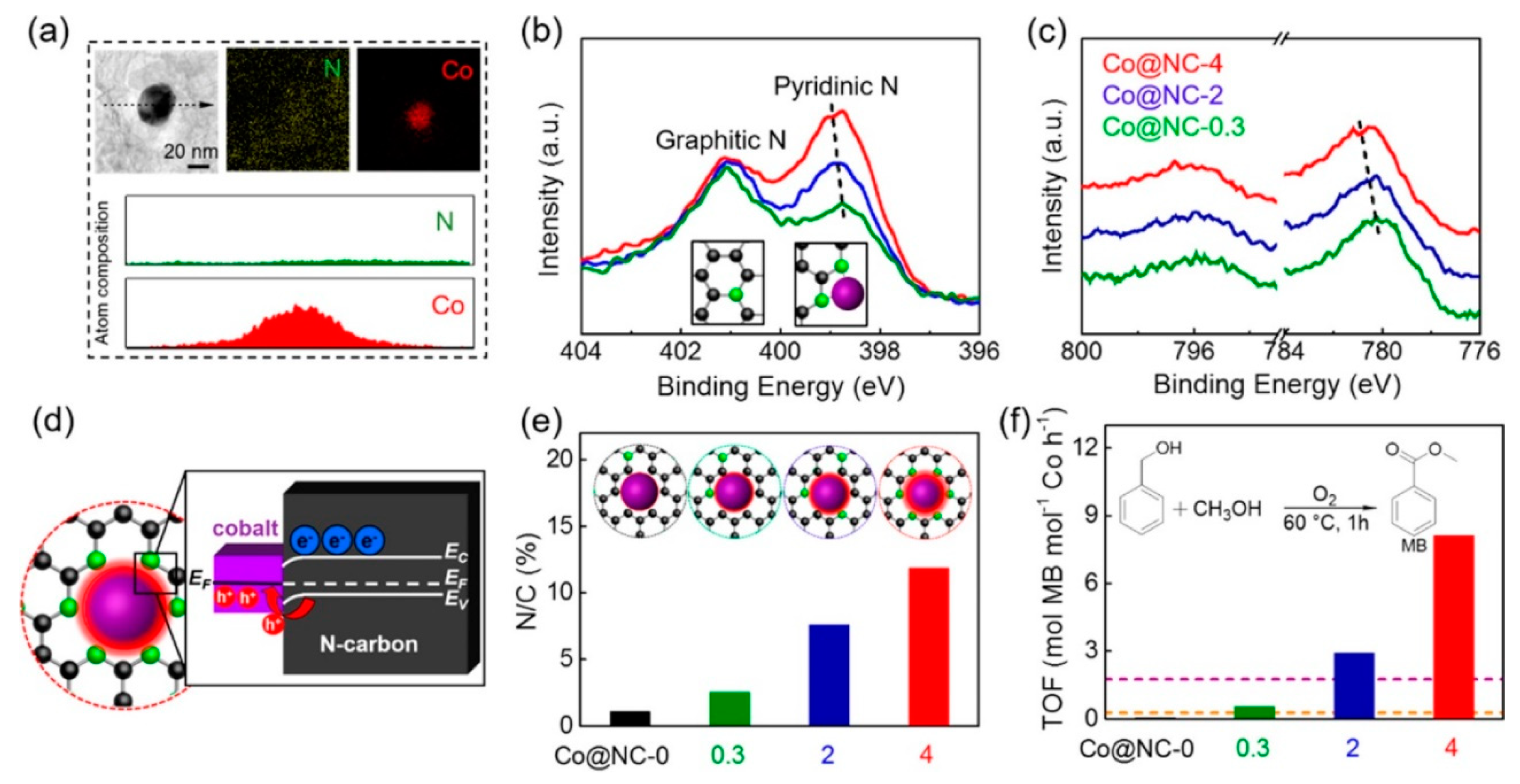

{kind=link}
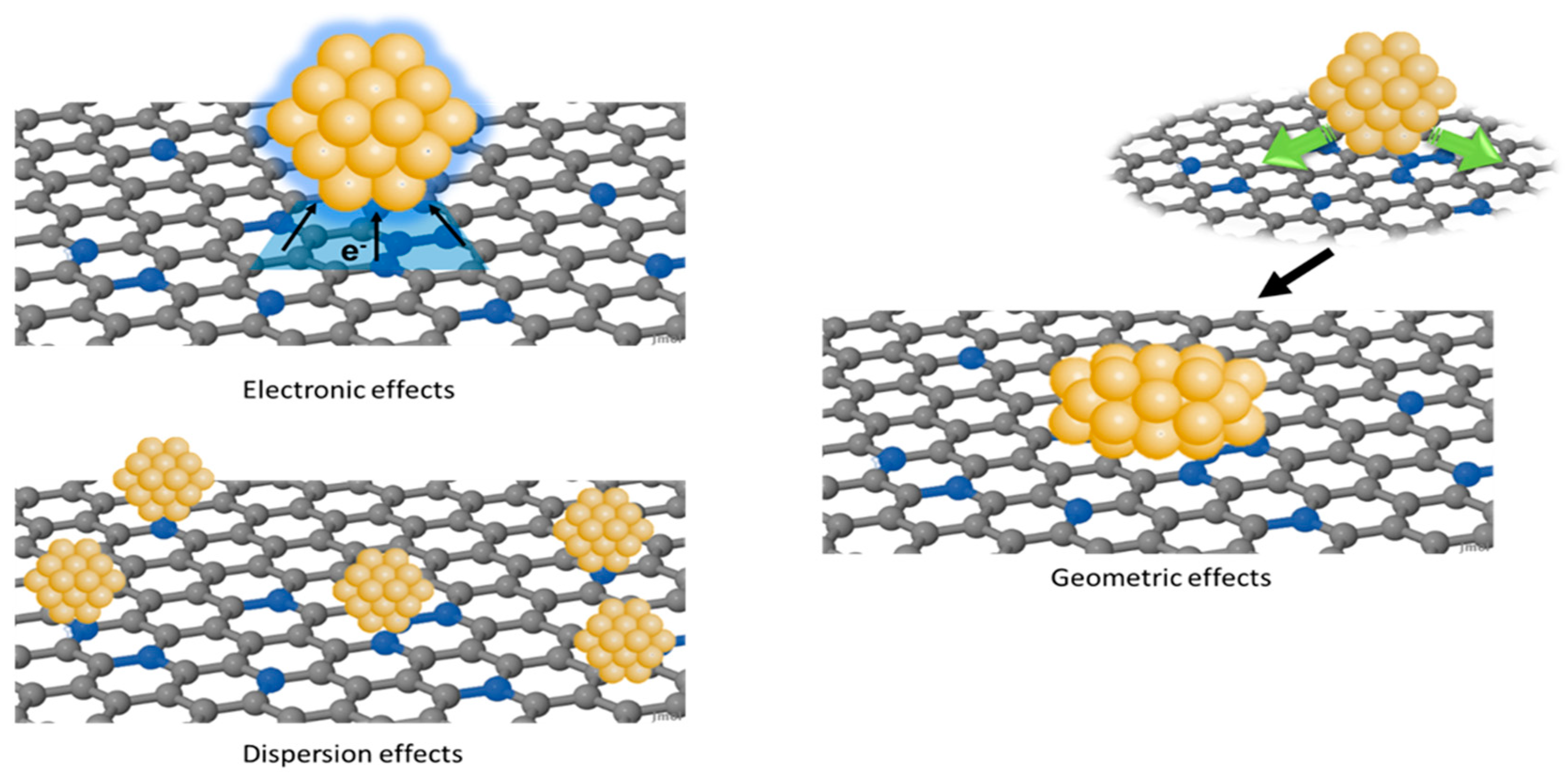{kind=link}
{kind=link}
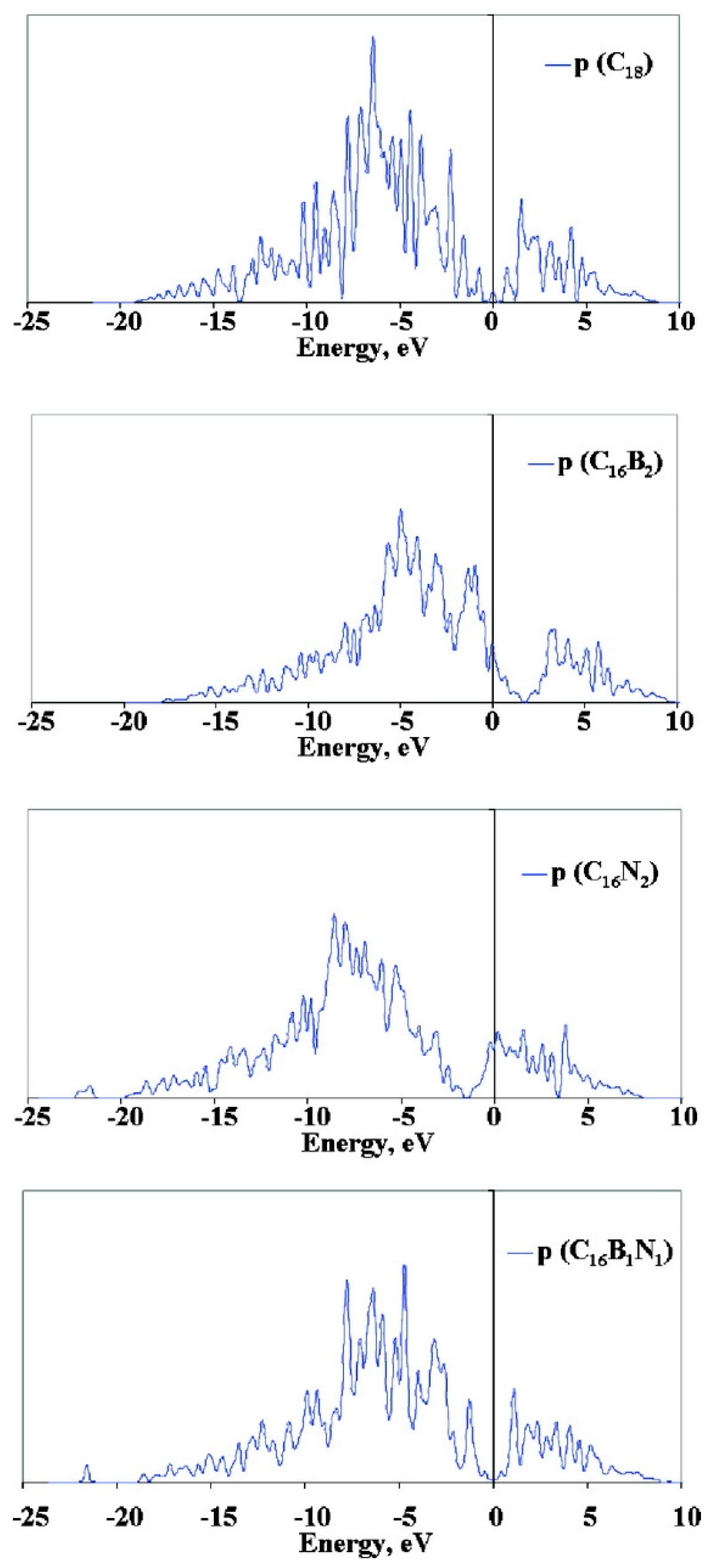{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Activity of metal supported on functionalized carbon catalysts in benzyl alcohol oxidation.
Table 1.
Activity of metal supported on functionalized carbon catalysts in benzyl alcohol oxidation.
| Entry | Catalyst | TOF (1/h) | Conversion (%) | Solvent | Alcohol/Metal (mol %) | T (°C) | pO2 (ATM) | Sel to Benzal-Dehyde | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Pd/PR24-PS | 7260 | n.d. | solventless | 35,000 | 120 | 1.5 | 62 | [101] |
| 2 | Pd/N-PR24-PS | 65,876 | n.d. | solventless | 35,000 | 120 | 1.5 | 64 | [101] |
| 3 | AuPd/PR24-PS | 6076 | n.d. | solventless | 35,000 | 120 | 1.5 | 74 | [101] |
| 4 | AuPd/N-PR24-PS | 52,638 | n.d. | solventless | 35,000 | 120 | 1.5 | 75 | [101] |
| 5 | Pd/NCNT873K | 11,457 | 95 (after 1 h) | solventless | 3000 | 80 | 2 | 76 | [52] |
| 6 | Pd/NCNT673K | 7865 | 88 (after 1 h) | solventless | 3000 | 80 | 2 | 73 | [52] |
| 7 | Pd/NCNT473K | 2387 | 35 (after 1 h) | solventless | 3000 | 80 | 2 | 68 | [52] |
| 8 | Pd/C | n.d. | 20 (after 5 h) | solventless | n.d. | 125 | 1 | 99 | [105] |
| 9 | Pd/B-C | n.d. | >99 (after 5 h) | solventless | n.d. | 125 | 1 | 99 | [105] |
| 10 | [email protected] | n.d. | 3 (after 1 h) | CH3OH | 180 | 60 | 1 | 12 | [107] |
| 11 | Co@NC-2 | n.d. | 31 (after 1 h) | CH3OH | 180 | 60 | 1 | 52 | [107] |
| 12 | Co@NC-4 | n.d. | 58 (after 1 h) | CH3OH | 180 | 60 | 1 | 77 | [107] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Campisi, S.; Chan-Thaw, C.E.; Villa, A. Understanding Heteroatom-Mediated Metal–Support Interactions in Functionalized Carbons: A Perspective Review. Appl. Sci. 2018, 8, 1159. https://doi.org/10.3390/app8071159
AMA Style
Campisi S, Chan-Thaw CE, Villa A. Understanding Heteroatom-Mediated Metal–Support Interactions in Functionalized Carbons: A Perspective Review. Applied Sciences. 2018; 8(7):1159. https://doi.org/10.3390/app8071159
Chicago/Turabian StyleCampisi, Sebastiano, Carine Edith Chan-Thaw, and Alberto Villa. 2018. "Understanding Heteroatom-Mediated Metal–Support Interactions in Functionalized Carbons: A Perspective Review" Applied Sciences 8, no. 7: 1159. https://doi.org/10.3390/app8071159
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.