Direct Delivery of Cas9-sgRNA Ribonucleoproteins into Cells Using a Nanoneedle Array

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Preparation of Cas9 Protein
2.3. Preparation of sgRNA
2.4. Evaluation of the Cleavage Activity of RNP
2.5. Fluorescent Observation of Cas9 Delivery to Cell
2.6. Knockout of EGFP Gene in Reporter Cells
2.7. Knockout of Nestin Gene in Mouse Breast Cancer Cells
3. Results and Discussion
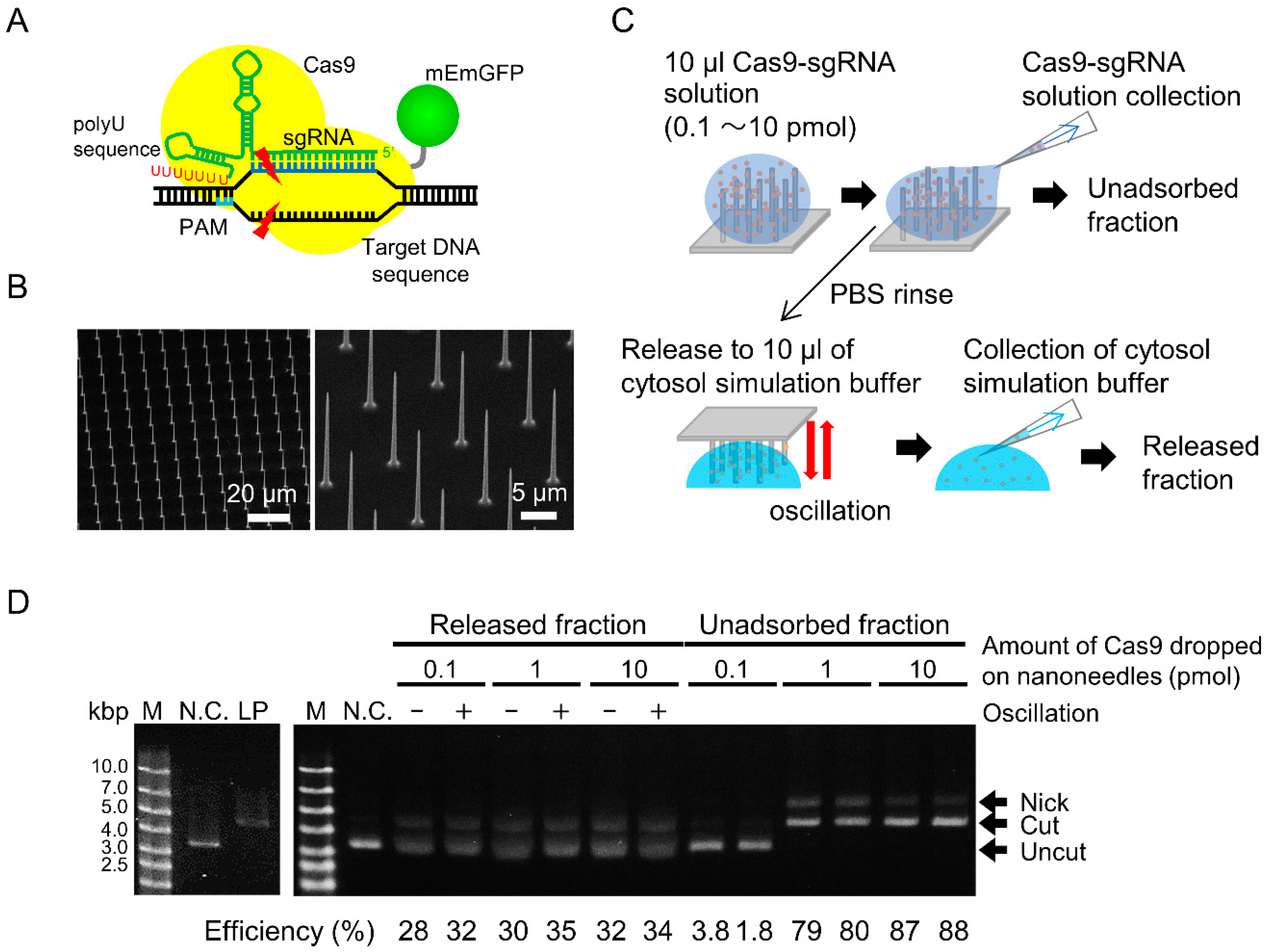
3.1. Cleavage Activity and Amount of Released RNP
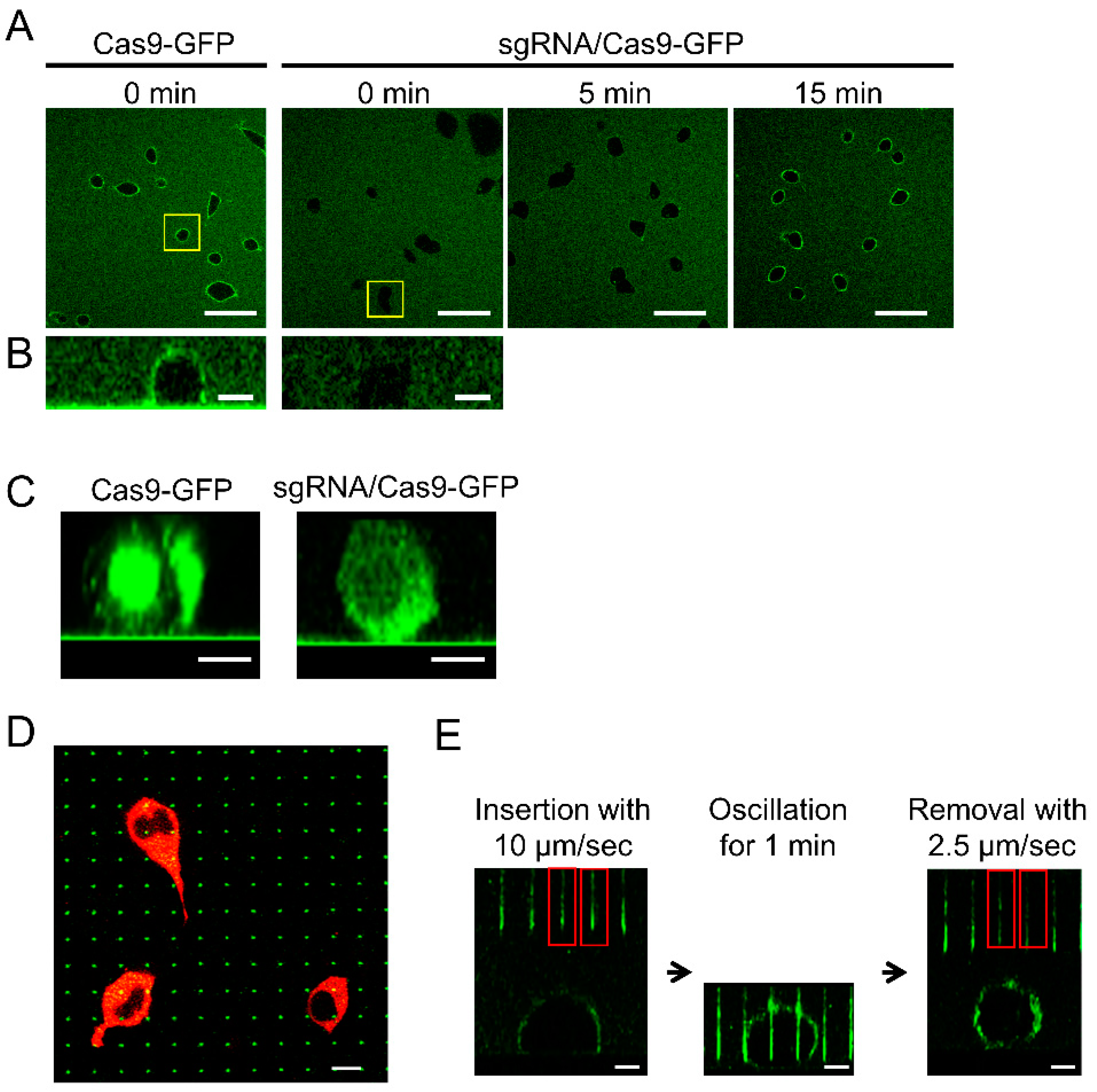
3.2. Delivery of Cas9-EmGFP to HEK293 Cell
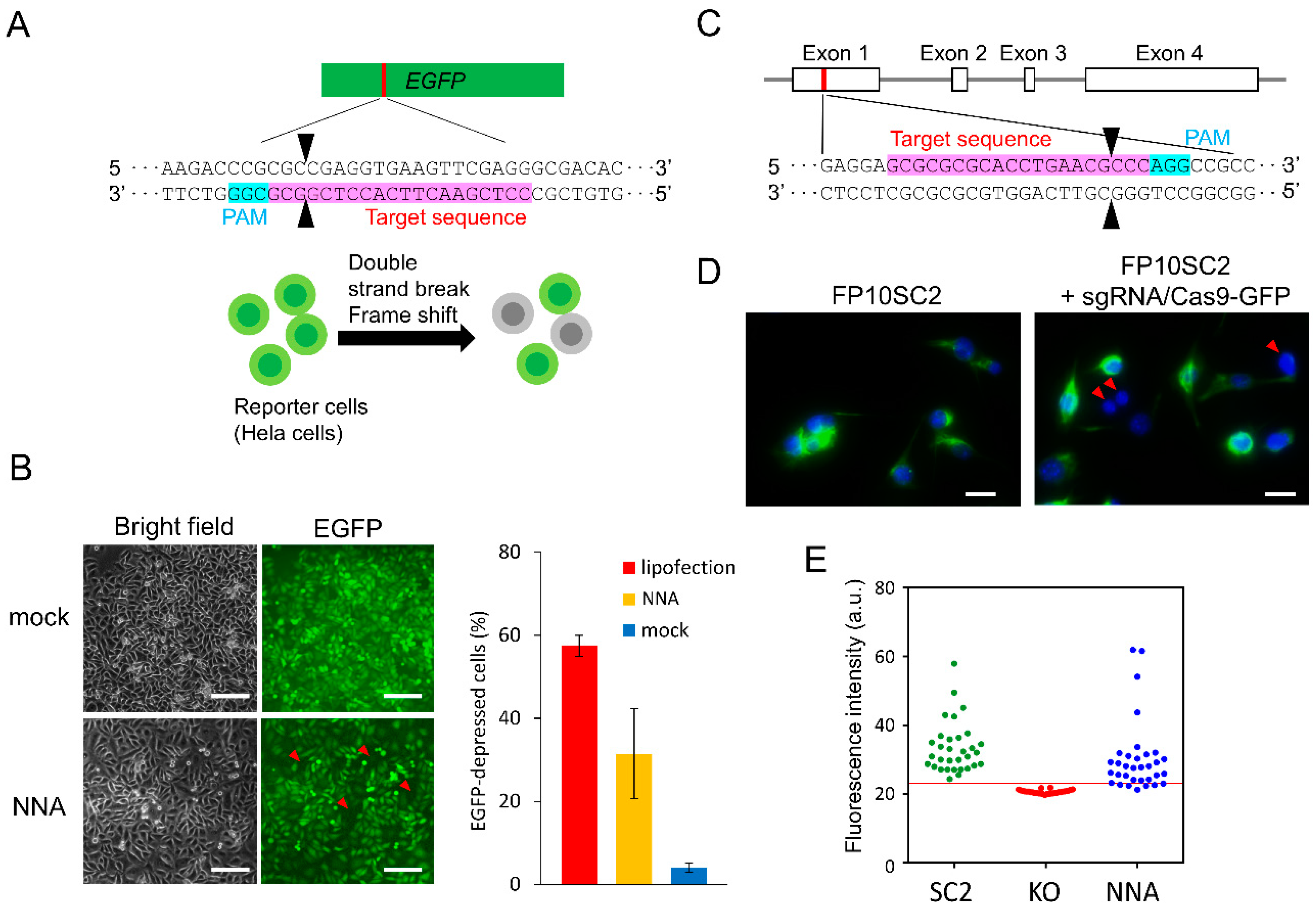
3.3. EGFP Knockout by Direct RNP Delivery
3.4. Nestin Knockout by Direct RNP Delivery
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 2012, 482, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.L.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.B.; Jiang, W.Y.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Jakimo, N.; Jacobson, J.M. Minimal PAM specificity of a highly similar SpCas9 ortholog. Sci. Adv. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Higashiyama, T. pKAMA-ITACHI vectors for highly efficient CRISPR/Cas9-mediated gene knockout in Arabidopsis thaliana. Plant Cell Physiol. 2017, 58, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuris, J.A.; Thompson, D.B.; Shu, Y.; Guilinger, J.P.; Bessen, J.L.; Hu, J.H.; Maeder, M.L.; Joung, J.K.; Chen, Z.Y.; Liu, D.R. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2015, 33, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zuris, J.A.; Meng, F.T.; Rees, H.; Sun, S.; Deng, P.; Han, Y.; Gao, X.; Pouli, D.; Wu, Q.; et al. Efficient delivery of genome-editing proteins using bioreducible lipid nanoparticles. Proc. Nat. Acad. Sci. USA 2016, 113, 2868–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Liang, X.Q.; Xie, H.M.; Kumar, S.; Ravinder, N.; Potter, J.; du Jeu, X.D.; Chesnut, J.D. Improved delivery of Cas9 protein/gRNA complexes using lipofectamine CRISPRMAX. Biotechnol. Lett. 2016, 38, 919–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKnight, T.E.; Melechko, A.V.; Griffin, G.D.; Guillorn, M.A.; Merkulov, V.I.; Serna, F.; Hensley, D.K.; Doktycz, M.J.; Lowndes, D.H.; Simpson, M.L. Intracellular integration of synthetic nanostructures with viable cells for controlled biochemical manipulation. Nanotechnology 2003, 14, 551–556. [Google Scholar] [CrossRef]
- Krivitsky, V.; Hsiung, L.C.; Lichtenstein, A.; Brudnik, B.; Kantaev, R.; Elnathan, R.; Pevzner, A.; Khatchtourints, A.; Patolsky, F. Si nanowires forest-based on-chip biomolecular filtering, separation and preconcentration devices: Nanowires do it all. Nano Lett. 2012, 12, 4748–4756. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, Y.; Yan, L.; Kwok, S.Y.; Li, W.; Wang, Z.; Zhu, X.; Zhu, G.; Zhang, W.; Chen, X.; et al. Poking cells for efficient vector-free intracellular delivery. Nat. Commun. 2014, 5, 4466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiappini, C.; De Rosa, E.; Martinez, J.O.; Liu, X.; Steele, J.; Stevens, M.M.; Tasciotti, E. Biodegradable silicon nanoneedles delivering nucleic acids intracellularly induce localized in vivo neovascularization. Nat. Mater. 2015, 14, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Obataya, I.; Nakamura, C.; Han, S.; Nakamura, N.; Miyake, J. Nanoscale operation of a living cell using an atomic force microscope with a nanoneedle. Nano Lett. 2005, 5, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, D.; Rao Sathuluri, R.; Kato, Y.; Silberberg, Y.R.; Kawamura, R.; Iwata, F.; Kobayashi, T.; Nakamura, C. Oscillating high-aspect-ratio monolithic silicon nanoneedle array enables efficient delivery of functional bio-macromolecules into living cells. Sci. Rep. 2015, 5, 15325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, D.; Nishio, M.; Kato, Y.; Yoshida, W.; Abe, K.; Fukazawa, K.; Ishihara, K.; Iwata, F.; Ikebukuro, K.; Nakamura, C. ATP-mediated release of a DNA-binding protein from a silicon nanoneedle array. Electrochemistry 2016, 84, 305–307. [Google Scholar] [CrossRef]
- Matsumoto, D.; Yamagishi, A.; Saito, M.; Sathuluri, R.R.; Silberberg, Y.R.; Iwata, F.; Kobayashi, T.; Nakamura, C. Mechanoporation of living cells for delivery of macromolecules using nanoneedle array. J. Biosci. Bioeng. 2016, 122, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Mieda, S.; Amemiya, Y.; Kihara, T.; Okada, T.; Sato, T.; Fukazawa, K.; Ishihara, K.; Nakamura, N.; Miyake, J.; Nakamura, C. Mechanical force-based probing of intracellular proteins from living cells using antibody-immobilized nanoneedles. Biosens. Bioelectron. 2012, 31, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, R.; Miyazaki, M.; Shimizu, K.; Matsumoto, Y.; Silberberg, Y.R.; Sathuluri, R.R.; Iijima, M.; Kuroda, S.; Iwata, F.; Kobayashi, T.; et al. A new cell separation method based on antibody-immobilized nanoneedle arrays for the detection of intracellular markers. Nano Lett. 2017, 17, 7117–7124. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Kurabayashi, A.; Akimitsu, N.; Furihata, M. Expression of cadherin-17 promotes metastasis in a highly bone marrow metastatic murine breast cancer model. BioMed Res. Int. 2017, 2017, 8494286. [Google Scholar] [CrossRef] [PubMed]
- Taki, M.; Kato, Y.; Miyagishi, M.; Takagi, Y.; Taira, K. Small-interfering-RNA expression in cells based on an efficiently constructed dumbbell-shaped DNA. Angew. Chem. Int. Ed. 2004, 43, 3160–3163. [Google Scholar] [CrossRef] [PubMed]
- Nagasaki, A.; Kato, Y.; Meguro, K.; Yamagishi, A.; Nakamura, C.; Uyeda, T.Q.P. A genome editing vector that enables easy selection and identification of knockout cells. Plasmid 2018, 98, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 2014, 156, 935–949. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Name | Sequence 5′-3′ | |
|---|---|---|
| Oligonucleotide | sgRNA-(7U) | AAAAAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC |
| T7sgRNA-GFP-g1 (R332) | TAATACGACTCACTATAGGCCTCGAACTTCACCTCGGCGGTTTTAGAGCTAGAAATAGCAAG | |
| T7sgRNA-mNes (F461) | TAATACGACTCACTATAGGGCGCGCGCACCTGAACGCCCGTTTTAGAGCTAGAAATAGCAAG | |
| sgRNA | sgRNA_GFPg1-7U | GGCCUCGAACUUCACCUCGGCGGUUUUAGAGCUAGAAAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCUUUUUUU |
| sgRNA_mNes (F461) | GGGCGCGCGCACCUGAACGCCCGUUUUAGAGCUAGAAAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCUUUUUUU |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamagishi, A.; Matsumoto, D.; Kato, Y.; Honda, Y.; Morikawa, M.; Iwata, F.; Kobayashi, T.; Nakamura, C. Direct Delivery of Cas9-sgRNA Ribonucleoproteins into Cells Using a Nanoneedle Array. Appl. Sci. 2019, 9, 965. https://doi.org/10.3390/app9050965
Yamagishi A, Matsumoto D, Kato Y, Honda Y, Morikawa M, Iwata F, Kobayashi T, Nakamura C. Direct Delivery of Cas9-sgRNA Ribonucleoproteins into Cells Using a Nanoneedle Array. Applied Sciences. 2019; 9(5):965. https://doi.org/10.3390/app9050965
Chicago/Turabian StyleYamagishi, Ayana, Daisuke Matsumoto, Yoshio Kato, Yuki Honda, Mone Morikawa, Futoshi Iwata, Takeshi Kobayashi, and Chikashi Nakamura. 2019. "Direct Delivery of Cas9-sgRNA Ribonucleoproteins into Cells Using a Nanoneedle Array" Applied Sciences 9, no. 5: 965. https://doi.org/10.3390/app9050965