Fetal Alcohol Spectrum Disorder (FASD) Associated Neural Defects: Complex Mechanisms and Potential Therapeutic Targets


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Alcohol Induced Craniofacial Defects
1.2. Alcohol Induced Central Nervous System Development Defects
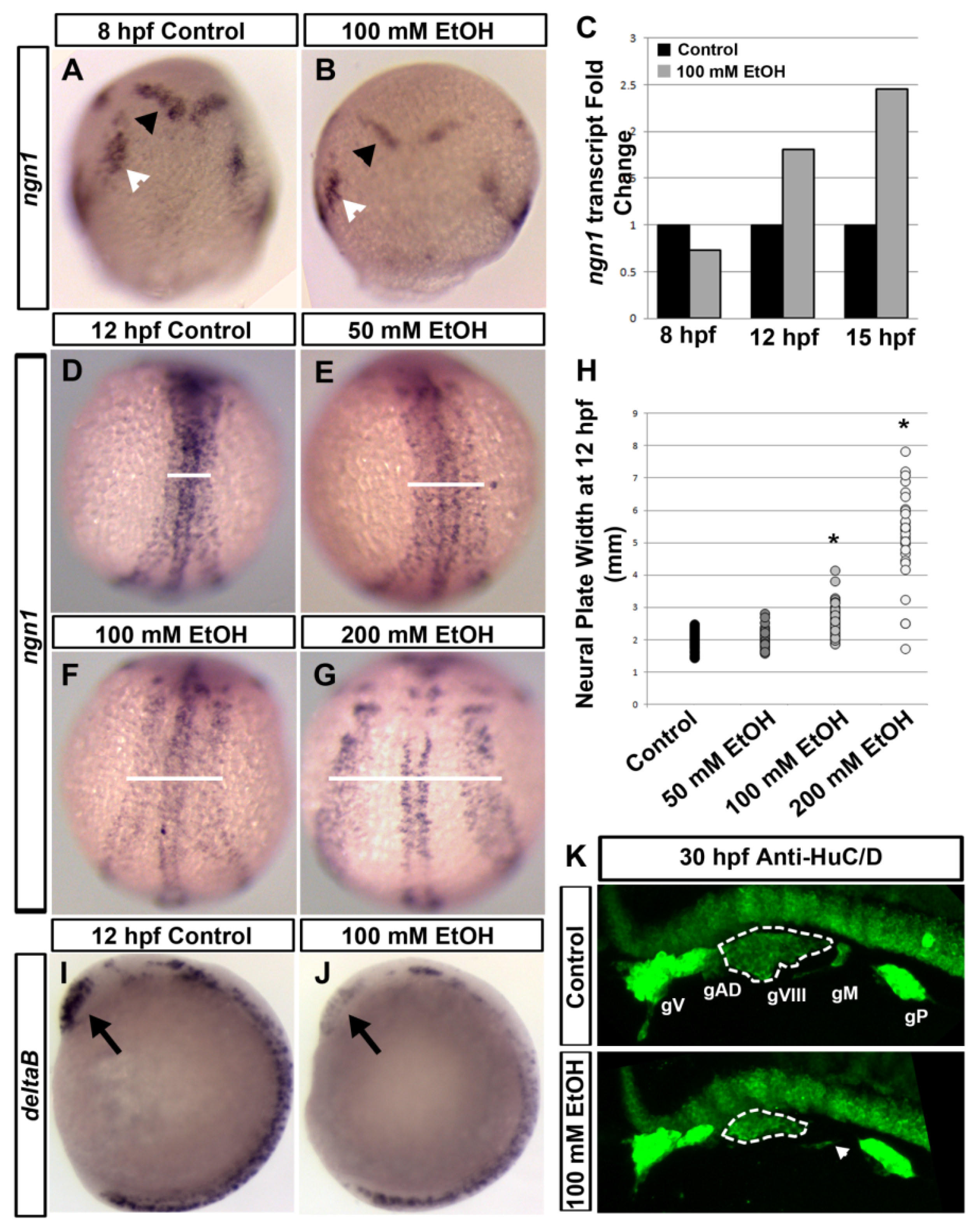
1.3. Ethanol Induced Defects in Sensory System Development
1.3.1. Auditory Defects
1.3.2. Olfactory Defects
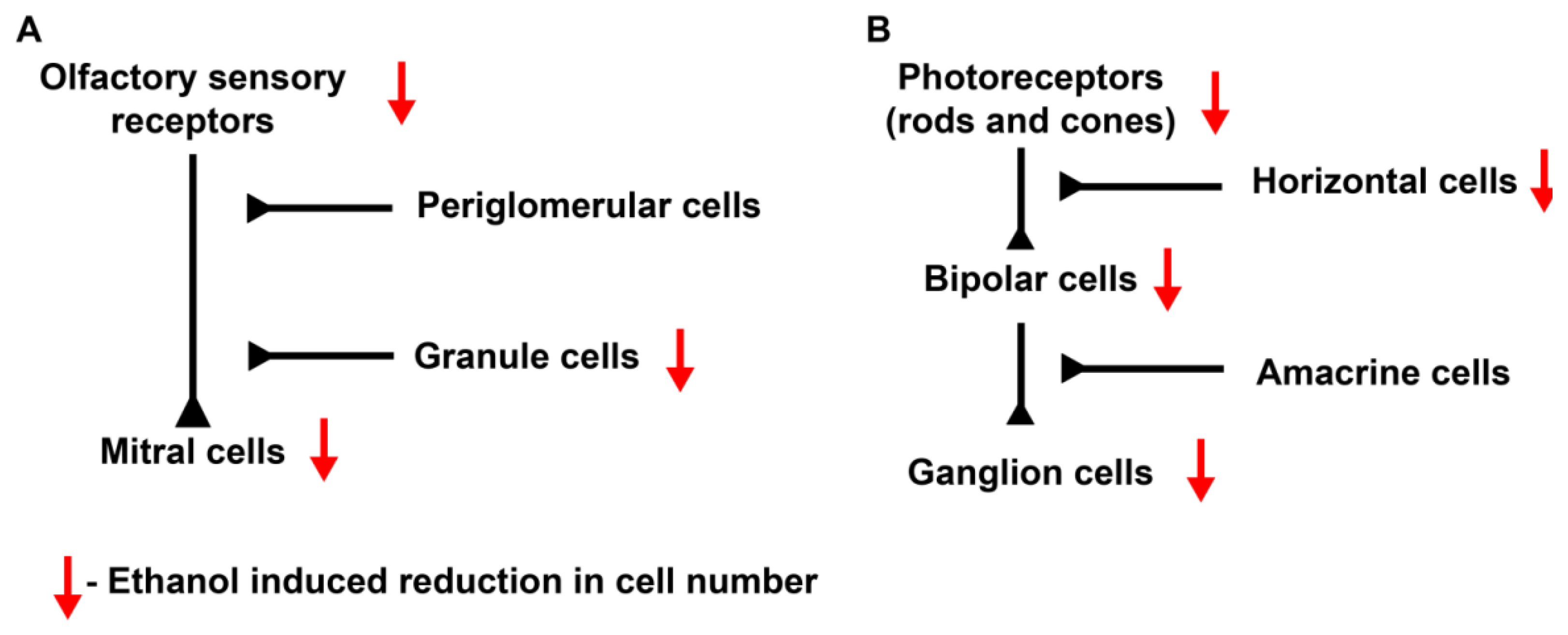
1.3.3. Ocular Defects
1.4. Ethanol and Neural Crest Cells
2. Potential Mechanisms Underlying Ethanol-Induced Neural Defects: Possible Therapeutic Targets
2.1. Ethanol-Induced Nutritional Deficiencies
2.1.1. Retinoic Acid
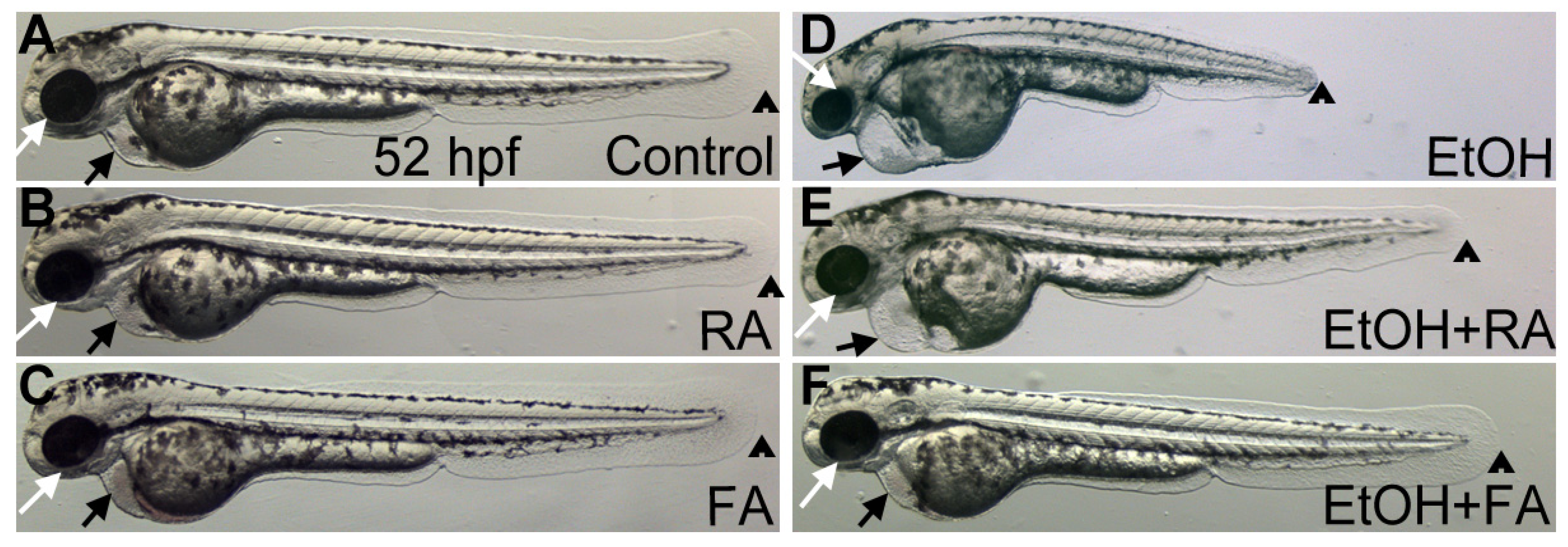
2.1.2. Folic Acid
2.1.3. Choline
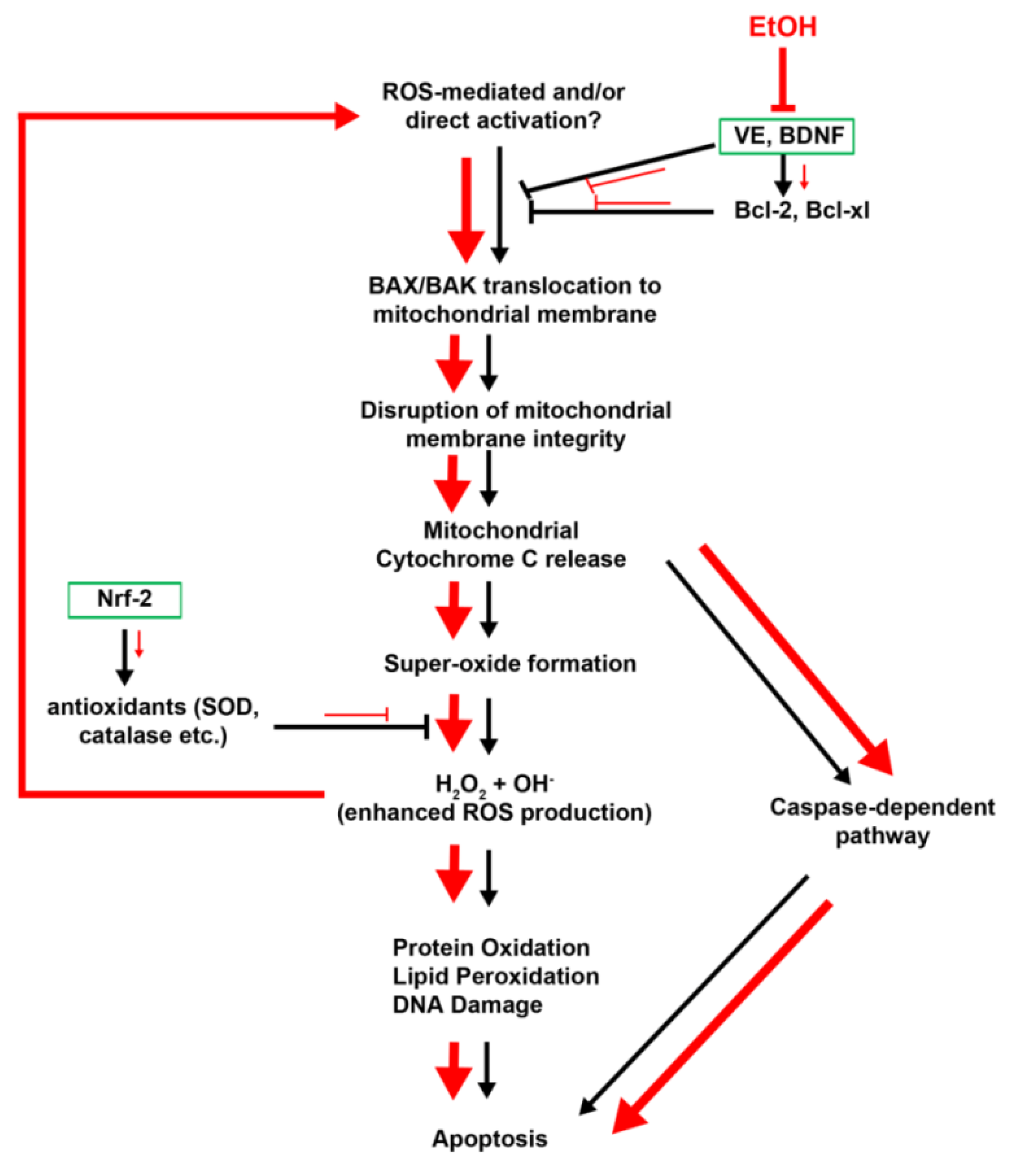
2.1.4. Vitamin E
2.2. Ethanol-Induced Oxidative Stress: ROS Generation and Cell Death
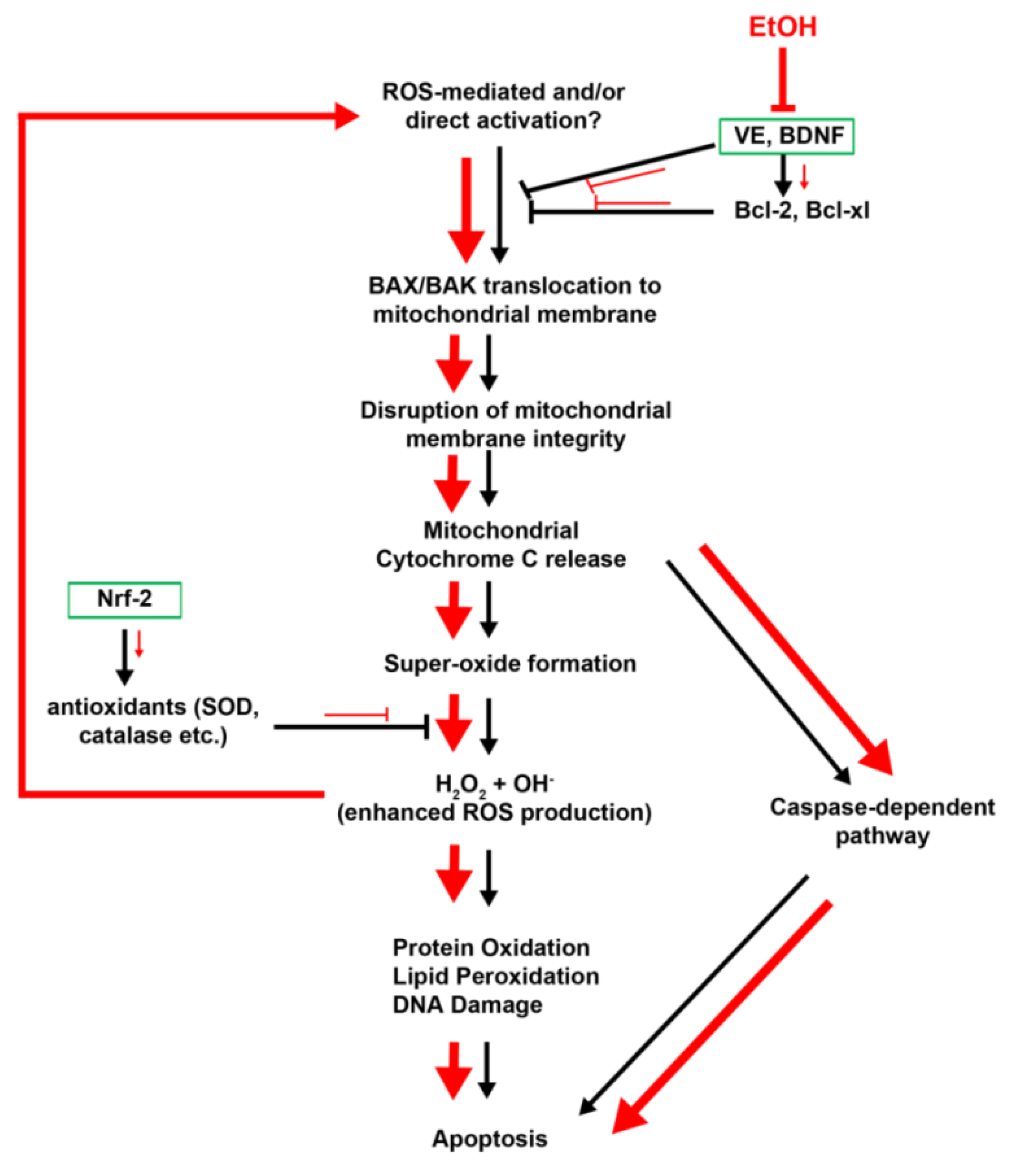
2.3. Cell Death
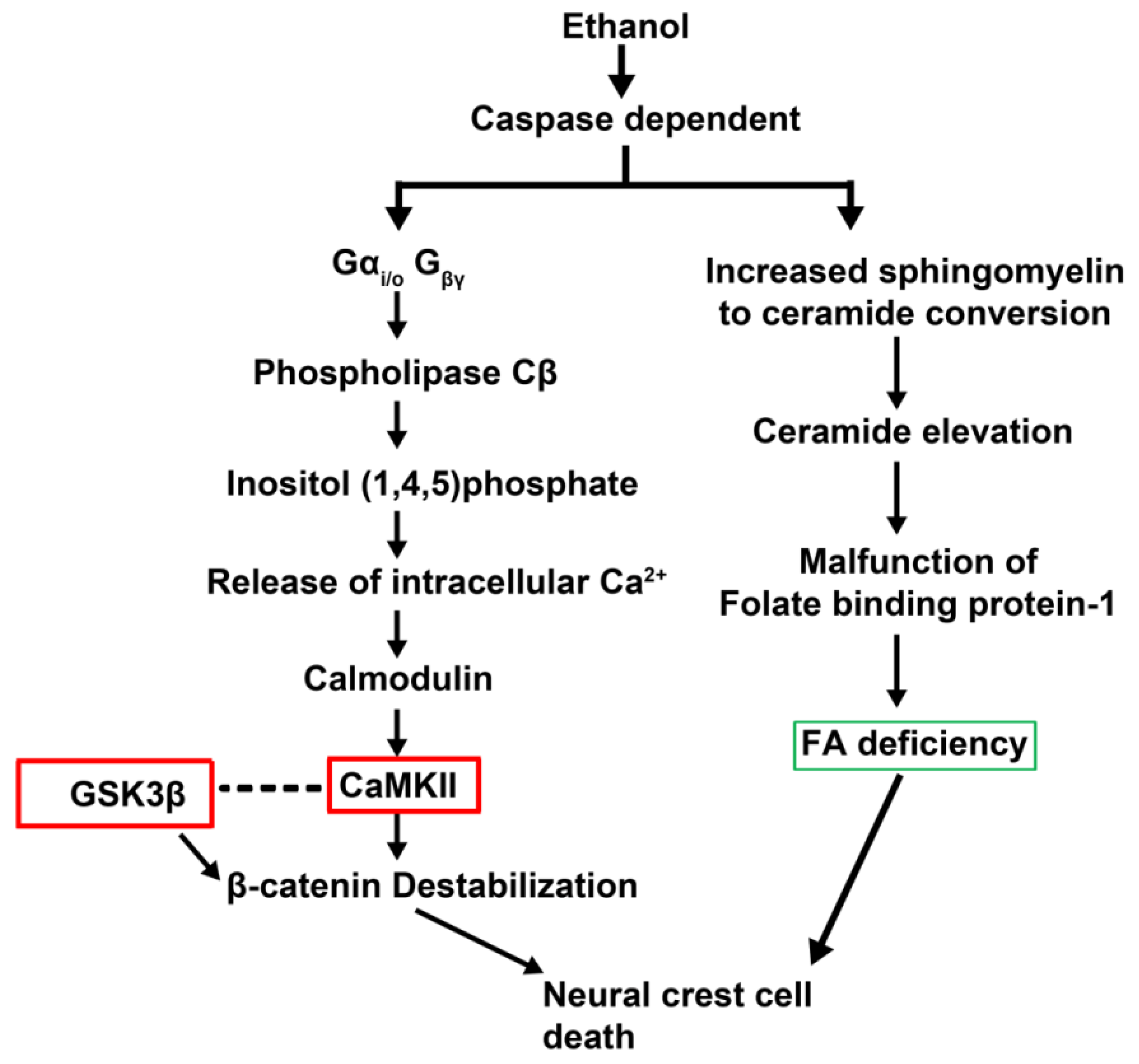
2.4. Epigenetics
2.5. Non-Coding RNA
3. Conclusions
Acknowledgements
Conflict of Interest
References
- May, P.A.; Gossage, J.P.; Kalberg, W.O.; Robinson, L.K.; Buckley, D.; Manning, M.; Hoyme, H.E. Prevalence and epidemiologic characteristics of FASD from various research methods with an emphasis on recent in-school studies. Dev. Disabil. Res. Rev. 2009, 15, 176–192. [Google Scholar] [CrossRef]
- Moore, E.S.; Ward, R.E.; Jamison, P.L.; Morris, C.A.; Bader, P.I.; Hall, B.D. The subtle facial signs of prenatal exposure to alcohol: An anthropometric approach. J. Pediatr. 2001, 139, 215–219. [Google Scholar] [CrossRef]
- Moore, E.S.; Ward, R.E.; Wetherill, L.F.; Rogers, J.L.; Autti-Ramo, I.; Fagerlund, A.; Jacobson, S.W.; Robinson, L.K.; Hoyme, H.E.; Mattson, S.N.; Foroud, T. Unique facial features distinguish fetal alcohol syndrome patients and controls in diverse ethnic populations. Alcohol. Clin. Exp. Res. 2007, 31, 1707–1713. [Google Scholar] [CrossRef]
- Webster, W.S.; Walsh, D.A.; Lipson, A.H.; McEwen, S.E. Teratogenesis after acute alcohol exposure in inbred and out-bred mice. Neurobehav. Toxicol. 1980, 2, 227–234. [Google Scholar]
- Sulik, K.K. Genesis of alcohol-induced craniofacial dysmorphism. Exp. Biol. Med. (Maywood) 2005, 230, 366–375. [Google Scholar]
- Sulik, K.K.; Cook, C.S.; Webster, W.S. Teratogens and craniofacial malformations: Relationships to cell death. Development 1988, 103, 213–231. [Google Scholar]
- Sulik, K.K.; Johnston, M.C.; Webb, M.A. Fetal alcohol syndrome: Embryogenesis in a mouse model. Science 1981, 214, 936–938. [Google Scholar]
- Blader, P.; Strahle, U. Ethanol impairs migration of the prechordal plate in the zebrafish embryo. Dev. Biol. 1998, 201, 185–201. [Google Scholar] [CrossRef]
- Lipinski, R.J.; Hammond, P.; O’Leary-Moore, S.K.; Ament, J.J.; Pecevich, S.J.; Jiang, Y.; Budin, F.; Parnell, S.E.; Suttie, M.; Godin, E.A.; et al. Ethanol-induced face-brain dysmorphology patterns are correlative and exposure-stage dependent. PLoS One 2012, 7, e43067. [Google Scholar]
- Anthony, B.; Vinci-Booher, S.; Wetherill, L.; Ward, R.; Goodlett, C.; Zhou, F.C. Alcohol-induced facial dysmorphology in C57BL/6 mouse models of fetal alcohol spectrum disorder. Alcohol 2010, 44, 659–671. [Google Scholar] [CrossRef]
- Nakatsuji, N. Craniofacial malformation in Xenopus laevis tadpoles caused by the exposure of early embryos to ethanol. Teratology 1983, 28, 299–305. [Google Scholar] [CrossRef]
- Pennington, S.N. Alcohol metabolism and fetal hypoplasia in chick brain. Alcohol 1988, 5, 91–94. [Google Scholar] [CrossRef]
- Roebuck, T.M.; Mattson, S.N.; Riley, E.P. A review of the neuroanatomical findings in children with fetal alcohol syndrome or prenatal exposure to alcohol. Alcohol. Clin. Exp. Res. 1998, 22, 339–344. [Google Scholar] [CrossRef]
- Coulter, C.L.; Leech, R.W.; Schaefer, G.B.; Scheithauer, B.W.; Brumback, R.A. Midline cerebral dysgenesis, dysfunction of the hypothalamic-pituitary axis, and fetal alcohol effects. Arch. Neurol. 1993, 50, 771–775. [Google Scholar]
- Lebel, C.; Roussotte, F.; Sowell, E.R. Imaging the impact of prenatal alcohol exposure on the structure of the developing human brain. Neuropsychol. Rev. 2011, 21, 102–118. [Google Scholar] [CrossRef]
- Zhou, D.; Lebel, C.; Lepage, C.; Rasmussen, C.; Evans, A.; Wyper, K.; Pei, J.; Andrew, G.; Massey, A.; Massey, D.; Beaulieu, C. Developmental cortical thinning in fetal alcohol spectrum disorders. NeuroImage 2011, 58, 16–25. [Google Scholar] [CrossRef]
- Yang, Y.; Roussotte, F.; Kan, E.; Sulik, K.K.; Mattson, S.N.; Riley, E.P.; Jones, K.L.; Adnams, C.M.; May, P.A.; O’Connor, M.J.; et al. Abnormal cortical thickness alterations in fetal alcohol spectrum disorders and their relationships with facial dysmorphology. Cereb. Cortex 2012, 22, 1170–1179. [Google Scholar] [CrossRef]
- Yang, Y.; Phillips, O.R.; Kan, E.; Sulik, K.K.; Mattson, S.N.; Riley, E.P.; Jones, K.L.; Adnams, C.M.; May, P.A.; O’Connor, M.J.; et al. Callosal thickness reductions relate to facial dysmorphology in fetal alcohol spectrum disorders. Alcohol. Clin. Exp. Res. 2012, 36, 798–806. [Google Scholar] [CrossRef]
- O’Leary-Moore, S.K.; Parnell, S.E.; Godin, E.A.; Dehart, D.B.; Ament, J.J.; Khan, A.A.; Johnson, G.A.; Styner, M.A.; Sulik, K.K. Magnetic resonance microscopy-based analyses of the brains of normal and ethanol-exposed fetal mice. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 953–964. [Google Scholar] [CrossRef]
- O’Leary-Moore, S.K.; Parnell, S.E.; Lipinski, R.J.; Sulik, K.K. Magnetic resonance-based imaging in animal models of fetal alcohol spectrum disorder. Neuropsychol. Rev. 2011, 21, 167–185. [Google Scholar] [CrossRef]
- Parnell, S.E.; O’Leary-Moore, S.K.; Godin, E.A.; Dehart, D.B.; Johnson, B.W.; Allan Johnson, G.; Styner, M.A.; Sulik, K.K. Magnetic resonance microscopy defines ethanol-induced brain abnormalities in prenatal mice: Effects of acute insult on gestational day 8. Alcohol. Clin. Exp. Res. 2009, 33, 1001–1011. [Google Scholar] [CrossRef]
- Godin, E.A.; O’Leary-Moore, S.K.; Khan, A.A.; Parnell, S.E.; Ament, J.J.; Dehart, D.B.; Johnson, B.W.; Allan Johnson, G.; Styner, M.A.; Sulik, K.K. Magnetic resonance microscopy defines ethanol-induced brain abnormalities in prenatal mice: Effects of acute insult on gestational day 7. Alcohol. Clin. Exp. Res. 2010, 34, 98–111. [Google Scholar] [CrossRef]
- Pierce, D.R.; Williams, D.K.; Light, K.E. Purkinje cell vulnerability to developmental ethanol exposure in the rat cerebellum. Alcohol. Clin. Exp. Res. 1999, 23, 1650–1659. [Google Scholar] [CrossRef]
- Ramadoss, J.; Lunde, E.R.; Chen, W.J.; West, J.R.; Cudd, T.A. Temporal vulnerability of fetal cerebellar Purkinje cells to chronic binge alcohol exposure: Ovine model. Alcohol. Clin. Exp. Res. 2007, 31, 1738–1745. [Google Scholar] [CrossRef]
- Ramadoss, J.; Lunde, E.R.; Pina, K.B.; Chen, W.J.; Cudd, T.A. All three trimester binge alcohol exposure causes fetal cerebellar purkinje cell loss in the presence of maternal hypercapnea, acidemia, and normoxemia: Ovine model. Alcohol. Clin. Exp. Res. 2007, 31, 1252–1258. [Google Scholar] [CrossRef]
- West, J.R.; Goodlett, C.R.; Bonthius, D.J.; Hamre, K.M.; Marcussen, B.L. Cell population depletion associated with fetal alcohol brain damage: Mechanisms of BAC-dependent cell loss. Alcohol. Clin. Exp. Res. 1990, 14, 813–818. [Google Scholar] [CrossRef]
- Olney, J.W. Fetal alcohol syndrome at the cellular level. Addict. Biol. 2004, 9, 137–149. [Google Scholar] [CrossRef]
- Sarmah, S.; Muralidharan, P.; Curtis, C.L.; McClintick, J.N.; Buente, B.B.; Holdgrafer, D.J.; Ogbeifun, O.; Olorungbounmi, O.C.; Patino, L.; Lucas, R.; et al. Ethanol exposure disrupts extraembryonic microtubule cytoskeleton and embryonic blastomere cell adhesion, producing epiboly and gastrulation defects. 2013; submitted for publication. [Google Scholar]
- Church, M.W. Chronic in utero alcohol exposure affects auditory function in rats and in humans. Alcohol 1987, 4, 231–239. [Google Scholar] [CrossRef]
- Cone-Wesson, B. Prenatal alcohol and cocaine exposure: Influences on cognition, speech, language, and hearing. J. Commun. Disord. 2005, 38, 279–302. [Google Scholar] [CrossRef]
- Kotch, L.E.; Sulik, K.K. Patterns of ethanol-induced cell death in the developing nervous system of mice; neural fold states through the time of anterior neural tube closure. Int. J. Dev. Neurosci. 1992, 10, 273–279. [Google Scholar] [CrossRef]
- Liang, Y.; Wang, Z.M.; Qu, W.D. Evaluation of embryonic alcoholism from auditory event-related potential in fetal rats. Chin. Med. J. (Engl.) 2004, 117, 1422–1424. [Google Scholar]
- Eade, A.M.; Sheehe, P.R.; Youngentob, S.L. Ontogeny of the enhanced fetal-ethanol-induced behavioral and neurophysiologic olfactory response to ethanol odor. Alcohol. Clin. Exp. Res. 2010, 34, 206–213. [Google Scholar] [CrossRef]
- Youngentob, S.L.; Glendinning, J.I. Fetal ethanol exposure increases ethanol intake by making it smell and taste better. Proc. Natl. Acad. Sci. USA 2009, 106, 5359–5364. [Google Scholar] [CrossRef]
- Bonthius, D.J.; Bonthius, N.E.; Napper, R.M.; West, J.R. Early postnatal alcohol exposure acutely and permanently reduces the number of granule cells and mitral cells in the rat olfactory bulb: A stereological study. J. Comp. Neurol. 1992, 324, 557–566. [Google Scholar] [CrossRef]
- Akers, K.G.; Kushner, S.A.; Leslie, A.T.; Clarke, L.; van der Kooy, D.; Lerch, J.P.; Frankland, P.W. Fetal alcohol exposure leads to abnormal olfactory bulb development and impaired odor discrimination in adult mice. Mol. Brain 2011, 4, 29. [Google Scholar] [CrossRef]
- Middleton, F.A.; Carrierfenster, K.; Mooney, S.M.; Youngentob, S.L. Gestational ethanol exposure alters the behavioral response to ethanol odor and the expression of neurotransmission genes in the olfactory bulb of adolescent rats. Brain Res. 2009, 1252, 105–116. [Google Scholar] [CrossRef]
- Stromland, K.; Pinazo-Duran, M.D. Ophthalmic involvement in the fetal alcohol syndrome: Clinical and animal model studies. Alcohol Alcohol. 2002, 37, 2–8. [Google Scholar] [CrossRef]
- Hug, T.E.; Fitzgerald, K.M.; Cibis, G.W. Clinical and electroretinographic findings in fetal alcohol syndrome. J. AAPOS 2000, 4, 200–204. [Google Scholar] [CrossRef]
- Katz, L.M.; Fox, D.A. Prenatal ethanol exposure alters scotopic and photopic components of adult rat electroretinograms. Invest. Ophthalmol. Vis. Sci. 1991, 32, 2861–2872. [Google Scholar]
- Matsui, J.I.; Egana, A.L.; Sponholtz, T.R.; Adolph, A.R.; Dowling, J.E. Effects of ethanol on photoreceptors and visual function in developing zebrafish. Invest. Ophthalmol. Vis. Sci. 2006, 47, 4589–4597. [Google Scholar] [CrossRef]
- Bilotta, J.; Saszik, S.; Givin, C.M.; Hardesty, H.R.; Sutherland, S.E. Effects of embryonic exposure to ethanol on zebrafish visual function. Neurotoxicol. Teratol. 2002, 24, 759–766. [Google Scholar] [CrossRef]
- Arenzana, F.J.; Carvan, M.J., III; Aijon, J.; Sanchez-Gonzalez, R.; Arevalo, R.; Porteros, A. Teratogenic effects of ethanol exposure on zebrafish visual system development. Neurotoxicol. Teratol. 2006, 28, 342–348. [Google Scholar]
- Yelin, R.; Kot, H.; Yelin, D.; Fainsod, A. Early molecular effects of ethanol during vertebrate embryogenesis. Differentiation 2007, 75, 393–403. [Google Scholar] [CrossRef]
- Kashyap, B.; Frederickson, L.C.; Stenkamp, D.L. Mechanisms for persistent microphthalmia following ethanol exposure during retinal neurogenesis in zebrafish embryos. Vis. Neurosci. 2007, 24, 409–421. [Google Scholar]
- Deng, J.X.; Liu, X.; Zang, J.F.; Huang, H.E.; Xi, Y.; Zheng, H.; Yao, H.L.; Yu, D.M.; Deng, J.B. The effects of prenatal alcohol exposure on the developmental retina of mice. Alcohol Alcohol. 2012, 47, 380–385. [Google Scholar] [CrossRef]
- Cartwright, M.M.; Smith, S.M. Stage-dependent effects of ethanol on cranial neural crest cell development: Partial basis for the phenotypic variations observed in fetal alcohol syndrome. Alcohol. Clin. Exp. Res. 1995, 19, 1454–1462. [Google Scholar] [CrossRef]
- Cartwright, M.M.; Tessmer, L.L.; Smith, S.M. Ethanol-induced neural crest apoptosis is coincident with their endogenous death, but is mechanistically distinct. Alcohol. Clin. Exp. Res. 1998, 22, 142–149. [Google Scholar] [CrossRef]
- Ahlgren, S.C.; Thakur, V.; Bronner-Fraser, M. Sonic hedgehog rescues cranial neural crest from cell death induced by ethanol exposure. Proc. Natl. Acad. Sci. USA 2002, 99, 10476–10481. [Google Scholar] [CrossRef]
- Smith, S.M. Alcohol-induced cell death in the embryo. Alcohol Health Res. World 1997, 21, 287–297. [Google Scholar]
- Bronner-Fraser, M. Neural crest cell formation and migration in the developing embryo. FASEB J. 1994, 8, 699–706. [Google Scholar]
- Knecht, A.K.; Bronner-Fraser, M. Induction of the neural crest: A multigene process. Nat. Rev. Genet. 2002, 3, 453–461. [Google Scholar] [CrossRef]
- Lieber, C.S. Relationships between nutrition, alcohol use, and liver disease. Alcohol Res. Health 2003, 27, 220–231. [Google Scholar]
- Durston, A.J.; Timmermans, J.P.; Hage, W.J.; Hendriks, H.F.; de Vries, N.J.; Heideveld, M.; Nieuwkoop, P.D. Retinoic acid causes an anteroposterior transformation in the developing central nervous system. Nature 1989, 340, 140–144. [Google Scholar] [CrossRef]
- Mezey, E.; Holt, P.R. The inhibitory effect of ethanol on retinol oxidation by human liver and cattle retina. Exp. Mol. Pathol. 1971, 15, 148–156. [Google Scholar] [CrossRef]
- Duester, G. A hypothetical mechanism for fetal alcohol syndrome involving ethanol inhibition of retinoic acid synthesis at the alcohol dehydrogenase step. Alcohol. Clin. Exp. Res. 1991, 15, 568–572. [Google Scholar] [CrossRef]
- Keir, W.J. Inhibition of retinoic acid synthesis and its implications in fetal alcohol syndrome. Alcohol. Clin. Exp. Res. 1991, 15, 560–564. [Google Scholar] [CrossRef]
- Marrs, J.A.; Clendenon, S.G.; Ratcliffe, D.R.; Fielding, S.M.; Liu, Q.; Bosron, W.F. Zebrafish fetal alcohol syndrome model: Effects of ethanol are rescued by retinoic acid supplement. Alcohol 2010, 44, 707–715. [Google Scholar] [CrossRef]
- Kot-Leibovich, H.; Fainsod, A. Ethanol induces embryonic malformations by competing for retinaldehyde dehydrogenase activity during vertebrate gastrulation. Dis. Model. Mech. 2009, 2, 295–305. [Google Scholar] [CrossRef]
- Yelin, R.; Schyr, R.B.; Kot, H.; Zins, S.; Frumkin, A.; Pillemer, G.; Fainsod, A. Ethanol exposure affects gene expression in the embryonic organizer and reduces retinoic acid levels. Dev. Biol. 2005, 279, 193–204. [Google Scholar] [CrossRef]
- Sarmah, S.; Marrs, J.A. Complex cardiac defects after ethanol exposure during discrete cardiogenic events in zebrafish: Prevention with folic acid. 2013; submitted for publication. [Google Scholar]
- Kashyap, B.; Frey, R.A.; Stenkamp, D.L. Ethanol-induced microphthalmia is not mediated by changes in retinoic acid or sonic hedgehog signaling during retinal neurogenesis. Alcohol. Clin. Exp. Res. 2011, 35, 1644–1661. [Google Scholar]
- Barak, A.J.; Beckenhauer, H.C.; Tuma, D.J.; Badakhsh, S. Effects of prolonged ethanol feeding on methionine metabolism in rat liver. Biochem. Cell Biol. 1987, 65, 230–233. [Google Scholar] [CrossRef]
- Mason, J.B.; Choi, S.W. Effects of alcohol on folate metabolism: Implications for carcinogenesis. Alcohol 2005, 35, 235–241. [Google Scholar] [CrossRef]
- Barak, A.J.; Beckenhauer, H.C.; Junnila, M.; Tuma, D.J. Dietary betaine promotes generation of hepatic S-adenosylmethionine and protects the liver from ethanol-induced fatty infiltration. Alcohol. Clin. Exp. Res. 1993, 17, 552–555. [Google Scholar] [CrossRef]
- Beaudin, A.E.; Abarinov, E.V.; Noden, D.M.; Perry, C.A.; Chu, S.; Stabler, S.P.; Allen, R.H.; Stover, P.J. Shmt1 and de novo thymidylate biosynthesis underlie folate-responsive neural tube defects in mice. Am. J. Clin. Nutr. 2011, 93, 789–798. [Google Scholar] [CrossRef]
- Hutson, J.R.; Stade, B.; Lehotay, D.C.; Collier, C.P.; Kapur, B.M. Folic acid transport to the human fetus is decreased in pregnancies with chronic alcohol exposure. PLoS One 2012, 7, e38057. [Google Scholar]
- Wang, L.L.; Zhang, Z.; Li, Q.; Yang, R.; Pei, X.; Xu, Y.; Wang, J.; Zhou, S.F.; Li, Y. Ethanol exposure induces differential microRNA and target gene expression and teratogenic effects which can be suppressed by folic acid supplementation. Hum. Reprod. 2009, 24, 562–579. [Google Scholar]
- Serrano, M.; Han, M.; Brinez, P.; Linask, K.K. Fetal alcohol syndrome: Cardiac birth defects in mice and prevention with folate. Am. J. Obstet. Gynecol. 2010, 203, 75 e77–75 e15. [Google Scholar]
- Fisher, M.C.; Zeisel, S.H.; Mar, M.H.; Sadler, T.W. Perturbations in choline metabolism cause neural tube defects in mouse embryos in vitro. FASEB J. 2002, 16, 619–621. [Google Scholar]
- Thomas, J.D.; Abou, E.J.; Dominguez, H.D. Prenatal choline supplementation mitigates the adverse effects of prenatal alcohol exposure on development in rats. Neurotoxicol. Teratol. 2009, 31, 303–311. [Google Scholar] [CrossRef]
- Thomas, J.D.; Idrus, N.M.; Monk, B.R.; Dominguez, H.D. Prenatal choline supplementation mitigates behavioral alterations associated with prenatal alcohol exposure in rats. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 827–837. [Google Scholar] [CrossRef]
- Monk, B.R.; Leslie, F.M.; Thomas, J.D. The effects of perinatal choline supplementation on hippocampal cholinergic development in rats exposed to alcohol during the brain growth spurt. Hippocampus 2012, 22, 1750–1757. [Google Scholar] [CrossRef]
- Otero, N.K.; Thomas, J.D.; Saski, C.A.; Xia, X.; Kelly, S.J. Choline supplementation and DNA methylation in the hippocampus and prefrontal cortex of rats exposed to alcohol during development. Alcohol. Clin. Exp. Res. 2012, 36, 1701–1709. [Google Scholar] [CrossRef]
- Miller, R.R., Jr.; Olson, B.M.; Rorick, N.; Wittingen, A.L.; Bullock, M. Embryonic exposure to exogenous alpha- and gamma-tocopherol partially attenuates ethanol-induced changes in brain morphology and brain membrane fatty acid composition. Nutr. Neurosci. 2003, 6, 201–212. [Google Scholar] [CrossRef]
- Mitchell, J.J.; Paiva, M.; Heaton, M.B. The antioxidants vitamin E and beta-carotene protect against ethanol-induced neurotoxicity in embryonic rat hippocampal cultures. Alcohol 1999, 17, 163–168. [Google Scholar] [CrossRef]
- Siler-Marsiglio, K.I.; Shaw, G.; Heaton, M.B. Pycnogenol and vitamin E inhibit ethanol-induced apoptosis in rat cerebellar granule cells. J. Neurobiol. 2004, 59, 261–271. [Google Scholar] [CrossRef]
- Miller, G.W.; Labut, E.M.; Lebold, K.M.; Floeter, A.; Tanguay, R.L.; Traber, M.G. Zebrafish (Danio rerio) fed vitamin E-deficient diets produce embryos with increased morphologic abnormalities and mortality. J. Nutr. Biochem. 2012, 23, 478–486. [Google Scholar] [CrossRef]
- Heaton, M.B.; Paiva, M.; Siler-Marsiglio, K. Ethanol influences on Bax translocation, mitochondrial membrane potential, and reactive oxygen species generation are modulated by vitamin E and brain-derived neurotrophic factor. Alcohol. Clin. Exp. Res. 2011, 35, 1122–1133. [Google Scholar] [CrossRef]
- Brocardo, P.S.; Gil-Mohapel, J.; Christie, B.R. The role of oxidative stress in fetal alcohol spectrum disorders. Brain Res. Rev. 2011, 67, 209–225. [Google Scholar]
- Peng, Y.; Yang, P.H.; Guo, Y.; Ng, S.S.; Liu, J.; Fung, P.C.; Tay, D.; Ge, J.; He, M.L.; Kung, H.F.; Lin, M.C. Catalase and peroxiredoxin 5 protect Xenopus embryos against alcohol-induced ocular anomalies. Invest. Ophthalmol. Vis. Sci. 2004, 45, 23–29. [Google Scholar] [CrossRef]
- Antonio, A.M.; Gillespie, R.A.; Druse-Manteuffel, M.J. Effects of lipoic acid on antiapoptotic genes in control and ethanol-treated fetal rhombencephalic neurons. Brain Res 2011, 1383, 13–21. [Google Scholar] [CrossRef]
- Parnell, S.E.; Sulik, K.K.; Dehart, D.B.; Chen, S.Y. Reduction of ethanol-induced ocular abnormalities in mice through dietary administration of N-acetylcysteine. Alcohol 2010, 44, 699–705. [Google Scholar] [CrossRef]
- Peng, Y.; Kwok, K.H.; Yang, P.H.; Ng, S.S.; Liu, J.; Wong, O.G.; He, M.L.; Kung, H.F.; Lin, M.C. Ascorbic acid inhibits ROS production, NF-kappa B activation and prevents ethanol-induced growth retardation and microencephaly. Neuropharmacology 2005, 48, 426–434. [Google Scholar] [CrossRef]
- Dong, J.; Sulik, K.K.; Chen, S.Y. Nrf2-mediated transcriptional induction of antioxidant response in mouse embryos exposed to ethanol in vivo: Implications for the prevention of fetal alcohol spectrum disorders. Antioxid. Redox Signal. 2008, 10, 2023–2033. [Google Scholar] [CrossRef]
- Kane, C.J.; Phelan, K.D.; Han, L.; Smith, R.R.; Xie, J.; Douglas, J.C.; Drew, P.D. Protection of neurons and microglia against ethanol in a mouse model of fetal alcohol spectrum disorders by peroxisome proliferator-activated receptor-gamma agonists. Brain Behav. Immun. 2011, 25, S137–S145. [Google Scholar] [CrossRef]
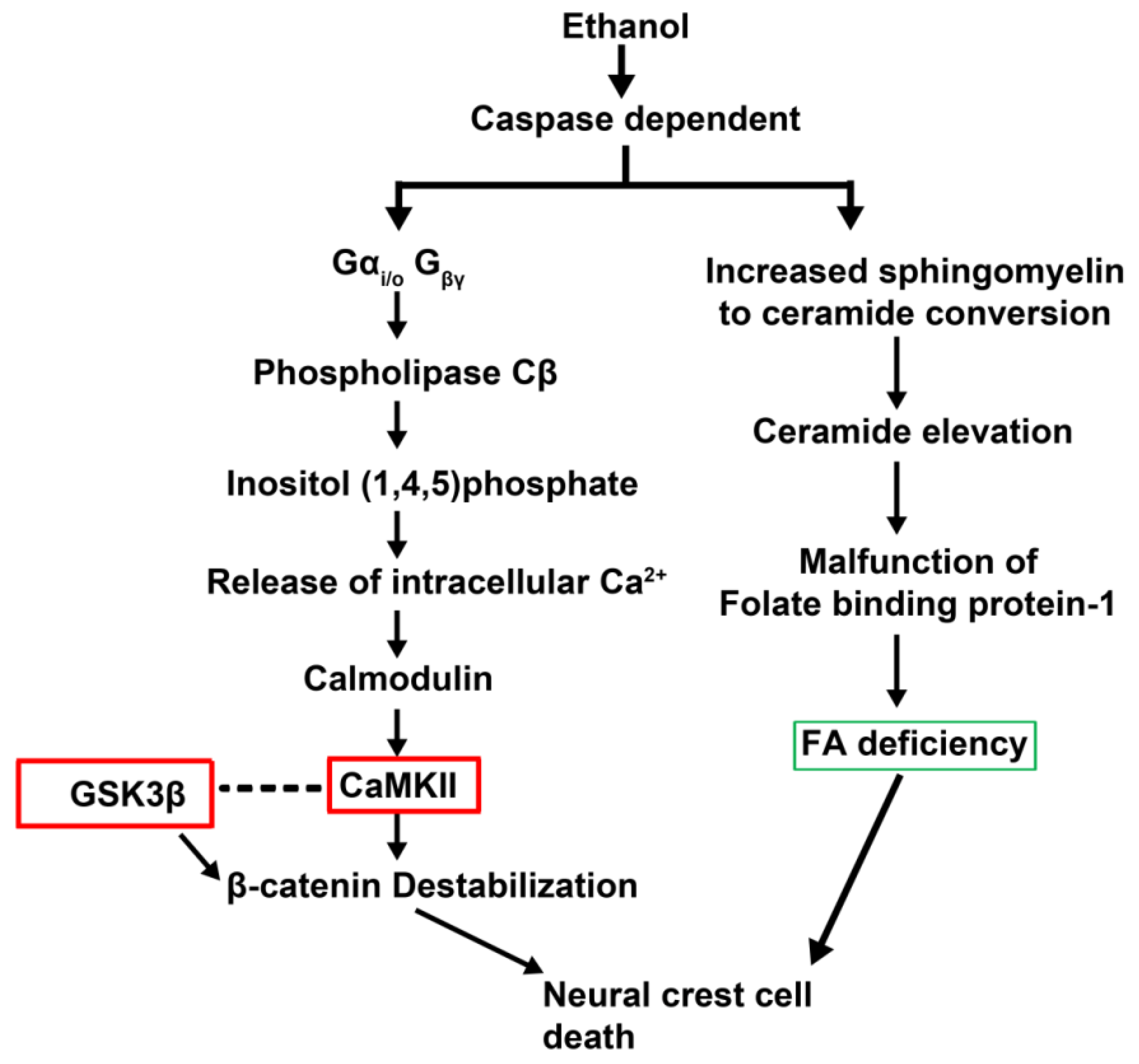
- Wang, G.; Bieberich, E. Prenatal alcohol exposure triggers ceramide-induced apoptosis in neural crest-derived tissues concurrent with defective cranial development. Cell Death Dis. 2010, 1, e46. [Google Scholar] [CrossRef]
- Flentke, G.R.; Garic, A.; Amberger, E.; Hernandez, M.; Smith, S.M. Calcium-mediated repression of beta-catenin and its transcriptional signaling mediates neural crest cell death in an avian model of fetal alcohol syndrome. Birth Defects Res. A Clin. Mol. Teratol. 2011, 91, 591–602. [Google Scholar] [CrossRef]
- Garic-Stankovic, A.; Hernandez, M.R.; Chiang, P.J.; Debelak-Kragtorp, K.A.; Flentke, G.R.; Armant, D.R.; Smith, S.M. Ethanol triggers neural crest apoptosis through the selective activation of a pertussis toxin-sensitive G protein and a phospholipase Cbeta-dependent Ca2+ transient. Alcohol. Clin. Exp. Res. 2005, 29, 1237–1246. [Google Scholar] [CrossRef]
- Vangipuram, S.D.; Grever, W.E.; Parker, G.C.; Lyman, W.D. Ethanol increases fetal human neurosphere size and alters adhesion molecule gene expression. Alcohol. Clin. Exp. Res. 2008, 32, 339–347. [Google Scholar] [CrossRef]
- Garic, A.; Flentke, G.R.; Amberger, E.; Hernandez, M.; Smith, S.M. CaMKII activation is a novel effector of alcohol’s neurotoxicity in neural crest stem/progenitor cells. J. Neurochem. 2011, 118, 646–657. [Google Scholar] [CrossRef]
- Luo, J. Lithium-mediated protection against ethanol neurotoxicity. Front. Neurosci. 2010, 4, 41. [Google Scholar]
- Zhou, F.C. DNA methylation program during development. Front. Biol. 2012, 485–494. [Google Scholar] [CrossRef]
- Mill, J.; Petronis, A. Pre- and peri-natal environmental risks for attention-deficit hyperactivity disorder (ADHD): The potential role of epigenetic processes in mediating susceptibility. J. Child Psychol. Psychiatry 2008, 49, 1020–1030. [Google Scholar] [CrossRef]
- Resendiz, M.; Chen, Y.; Ozturk, N.C.; Zhou, F.C. Epigenetic medicine and fetal alcohol spectrum disorders. Epigenomics 2013, 5, 73–86. [Google Scholar] [CrossRef]
- Haycock, P.C. Fetal alcohol spectrum disorders: The epigenetic perspective. Biol. Reprod. 2009, 81, 607–617. [Google Scholar] [CrossRef]
- Garro, A.J.; McBeth, D.L.; Lima, V.; Lieber, C.S. Ethanol consumption inhibits fetal DNA methylation in mice: Implications for the fetal alcohol syndrome. Alcohol. Clin. Exp. Res. 1991, 15, 395–398. [Google Scholar] [CrossRef]
- Kaminen-Ahola, N.; Ahola, A.; Maga, M.; Mallitt, K.A.; Fahey, P.; Cox, T.C.; Whitelaw, E.; Chong, S. Maternal ethanol consumption alters the epigenotype and the phenotype of offspring in a mouse model. PLoS Genet. 2010, 6, e1000811. [Google Scholar] [CrossRef]
- Bonsch, D.; Lenz, B.; Fiszer, R.; Frieling, H.; Kornhuber, J.; Bleich, S. Lowered DNA methyltransferase (DNMT-3b) mRNA expression is associated with genomic DNA hypermethylation in patients with chronic alcoholism. J. Neural. Transm. 2006, 113, 1299–1304. [Google Scholar] [CrossRef]
- Zhou, F.C.; Balaraman, Y.; Teng, M.; Liu, Y.; Singh, R.P.; Nephew, K.P. Alcohol alters DNA methylation patterns and inhibits neural stem cell differentiation. Alcohol. Clin. Exp. Res. 2011, 35, 735–746. [Google Scholar] [CrossRef]
- Liu, Y.; Balaraman, Y.; Wang, G.; Nephew, K.P.; Zhou, F.C. Alcohol exposure alters DNA methylation profiles in mouse embryos at early neurulation. Epigenetics 2009, 4, 500–511. [Google Scholar] [CrossRef]
- Chen, Y.; Ozturk, N.C.; Zhou, F.C. DNA methylation program in developing hippocampus and its alteration by alcohol. PLoS One 2013, 8, e60503. [Google Scholar] [CrossRef]
- Zhou, F.C.; Chen, Y.; Love, A. Cellular DNA methylation program during neurulation and its alteration by alcohol exposure. Birth Defects Res. A Clin. Mol. Teratol. 2011, 91, 703–715. [Google Scholar] [CrossRef]
- Singh, R.P.; Shiue, K.; Schomberg, D.; Zhou, F.C. Cellular epigenetic modifications of neural stem cell differentiation. Cell Transplant. 2009, 18, 1197–1211. [Google Scholar] [CrossRef]
- Subbanna, S.; Shivakumar, M.; Umapathy, N.S.; Saito, M.; Mohan, P.S.; Kumar, A.; Nixon, R.A.; Verin, A.D.; Psychoyos, D.; Basavarajappa, B.S. G9a-mediated histone methylation regulates ethanol-induced neurodegeneration in the neonatal mouse brain. Neurobiol. Dis. 2013, 54, 475–485. [Google Scholar] [CrossRef]
- Bekdash, R.A.; Zhang, C.; Sarkar, D.K. Gestational choline supplementation normalized fetal alcohol-induced alterations in histone modifications, DNA methylation, and proopiomelanocortin (POMC) gene expression in beta-endorphin-producing POMC neurons of the hypothalamus. Alcohol. Clin. Exp. Res. 2013. [Google Scholar] [CrossRef]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Wang, G.; Wang, X.; Wang, Y.; Yang, J.Y.; Li, L.; Nephew, K.P.; Edenberg, H.J.; Zhou, F.C.; Liu, Y. Identification of transcription factor and microRNA binding sites in responsible to fetal alcohol syndrome. BMC Genomics 2008, 9, S19. [Google Scholar]
- Sathyan, P.; Golden, H.B.; Miranda, R.C. Competing interactions between micro-RNAs determine neural progenitor survival and proliferation after ethanol exposure: Evidence from an ex vivo model of the fetal cerebral cortical neuroepithelium. J. Neurosci. 2007, 27, 8546–8557. [Google Scholar] [CrossRef]
- Soares, A.R.; Pereira, P.M.; Ferreira, V.; Reverendo, M.; Simoes, J.; Bezerra, A.R.; Moura, G.R.; Santos, M.A. Ethanol exposure induces upregulation of specific microRNAs in zebrafish embryos. Toxicol. Sci. 2012, 127, 18–28. [Google Scholar] [CrossRef]
- Shirasaka, T.; Hashimoto, E.; Ukai, W.; Yoshinaga, T.; Ishii, T.; Tateno, M.; Saito, T. Stem cell therapy: Social recognition recovery in a FASD model. Transl. Psychiatry 2012, 2, e188. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Muralidharan, P.; Sarmah, S.; Zhou, F.C.; Marrs, J.A. Fetal Alcohol Spectrum Disorder (FASD) Associated Neural Defects: Complex Mechanisms and Potential Therapeutic Targets. Brain Sci. 2013, 3, 964-991. https://doi.org/10.3390/brainsci3020964
Muralidharan P, Sarmah S, Zhou FC, Marrs JA. Fetal Alcohol Spectrum Disorder (FASD) Associated Neural Defects: Complex Mechanisms and Potential Therapeutic Targets. Brain Sciences. 2013; 3(2):964-991. https://doi.org/10.3390/brainsci3020964
Chicago/Turabian StyleMuralidharan, Pooja, Swapnalee Sarmah, Feng C. Zhou, and James A. Marrs. 2013. "Fetal Alcohol Spectrum Disorder (FASD) Associated Neural Defects: Complex Mechanisms and Potential Therapeutic Targets" Brain Sciences 3, no. 2: 964-991. https://doi.org/10.3390/brainsci3020964