3.1. Pore Intrusion Identification in PBM-coated Membranes after UV-LED Polymerization
As stated in the introduction, PBM polymerization induced by UV-LED light led to a lower filtration capacity of the PBM-coated membrane in model foulant tests compared to redox-polymerized PBM-coated membranes [
7]. The two polymerization attempts differ with regard to the process sequence. Specifically, the redox-polymerized-PBM was spread onto the membrane surface after 5 min of the polymerization initiation. Hence, already formed long-chain polymers increased the viscosity which changed the membrane wetting properties and thus the coating film characteristic. Additionally, according to Equation (1), a higher liquid viscosity reduces the capillary of a liquid–solid interface at a given pore size. In contrast, the UV-LED-PBM was spread onto the membrane prior to the polymerization initiation (start of UV-LED exposure) thus being more liquid in the casting process. Comparable performance for the UV-LED-polymerized-PBM should be achieved with an appropriate adjusted viscosity.
To verify that this was actually due to a higher intrusion into the membrane pores for UV-LED-polymerized PBM, we performed pore intrusion quantification experiments by visualizing fluorescent nanoparticles incorporated into the membrane. The manufacturer specifications give a nominal pore diameter of 35 nm and a maximum possible 0.1 µm pore size (Martin Systems). The total membrane thickness, as delivered, is indicated as 230 ± 20 µm (Martin Systems).
Figure 3 shows the cross-section of the UV-LED-polymerized PBM-coated membrane. Since the cast wet layer thicknesses were 4 ± 2 µm, the potential pore intrusion was calculated by subtracting the coated layer fraction from the total intrusion depth. This resulted in a maximum 60–80 µm intrusion depth along the cutting edge (
Figure 3, green layer). The minimum pore intrusion was determined at around 26 µm.
The calculated values are based only on the visual observations using the confocal light microscope. Therefore, the values are just a rough estimate and provide qualitative prove of the high level of pore intrusion of the liquid PBM prior to the casting process. As the PBM tended to penetrate into the porous PES structure, the UV-LED polymerization caused partial or complete pore blockage. Aside from the increased membrane resistance through a higher membrane thickness, the decreased nominal pore diameter elevated the total membrane resistance substantially, leading to a lower permeability.
These observations prompted us to establish an experimental polymerization protocol that could suitably modulate the PBM viscosity prior to membrane casting and UV-LED curing in order to reduce the capillarity between liquid and solid porous membranes.
3.2. Controlled Radical Polymerization (CRP) of PBM
The basic premise was to verify the possibility of controlling the PBM viscosity by subjecting the PBM to a controlled radical polymerization stage (CRP) before it was cast on the surface of a commercial membrane and subjected to curing by UV-LED.
The goal was to develop a reproducible protocol for stopping the PBM polymerization within a defined time window prior to membrane coating. In this way the viscosity could be suitably increased, in order to reduce membrane pore intrusion during the coating stage. The method of adding TEMPO in order to retard and ultimately stop polymerization was adapted from Platkowski and Reichert [
8]. The polymerization was stopped almost instantaneously after the addition of the very finely ground inhibitor TEMPO to the test tube. The inhibitor amount was gradually reduced in order to find the lowest possible amount required to stop the reaction while still allowing for an effective re-initiation by UV-LED curing with a reasonable amount of initiator. Since the initiator and the inhibitor were mutually influential to the polymerization reaction, it was vital to optimize the ratio of these two materials.
The application time of the inhibitor dosage to the PBM solution was defined as the CRP time Δti and varied between 7, 8, 9 and 10 min, relative to the initial starting value after the first redox initiation. Adding TEMPO too early caused an interruption leading to insufficiently low viscosities, whereas a late dosage had no effect on the self-accelerating polymerization. Successful reaction termination allowed a controlled radical polymerized PBM (CRP-PBM) to be obtained.
Re-initiation was completed first with APS and TMEDA to determine the feasibility of re-starting the chemical reaction. Consequently, the interim periods between stopping and re-starting were kept short and the material input was reduced successively. Re-initiation was then carried out after casting the produced CRP-PBM on the commercial membrane surface, by using the photo-initiator Irgacure 184 and UV-LED light for activation. Small amounts of APS could help eliminate excess TEMPO radicals and thus improve the curing efficacy.
The self-accelerating nature of exothermal polymerization reactions is well known [
13].
Figure 4a–c shows the exothermal temperature profiles recorded in a flask containing 10 g PBM. The data highlights the influence of turbulences inside the test tube at passive (ambient) cooling for a stagnant (0 rpm (a)) and stirred sample at 1000 (b) and 1500 rpm (c) mixing speed.
Much higher temperature deviations in the space (yellow area) for the stagnant PBM solution (max. ± 14.5 K) indicated a highly inhomogeneous polymerization, whereas the temperature distribution was reduced at 1000 rpm (max. ± 6.5 K).
The wider temperature distribution deviation band for less turbulent flow conditions could be explained by the self-accelerating chemical reaction enhanced by the Trommsdorff–Norrish effect [
21]. During polymerization, the heat transfer coefficient for the solid PBM phase dropped compared to the liquid phase, which reduced the heat dissipation.
Moreover, the chances of processes being terminated by recombination became lower due to decreased movement ability of the growing polymers with increasing viscosity. Furthermore, higher temperatures accelerated the radical decay and the heat dissipation was reduced by an increased viscosity with a lower heat conduction coefficient [
21]. These effects caused an inhomogeneous polymerization with higher spatial temperature deviations (
Figure 4, orange deviation band) and partial polymer agglomerates for low turbulences. On the contrary,
Figure 4 ((d), (e) and (f)) depicts the polymerization inside a 1.5 g PBM test tube. The significant lower peak temperatures and the small deviation band were the result of the lower PBM mass. The total reaction enthalpy was much lower and the higher surface-to-volume ratio of the test tube substantially reduced both the Trommsdorff–Norrish effect and temperature increase due to improved heat dissipation into the surroundings. Consequently, more even spatial temperature distributions (max. ± 1 K) were obtained, beneficial for the desired homogeneous polymerization and the result of an even coating layer using casting coating techniques.
Complete polymerization at high turbulences (1500 rpm) did not occur for either PBM masses. This behavior was demonstrated by the near constant temperature observed until minutes 56 and 60, respectively (
Figure 4, bottom graphs (c) and (f)). After reducing the turbulence to 1000 rpm, the PBM polymerized spontaneously in line with the propagation of the other experiments. However, it was noticeable that the temperature development of the delayed polymerization showed lower values, for instance, given a 55 °C temperature compared with 69.6 °C of the 1000 rpm polymerization (10 g PBM,
Figure 4b,c, respectively). It was assumed that the radical diffusion rate was strongly influenced by the high convection conditions, thus inhibiting molecule propagation and chain transfer reactions. Nevertheless, very few radical reactions occurred, reducing the energy release at 1000 rpm.
Spatial distribution of temperature for the 1.5 g water-cooled flask was within the measuring error range of ± 0.5 K as the heat dissipation was further heightened by the high heat convection of the water bath. This further reduced the impact of the Trommsdorff–Norrish effect during polymerization. Consequently, the peak temperatures were lower (
Figure 4e). Therefore, subsequent polymerization experiments were conducted with the smaller PBM sample volume of 1.5 g with active cooling inside the water bath.
Feasibility studies on successful polymerization inhibition were performed. The finely ground TEMPO radical inhibitor was then added to the polymerizing PBM solution.
Figure 5 shows two different experiments using different amounts of TEMPO. A 0.5 mg amount of TEMPO did not completely inhibit polymerization as polymerization restarted spontaneously at minute 27 (
Figure 5, left). Doubling the TEMPO amount to 1 mg resulted in the polymerization stopping successfully (
Figure 5, right), validated by a long-term dormancy. Initiator and inhibitor were mutually influential and minimizing the material input was of prime importance. A 0.9 mg amount of TEMPO was found to be the threshold for a successful, sustained polymerization inhibition.
Based on these findings, a PBM viscosity modification protocol could be established by PBM controlled radical polymerization. This protocol comprises the following sequence of actions, as depicted in
Figure 6 (Temperature measurements are highlighted for the passive cooled PBM):
- (1)
Polymerization initiation using 1.8 mg APS activated by 40.5 µL TMEDA in a water bath at a 20 °C constant temperature
- (2)
Polymerization inhibition with 0.9 mg of TEMPO-dosage administered after a defined time from initiation (controlled radical polymerization time, Δti = 7, 8, 9 and 10 min). This leads to controlled radical polymerized PBMs (CRP-PBMs) of different viscosities depending on the polymerization time Δt.
The CRP-PBM (1 g) thus obtained was then cast on the commercial membrane and cured by UV-LED using 75 mg Irgacure 184 as the photo-initiator, together with APS (5 mg), which was necessary to eliminate any excess of TEMPO radicals that could inhibit the UV-LED—induced curing reaction.
3.3. Rheological Examination
As previously discussed, the viscosity has a significant impact on the capillary and should thus affect the pore intrusion of the PBM into the membrane pores. The dynamic viscosity describes the internal resistance of fluids and gases to flow and shear conditions, depending on temperature and pressure [
22]. The higher the viscosity, the higher the friction forces between the lattice molecules of individual molecular layers.
The potential effect of a higher viscosity of the controlled radical polymerized PBM (CRP-PBM) was observed by thickening the PBM with the water soluble, biodegradable polyethylene glycol (PEG). Visible examinations validated a lower intrusion level through the reduced transparency of the coated substrate. It is known that a higher viscosity reduces the capillarity between a liquid and a solid substrate using the universal Equation (1).
Equation (1) represents the capillarity in mm equal to the ratio of the fluid’s surface tension σ (mN·m
−1) and the fluid density ρ in g·cm
−3, the acceleration of gravity (m·s
−2) and capillary radius
r (mm). Since the density ρ is proportional to the dynamic viscosity (η~ρ) [
23], lower capillarity can be achieved with a higher viscous fluid.
The results of rheological investigations are highlighted in
Table 2 for the original unpolymerized PBM (Δt = 0) and CRP-PBMs at different polymerization times (Δt = 5.5, 7, 8 and 9 min). Frictional variance produced from the under-filling or over-filling of the gap between the cone and the measuring plate could have caused these deviations. The results clearly indicate a successful modification from lower (ca. 7 mPa·s, for unpolymerized PBM) to higher viscosity values (8.1–9, 6 mPa·s, for CRP-PBM) thanks to the controlled radical polymerization (CRP) protocol.
3.4. Nuclear Magnetic Resonance Spectroscopy
Figure 7 shows the typical
13C cross polarization magic angle spinning (CPMAS) spectra recorded for the materials obtained after casting PBMs on a commercial membrane followed by UV-LED curing or redox polymerization, namely: (a) CRP-PBM-9 (∆t = 9 min) (material obtained after CRP of PBM for 9 min followed by UV-LED curing with 5 w% Irgacure 184 as photo-initiator); (b) UV-LED-1.8 (material obtained after polymerization of the PBM with UV-LED using 1.8 w% Irgacure 184 as photo-initiator); (c) UV-LED-5 (material obtained after polymerization of the PBM with UV-LED using 5 w% Irgacure 184 as photo-initiator); and (d) REDOX-PBM (material obtained after redox polymerization of the PBM for 5.5 min).
In all samples, the two resonances at 8.5 ppm and between 22–29 ppm are uniquely attributed to polymerized AUTEAB [
1]. Meanwhile, the signals around 18.1 ppm are associated with polymerized MMA (PMMA), HEMA (PHEMA) and EGDMA.
CPMAS spectra could be interpreted qualitatively given that the intensity of the resonance is a function of the strength of the dipolar coupling between observing carbons and neighboring protons.
Furthermore, dipolar couplings are modulated by local mobility; thus, the incorporated carbonyl groups (C=O) of the components are under-represented due to the absence of directly bonded protons [
24]. Moreover, methylene and methyl groups show different cross-polarization dynamics. However, since all the spectra are recorded with the same parameters, variations in intensity of these groups give a qualitative idea of the variation in the composition of the copolymer [
25,
26].
The intensity ratios between the 8.5 and 22–29 ppm signals were compared across all samples (see
Table 3, Integrals of CP spectra) and a significant variation is seen. This result is counterintuitive, since these signals all originate from polymerized AUTEAB. At 8.5 ppm the methyl groups resonate; 22–29 ppm all the methylene groups of the undecyl chain resonate except the last one bonded to oxygen, thus their relative intensity should not vary from one sample to the other. One possible explanation is that the distribution of stereo sequences of PMMA of PHEMA could vary within the different polymerization approaches, thereby modifying the line shape of the methyl group resonance from 16 to 22 ppm and partially overlapping with methylenes of the undecyl chain. As shown by Wilhelm et al. [
27] in the case of pure PMMA, the broadening of
13C NMR peaks in the solid state can be directly assigned to the effects of conformational disorder within the amorphous PMMA polymer. A variation in microstructure (distribution of stereo sequences) can significantly change the line shape. However, this should be ruled out eventuality since considerable changes in the stereo control of the polymerization requires a large variation in polymerization temperatures, polarity of the solvent or ability of the catalyst to control the stereochemistry of the polymerization [
28]. When comparing the polymerization conditions used for the different PBM samples, it is reasonable to assume that the obtained copolymers are atactic. Thus, the other only explanation for the variation of the internal ratio of the polymerized AUTEAB signal is that the different samples were characterized by a slightly different dynamic of the undecyl chain.
In a truly quantitative spectrum, the intensity ratio of methyls/methylenes for the undecyl chains of polymerized AUTEAB is 3:9. In fact, no sample shows this ratio, as methylene signals are always more intense than expected, because usually CH2 groups cross-polarize more efficiently than CH3. For the REDOX–BPM coated sample, the signals at 22–29 ppm are very intense. This is a possible indication of a very strong dipolar coupling typical of ordered (almost crystalline) systems (vide infra HPDEC spectra with a 20 s recycle delay). Although the 3:9 ratio was never observed because CPMAS is not quantitative, in the spectra of UV-LED-1.8 and UV-LED-5 the methylene signals are lower although very similar in the two samples. The spectrum of CRP-PBM-9 is analogous to those of the last two systems and presents an even lower methylene peak. Interestingly, the CRP-PBM-9 and UV-LED 5 samples distinctly showed detectable signals in the range of 120 to 140 ppm, due to the presence of Irgacure residues inside the polymers.
The HPDEC spectra were recorded with 20 s of recycle delay. Amorphous polymers with a low glass transition temperature, in general, can be analyzed more quantitatively with this technique [
29]. This was also confirmed by the nearly constant 1:3 ratio between the methyl groups and the non-direct bonding to nitrogen or oxygen methylenes of the polymerized AUTEAB molecules (8.5 and 22–29 ppm, respectively) for all samples. The same trend was found for the acrylate signals at 18.1 and 177 ppm. Under stoichiometric conditions, the peak intensity at 177 ppm should be equal to the sum of 1/3 of the integral of signal at 8.5 ppm plus the integral of the signal at 18.1 ppm. The agreement in this case is acceptable. However, the ratio between acrylates (MMA, HEMA and EGDMA) and the polymerized AUTEAB is more adequately quantified by the direct ratio between the methyl signals at 8.5 and 18.1 ppm. The highest amounts were obtained by the REDOX-BPM sample, whereas UV-LED-5 showed the lowest values.
MAS-NMR data demonstrate that while in REDOX-BPM the AUTEAB chains are arranged in rigid structures, possibly ordered, this feature is less pronounced in the other polymerized PBMs. Considering that the feed composition is the same for all the samples, this altered behavior should be attributed to the different reactivity of the monomers under the diverse polymerization conditions. In fact, it seems that in the REDOX polymerization the AUTEAB monomers have a slightly higher preference to react with the growing chain if the reactive monomeric unit is an AUTEAB unit, since homosequences of AUTEAB can favor the formation of ordered domains.
3.5. Model Foulant Tests and the Relationship Between the Individual Parameters
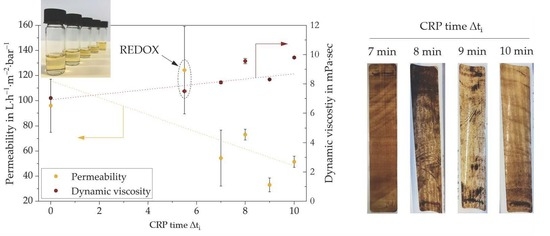
Figure 8a illustrates the permeability of the membranes obtained after UV-LED curing of CRP-PBM‒cast membranes (deriving from CRP-PBMs obtained after different CRP times) and of the membranes obtained after casting with unpolymerized PBM followed by UV-LED polymerization (Δt
i = 0) and by redox polymerization for 5.5 min (REDOX-PBM). The viscosities of the uncast PBMs, as obtained by rheological measurements (see
Section 3.3) are also shown.
As can be seen, for UV-LED cured CRP-PBM‒cast membranes, the permeability (orange dotted line) was decreased by increasing the dynamic viscosity of the uncast CRP-PBMs (brown dotted line), which, in turn, increased with the CRP time. Moreover, the permeability of the REDOX-PBM membrane was significantly higher with respect to all UV-LED polymerized coated membranes.
Since the casting coating procedure was the same for all PBMs (using the spiral-casting knife with a 4 µm wet coating thickness), these quite unexpected results are conceivably due to the modification of the polymer structure of the coating layers obtained upon polymerization under different conditions, as observed by solid state NMR measurements (
Section 3.4).
Figure 8b shows the same membranes after the model foulant tests. The brownish color illustrates the fouling propensity for each coated membrane. As previously mentioned, a lighter color did not necessarily lead to a better fouling mitigation per se, as the membrane tests were conducted at constant TMP. Yet, the results are further evidence that the intervention of CRP followed by UV curing caused the porous coating layer to become a relatively dense structure. Accordingly, a greater change in CRP time Δt
i resulted in a denser layer for water filtration application. This in turn led to lower fouling propensity although the filtration capacity was reduced. With this learned knowledge, membrane coating layers with different filtration properties could be produced for a specific wastewater composition without changing the coating equipment or the coating material input.
All in all, the redox-polymerized PBM-coated membrane showed the highest permeability at 0.5 bar TMP (
Figure 8a). As confirmed by NMR, the redox polymerization gave a different polymeric structure of polymerized PBM compared to the materials obtained by CRP followed by UV-LED curing, which was more favorable in terms of permeability for the filtration of humic acid solutions (100 mg·L
−1, pH 9). On the other hand, the membrane coated with UV-LED polymerized PBM (Δt
i = 0) possessed a higher pore intrusion and thus the pore blockage was more extended with respect to the redox-polymerized PBM-coated membrane.

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}