
New Meloxicam Derivatives—Synthesis and Interaction with Phospholipid Bilayers Measured by Differential Scanning Calorimetry and Fluorescence Spectroscopy

 , ,
, ,
Abstract
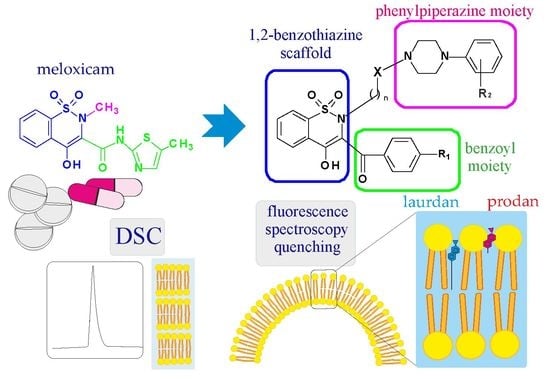
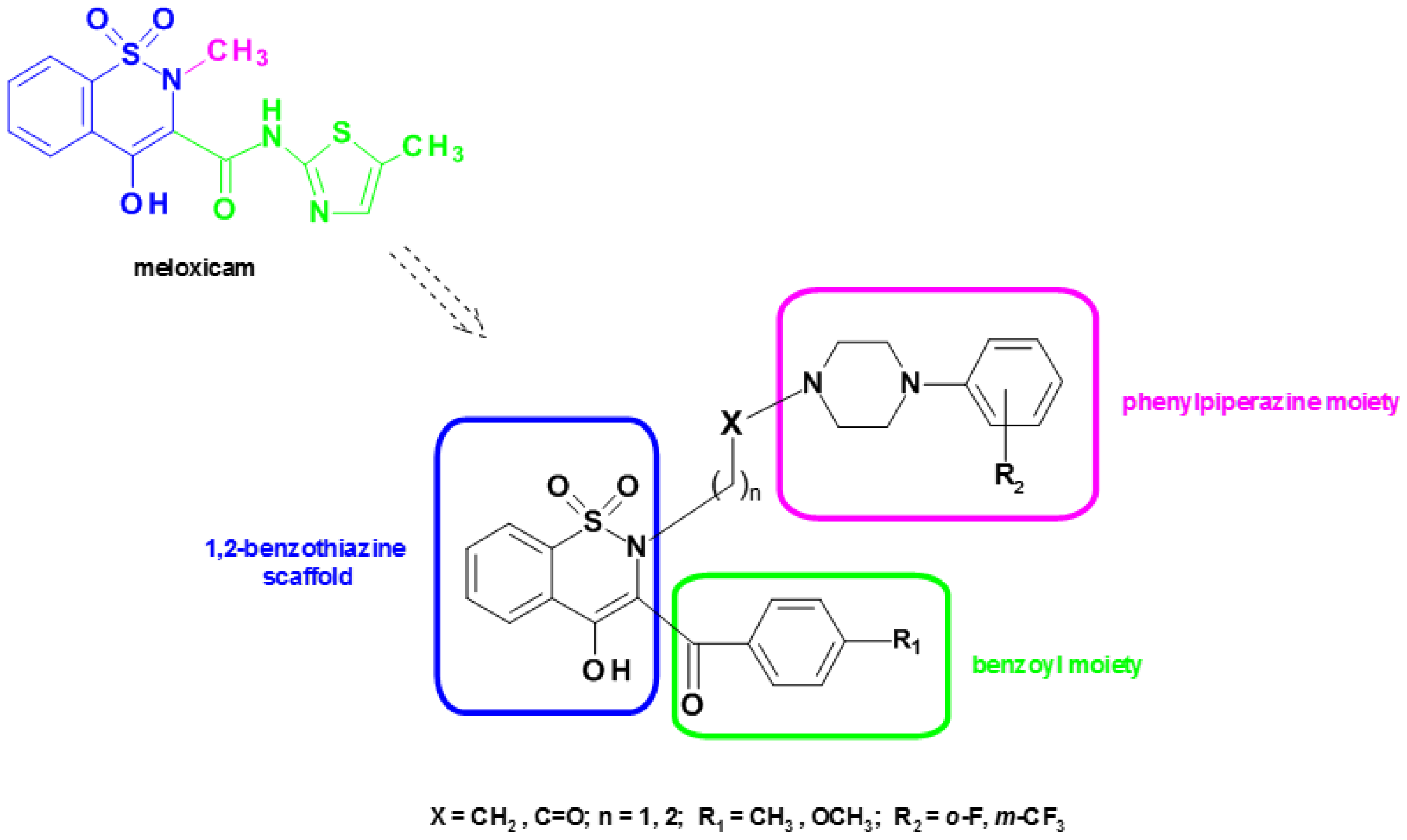
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Experimental
2.2.1. Synthesis
2.2.2. Differential Scanning Calorimetry (DSC)
2.2.3. Fluorescence Spectroscopy
2.2.4. Prediction of ADMET Properties
3. Results
3.1. Synthesis
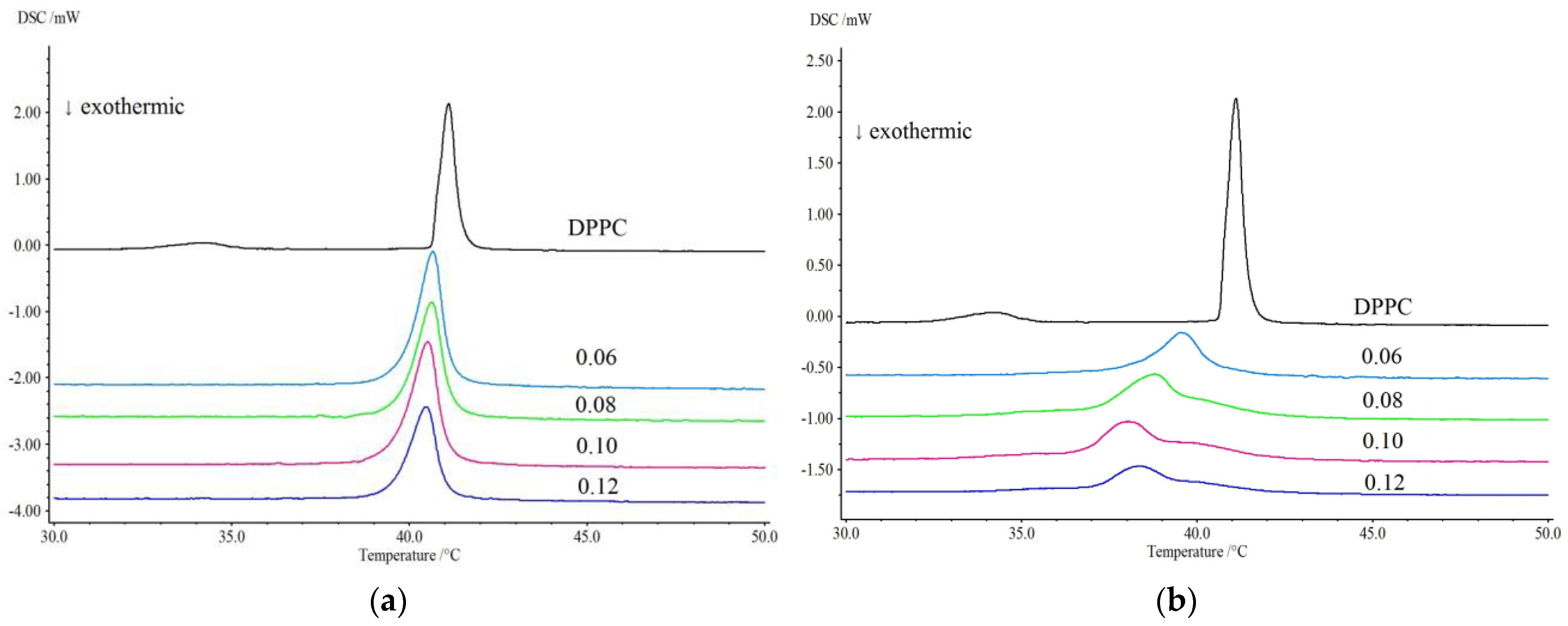
3.2. Differential Scanning Calorimetry (DSC)
3.3. Fluorescence Spectroscopy
3.4. Prediction of ADMET Properties
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seddon, A.M.; Casey, D.; Law, R.V.; Gee, A.; Templer, R.H.; Ces, O. Drug interactions with lipid membranes. Chem. Soc. Rev. 2009, 38, 2509. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Moghimianavval, H.; Hwang, S.W.; Liu, A.P. Synthetic cell as a platform for understanding membrane-membrane interactions. Membranes 2021, 11, 912. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Leite, C.; Nunes, C.; Reis, S. Interaction of nonsteroidal anti-inflammatory drugs with membranes: In vitro assessment and relevance for their biological actions. Prog. Lipid Res. 2013, 52, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Lúcio, M.; Nunes, C.; Gaspar, D.; Gołebska, K.; Wisniewski, M.; Lima, J.L.F.C.; Brezesinski, G.; Reis, S. Effect of anti-inflammatory drugs in phosphatidylcholine membranes: A fluorescence and calorimetric study. Chem. Phys. Lett. 2009, 471, 300–309. [Google Scholar] [CrossRef]
- Chen, Y.-F.; Jobanputra, P.; Barton, P.; Bryan, S.; Fry-Smith, A.; Harris, G.; Taylor, R.S. Cyclooxygenase-2 selective non-steroidal anti-inflammatory drugs (etodolac, meloxicam, celecoxib, rofecoxib, etoricoxib, valdecoxib and lumiracoxib) for osteoarthritis and rheumatoid arthritis: A systematic review and economic evaluation. Health Technol. Assess. 2008, 12, 1–278. [Google Scholar] [CrossRef] [Green Version]
- Luckey, M. Membrane Structural Biology; Cambridge University Press: Cambridge, UK, 2008. [Google Scholar]
- Mitchell, J.A.; Warner, T.D. COX isoforms in the cardiovascular system: Understanding the activities of non-steroidal anti-inflammatory drugs. Nat. Rev. Drug Discov. 2006, 5, 75–86. [Google Scholar] [CrossRef]
- Lucio, M.; Lima, J.L.F.C.; Reis, S. Drug-Membrane Interactions: Significance for Medicinal Chemistry. Curr. Med. Chem. 2010, 17, 1795–1809. [Google Scholar] [CrossRef]
- Bourgaux, C.; Couvreur, P. Interactions of anticancer drugs with biomembranes: What can we learn from model membranes? J. Control. Release 2014, 190, 127–138. [Google Scholar] [CrossRef]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Suwalsky, M.; Villena, F.; Sotomayor, C.P.; Bolognin, S.; Zatta, P. Human cells and cell membrane molecular models are affected in vitro by chlorpromazine. Biophys. Chem. 2008, 135, 7–13. [Google Scholar] [CrossRef]
- Wesołowska, O.; Michalak, K.; Maniewska, J.; Hendrich, A.B. Giant unilamellar vesicles-a perfect tool to visualize phase separation and lipid rafts in model systems. Acta Biochim. Pol. 2009, 56, 33–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, H.; Banerjee, R.; Sarkar, M. Incorporation of NSAIDs in micelles: Implication of structural switchover in drug–membrane interaction. Biophys. Chem. 2003, 104, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Leite, C.; Figueiredo, M.; Burdach, K.; Nunes, C.; Reis, S. Membranes Unraveling the Role of Drug-Lipid Interactions in NSAIDs-Induced Cardiotoxicity. Membranes 2020, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Ehehalt, R.; Braun, A.; Karner, M.; Füllekrug, J.; Stremmel, W. Biochimica et Biophysica Acta Phosphatidylcholine as a constituent in the colonic mucosal barrier—Physiological and clinical relevance. BBA Mol. Cell Biol. Lipids 2010, 1801, 983–993. [Google Scholar] [CrossRef]
- Malinka, W.; Kaczmarz, M.; Filipek, B.; Sapa, J.; Glod, B. Preparation of novel derivatives of pyridothiazine-1,1-dioxide and their CNS and antioxidant properties. Farmaco 2002, 57, 737–746. [Google Scholar] [CrossRef]
- Szczęśniak-Sięga, B.M.; Mogilski, S.; Wiglusz, R.J.; Janczak, J.; Maniewska, J.; Malinka, W.; Filipek, B. Synthesis and pharmacological evaluation of novel arylpiperazine oxicams derivatives as potent analgesics without ulcerogenicity. Bioorganic Med. Chem. 2019, 27, 1619–1628. [Google Scholar] [CrossRef]
- Xu, S.; Hermanson, D.J.; Banerjee, S.; Ghebreselasie, K.; Clayton, G.M.; Garavito, R.M.; Marnett, L.J. Oxicams bind in a novel mode to the cyclooxygenase active site via a two-water-mediated h-bonding network. J. Biol. Chem. 2014, 289, 6799–6808. [Google Scholar] [CrossRef] [Green Version]
- Krzyżak, E.; Szczȩåniak-Siȩga, B.; Malinka, W. Synthesis and thermal behaviour of new benzo-1,2-thiazine long-chain aryl-piperazine derivatives. J. Therm. Anal. Calorim. 2014, 115, 793–802. [Google Scholar] [CrossRef] [Green Version]
- Muszalska, I.; Lesniewska, M.A.; Ciemniejewski, M.; Szkatuła, D.; Malinka, W. Forced Degradation and Photodegradation Studies of Pyrrolo[3,4-c]pyridine-1,3-dione Derivatives as Analgesic Active Compounds Using HPLC, UV and IR Spectrometry, and HPLC/MS Methods. J. AOAC Int. 2015, 98, 1248–1259. [Google Scholar] [CrossRef]
- Malinka, A.; Sieklucka-Dziuba, M.; Rajtar, Y.; Rubaj, A.; Kleinrok, A. Synthesis and Pharmacological Screening of Some N-(4-substituted-Piperazin-1-Ylalkyl)-3,4-Pyrroledicarboximides; Elsevier: Amsterdam, The Netherlands, 1999. [Google Scholar]
- Szcześniak-sięga, B.; Maniewska, J.; Poła, A.; Środa-Pomianek, K.; Malinka, W.; Michalak, K. Synthesis of new piroxicam derivatives and their influence on lipid bilayers. Acta Pol. Pharm. Drug Res. 2014, 71, 1045–1050. [Google Scholar]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Quenching of Fluorescence. In Principles of Fluorescence Spectroscopy; Springer: Berlin/Heidelberg, Germany, 1999; pp. 237–265. [Google Scholar]
- Parasassi, T.; Krasnowska, E.K.; Bagatolli, L.; Gratton, E. Laurdan and Prodan as Polarity-Sensitive Fluorescent Membrane Probes. J. Fluoresc. 1998, 8, 365–373. [Google Scholar] [CrossRef]
- Bagatolli, L.A.; Parasassi, T.; Fidelio, G.D.; Gratton, E. A Model for the Interaction of 6-Lauroyl-2-(N,N-dimethylamino)naphthalene with Lipid Environments: Implications for Spectral Properties. Photochem. Photobiol. 1999, 70, 557. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, 1074–1082. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 54677470, Meloxicam. 2022. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Meloxicam (accessed on 14 November 2022).
- Lipinski, C.A. Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv. Drug Deliv. Rev. 2016, 101, 34–41. [Google Scholar] [CrossRef]
- Clark, D.E. In silico prediction of blood-brain barrier permeation. Drug Discov. Today 2003, 8, 927–933. [Google Scholar] [CrossRef]
- Waring, M.J. Lipophilicity in drug discovery. Expert Opin. Drug Discov. 2010, 5, 235–248. [Google Scholar] [CrossRef]
- Peng, L.; He, Z.; Chen, W.; Holzman, I.R.; Lin, J. Effects of butyrate on intestinal barrier function in a caco-2 cell monolayer model of intestinal barrier. Pediatr. Res. 2007, 61, 37–41. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.C.; Broccatelli, F.; Plise, E.; Chen, B.; Liu, L.; Cheong, J.; Zhang, S.; Jorski, J.; Ga, K.; Umemoto, K.K.; et al. Evaluating the Utility of Canine Mdr1 Knockout Madin-Darby Canine Kidney I Cells in Permeability Screening and E ffl ux Substrate Determination. Mol. Pharm. 2018, 15, 5103–5113. [Google Scholar] [CrossRef]
- Mollazadeh, S.; Sahebkar, A.; Hadizadeh, F.; Behravan, J. Structural and functional aspects of P-glycoprotein and its inhibitors. Life Sci. 2018, 214, 118–123. [Google Scholar] [CrossRef]
- Lambris, J.D. Advances in Experimental Medicine and Biology; Plenum Press: New York, NY, USA, 1967. [Google Scholar]
- Daly, A.K. Pharmacogenomics of CYP2C9: Functional and Clinical Considerations. J. Peresonalized Med. 2018, 8, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Yang, H.; Wu, Z.; Wang, T.; Li, W.; Tang, Y. In Silico Prediction of Blood–Brain Barrier Permeability of Compounds by Machine Learning and Resampling Methods. ChemMedChem 2018, 13, 2189–2201. [Google Scholar] [CrossRef] [PubMed]
- Maniewska, J.; Czyżnikowska, Ż.; Szczęśniak-Sięga, B.M.; Michalak, K. Interaction of Oxicam Derivatives with the Artificial Models of Biological Membranes—Calorimetric and Fluorescence Spectroscopic Study. Membranes 2022, 12, 791. [Google Scholar] [CrossRef] [PubMed]
- Maniewska, J.; Gąsiorowska, J.; Szczęśniak-Sięga, B.; Michalak, K. The interaction of new oxicam derivatives with lipid bilayers as measured by calorimetry and fluorescence spectroscopy. Acta Biochim. Pol. 2018, 65, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Heimburg, T. A model for the lipid pretransition: Coupling of ripple formation with the chain-melting transition. Biophys. J. 2000, 78, 1154–1165. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, T.J.; Ross, P.D.; Levin, I.W. Effects of anesthetic tetradecenols on phosphatidylcholine phase transitions. Implications for the mechanism of the bilayer pretransition. Biophys. J. 1986, 50, 1053–1059. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.K.; Wu, N.M. Effect of small molecules on the dipalmitoyl lecithin liposomal bilayer: III. Phase transition in lipid bilayer. J. Membr. Biol. 1977, 34, 157–201. [Google Scholar] [CrossRef]
- Kyrikou, I.; Hadjikakou, S.K.; Kovala-Demertzi, D.; Viras, K.; Mavromoustakos, T. Effects of non-steroid anti-inflammatory drugs in membrane bilayers. Chem. Phys. Lipids 2004, 132, 157–169. [Google Scholar] [CrossRef]
- Mondal Roy, S.; Bansode, A.S.; Sarkar, M. Effect of increase in orientational order of lipid chains and head group spacing on non steroidal anti-inflammatory drug induced membrane fusion. Langmuir 2010, 26, 18967–18975. [Google Scholar] [CrossRef]
- Lúcio, M.; Ferreira, H.; Lima, J.L.F.C.; Reis, S. Interactions between oxicams and membrane bilayers: An explanation for their different COX selectivity. Med. Chem. 2006, 2, 447–456. [Google Scholar] [CrossRef]
- Maniewska, J.; Szczęśniak-Sięga, B.; Poła, A.; Środa-Pomianek, K.; Malinka, W.; Michalak, K. The interaction of new piroxicam analogues with lipid bilayers-A calorimetric and fluorescence spectroscopic study. Acta Pol. Pharm. Drug Res. 2014, 71, 1004–1012. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter/Optimal Value | Compound | |||||
|---|---|---|---|---|---|---|
| PR23 | PR24 | PR25 | PR49 | PR50 | MLX | |
| MW (molecular weight) optimal 100–600 | 551.1 | 535.2 | 535.2 | 585.1 | 585.2 | 351.4 |
| nHA (number of hydrogen bond acceptors) optimal 0–12 | 9 | 7 | 8 | 8 | 7 | 5 |
| nHD (number of hydrogen bond donors) optimal 0–7 | 0 | 0 | 0 | 0 | 0 | 2 |
| TPSA (topological polar surface area) optimal 0–140 | 104 | 78 | 95 | 95 | 78 | 100 |
| nRot (number of rotatable bonds) optimal 0–11 | 7 | 7 | 6 | 7 | 8 | 2 |
| nRing (number of rings) optimal 0–6 | 5 | 5 | 5 | 5 | 5 | 3 |
| nHet (number of heteroatoms) optimal 1–15 | 11 | 9 | 10 | 12 | 11 | 9 |
| logP (log of the octanol/water partition coefficient) optimal 0–3 | 3.4 | 4.3 | 3.7 | 4.9 | 4.8 | 2.3 |
| logD (logP at physiological pH) optimal 1–3 | 2.4 | 3.5 | 2.8 | 3.5 | 4.1 | 0.6 |
| Parameter/Optimal Value | Compound | |||||
|---|---|---|---|---|---|---|
| PR23 | PR24 | PR25 | PR49 | PR50 | MLX | |
| QED (measure of drug likeness based on the concept of desirability; attractive > 0.67, unattractive 0.49–0.67, too complex < 0.34) | 0.3 | 0.3 | 0.4 | 0.3 | 0.3 | 0.7 |
| SA score (synthetic accessibility score is designed to estimate ease of synthesis of drug-like molecules; ≥6—difficult, <6—easy to synthesize | 3.1 | 3.1 | 3.1 | 3.2 | 3.3 | 4.0 |
| Fsp3 (number of sp3 hybridized carbons/total carbon count, correlating with melting point and solubility; ≥0.42 is considered a suitable value) | 0.2 | 0.3 | 0.2 | 0.3 | 0.3 | 0.2 |
| Lipinski Rule (MW ≤ 500; logP ≤ 5; Hacc ≤ 10; Hdon ≤ 5; if two properties are out of range, a poor absorption or permeability is possible, one is acceptable) | Accepted | Accepted | Accepted | Accepted | Accepted | Accepted |
| Pfizer Rule (compounds with a high logP (>3) and low TPSA (<75) are likely to be toxic) | Rejected | Accepted | Accepted | Accepted | Accepted | Accepted |
| GSK Rule (MW ≤ 400; logP ≤ 4; compounds satisfying the GSK rule may have a more favorable ADMET profile) | Rejected | Rejected | Rejected | Rejected | Rejected | Accepted |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maniewska, J.; Gąsiorowska, J.; Czyżnikowska, Ż.; Michalak, K.; Szczęśniak-Sięga, B.M. New Meloxicam Derivatives—Synthesis and Interaction with Phospholipid Bilayers Measured by Differential Scanning Calorimetry and Fluorescence Spectroscopy. Membranes 2023, 13, 416. https://doi.org/10.3390/membranes13040416
Maniewska J, Gąsiorowska J, Czyżnikowska Ż, Michalak K, Szczęśniak-Sięga BM. New Meloxicam Derivatives—Synthesis and Interaction with Phospholipid Bilayers Measured by Differential Scanning Calorimetry and Fluorescence Spectroscopy. Membranes. 2023; 13(4):416. https://doi.org/10.3390/membranes13040416
Chicago/Turabian StyleManiewska, Jadwiga, Justyna Gąsiorowska, Żaneta Czyżnikowska, Krystyna Michalak, and Berenika M. Szczęśniak-Sięga. 2023. "New Meloxicam Derivatives—Synthesis and Interaction with Phospholipid Bilayers Measured by Differential Scanning Calorimetry and Fluorescence Spectroscopy" Membranes 13, no. 4: 416. https://doi.org/10.3390/membranes13040416