
Multiphoton Imaging of Ca2+ Instability in Acute Myocardial Slices from a RyR2R2474S Murine Model of Catecholaminergic Polymorphic Ventricular Tachycardia


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Models
2.2. Isolation of Cardiomyocytes from Adult Mouse Hearts
2.3. Preparation of Acute Heart Slices and Loading with Fluorescent Ca2+ Dye
2.4. Two-Photon Emission Microscope
2.5. Two-Photon Ca2+ Imaging on Heart Slices
2.6. Image Analysis
2.7. Statistical Analysis
3. Results
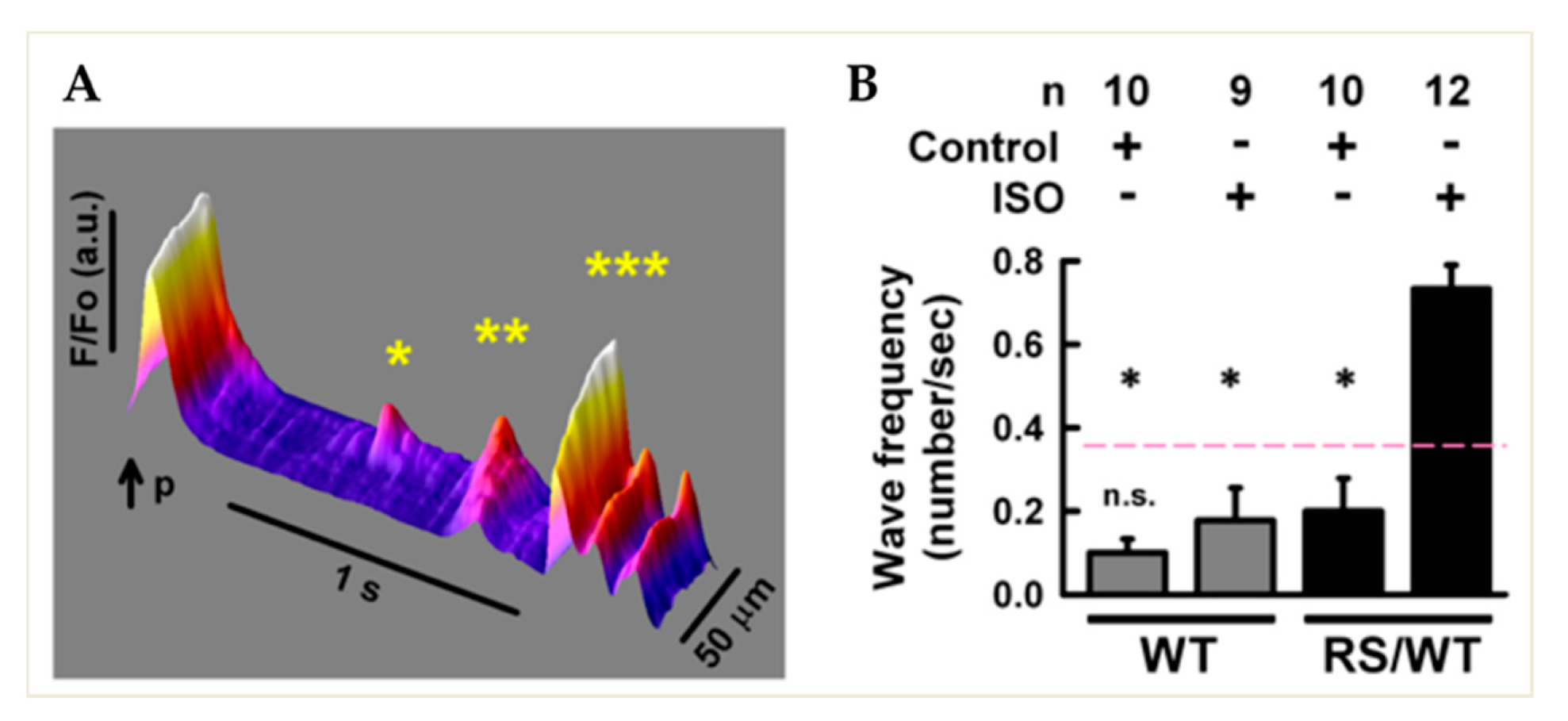
3.1. Diastolic Sarcoplasmic Reticulum Ca2+ Leak in Isolated RyR2RS/wt CPVT Cardiomyocytes
3.2. Preparation of Acute Myocardial Slices Suited for Ca2+ Imaging in Multicellular Heart Tissue
3.3. Multiphoton Ca2+ Imaging in RyR2RS/wt Acute Myocardial Slices Shows ‘Leaky’ RyR2 Behavior
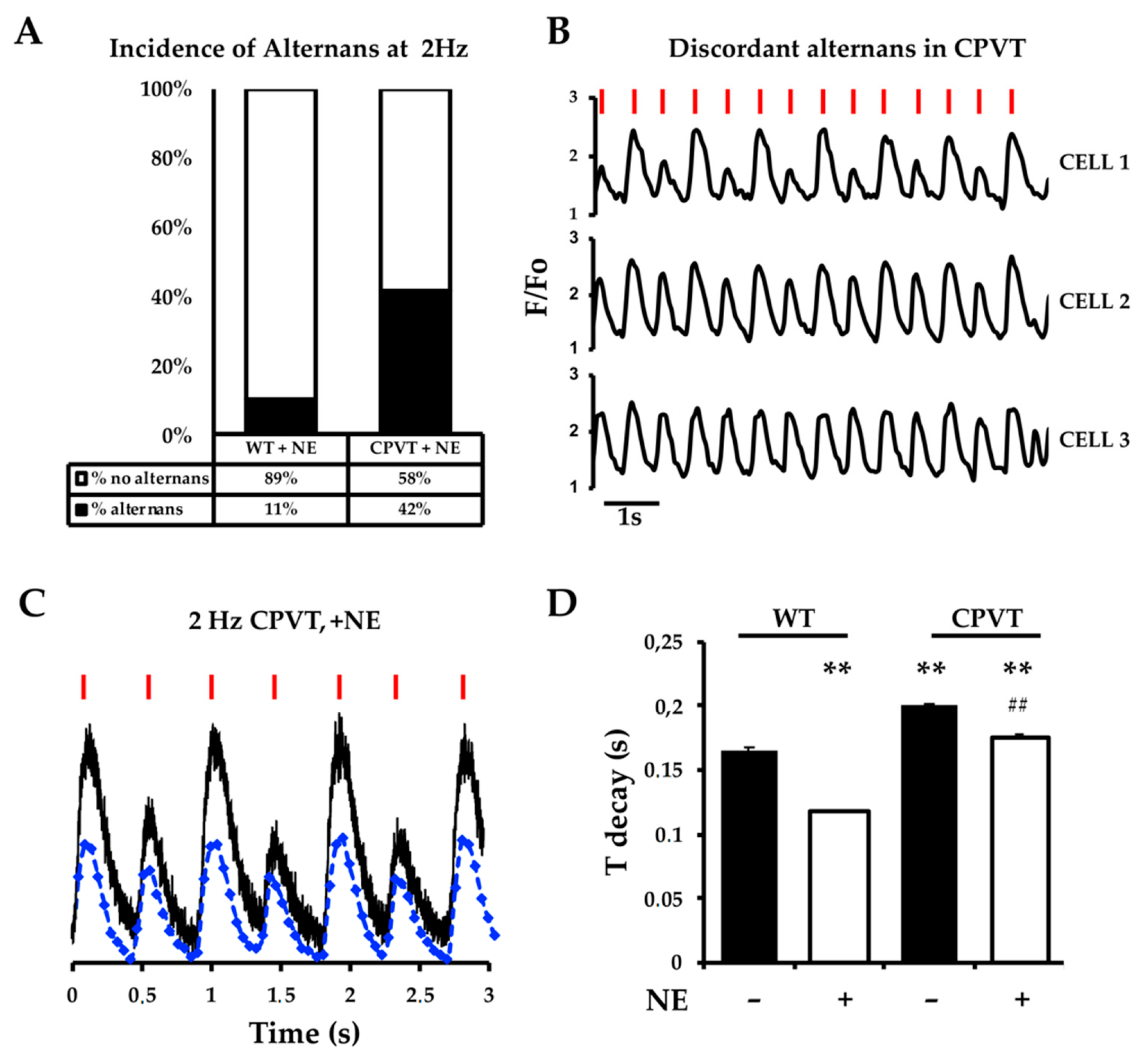
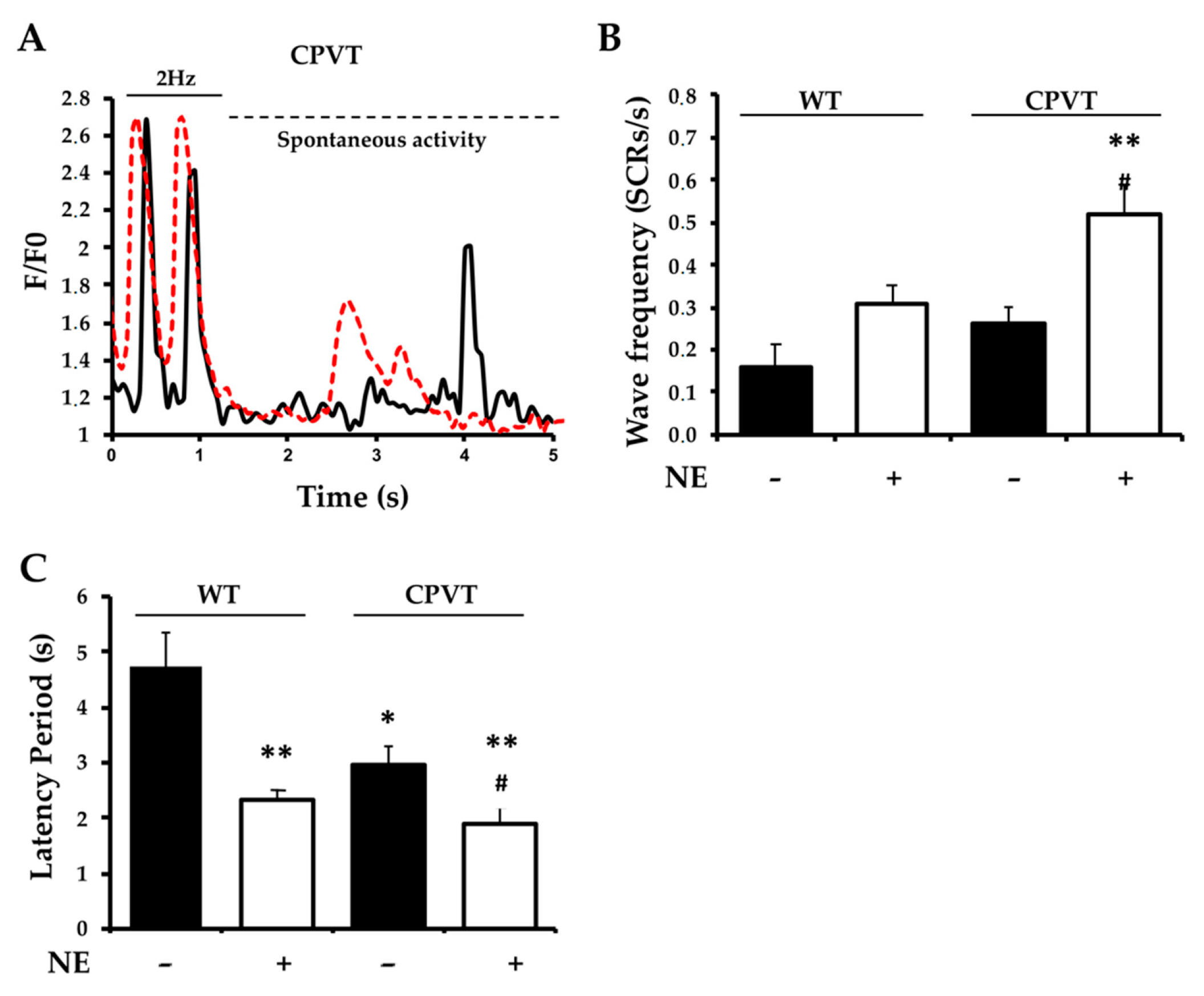
3.4. Pace-Stop Protocol Uncovers Spontaneous Diastolic Ca2+ Leak in CPVT Slices
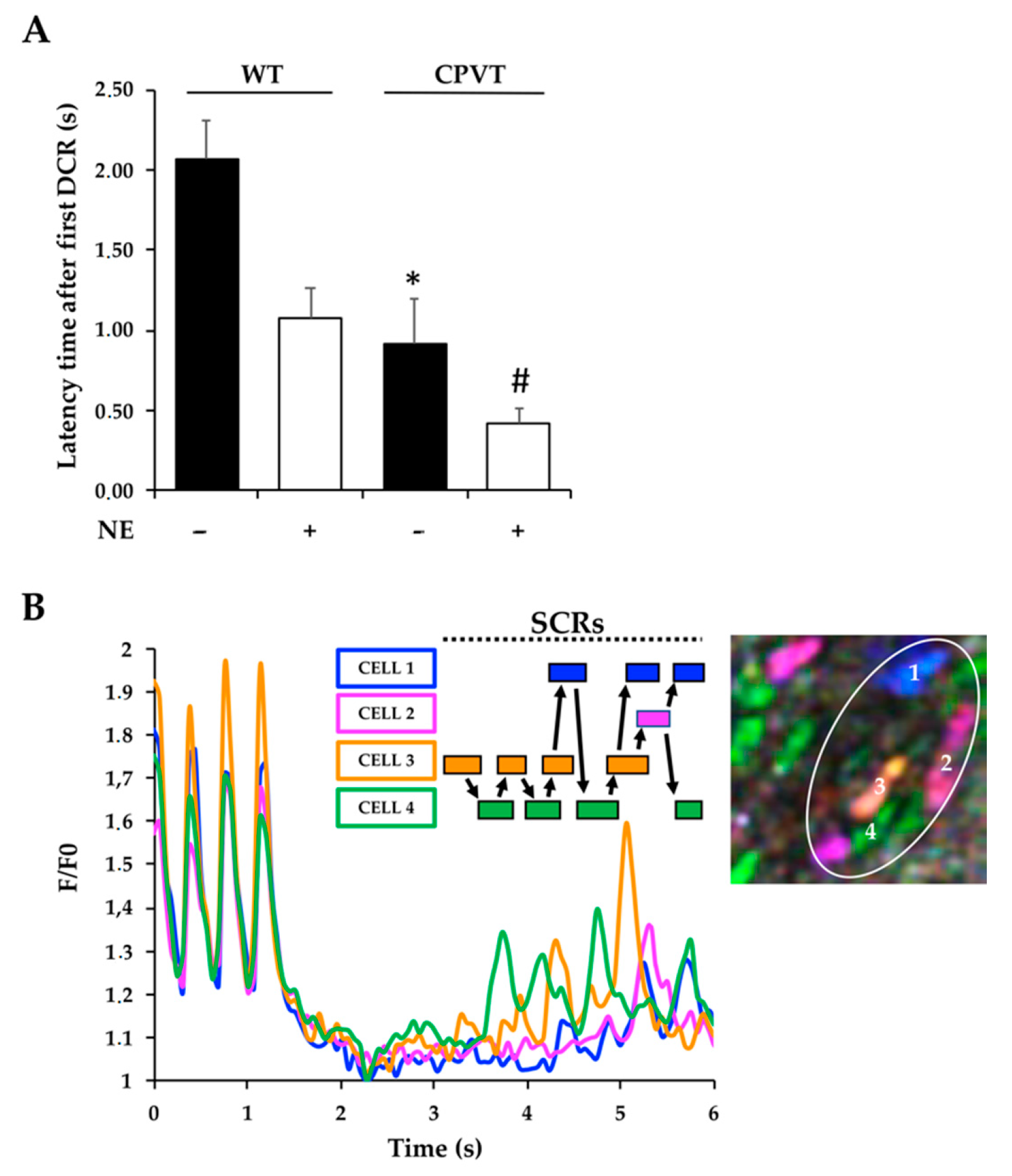
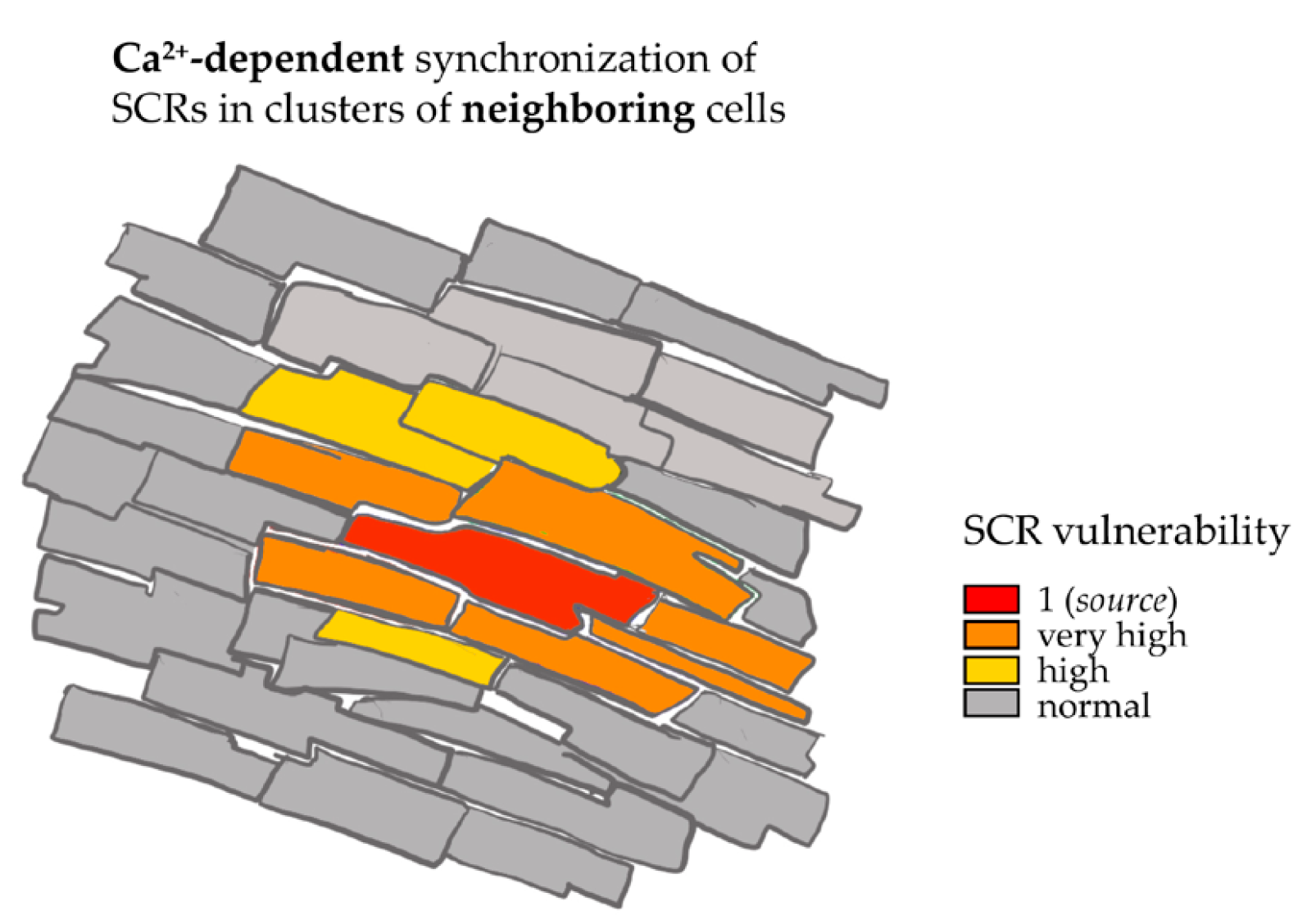
3.5. Intercellular Trasmission of Ca2+ Instability in Spatially Defined Myocardial Cell Clusters through Enhancement of SCR Vulnerability in Neighboring Cells
4. Discussion
5. Conclusions
6. Note
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Houser, S.R.; Carnell, L.H. Ca2+ Signaling Domains Responsible for Cardiac Hypertrophy and Arrhythmias. Circ. Res. 2009, 104, 413–415. [Google Scholar] [CrossRef] [Green Version]
- Kho, C.; Lee, A.; Hajjar, R.J. Altered sarcoplasmic reticulum calcium cycling—Targets for heart failure therapy. Nat. Rev. Cardiol. 2012, 9, 717–733. [Google Scholar] [CrossRef] [Green Version]
- Marks, A.R. Calcium cycling proteins and heart failure: Mechanisms and therapeutics. J. Clin. Investig. 2013, 123, 46–52. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac Sarcoplasmic Reticulum Calcium Leak: Basis and Roles in Cardiac Dysfunction. Annu. Rev. Physiol. 2014, 76, 107–127. [Google Scholar] [CrossRef] [Green Version]
- Marks, A.R.; Priori, S.; Memmi, M.; Kontula, K. Involvement of the cardiac ryanodine receptor/calcium release channel in catecholaminergic polymorphic ventricular tachycardia. J. Cell. Physiol. 2002, 190, 1–6. [Google Scholar] [CrossRef]
- Liu, N.; Ruan, Y.; Priori, S.G. Catecholaminergic Polymorphic Ventricular Tachycardia. Prog. Cardiovasc. Dis. 2008, 51, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Lehnart, S.E.; Mongillo, M.; Bellinger, A.; Lindegger, N.; Chen, B.-X.; Hsueh, W.; Reiken, S.; Wronska, A.; Drew, L.J.; Ward, C.W.; et al. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J. Clin. Investig. 2008, 118, 2230–2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiene, G.; Nava, A.; Corrado, D.; Rossi, L.; Pennelli, N. Right Ventricular Cardiomyopathy and Sudden Death in Young People. N. Engl. J. Med. 1988, 318, 129–133. [Google Scholar] [CrossRef]
- Basso, C.; Thiene, G.; Corrado, D.; Angelini, A.; Nava, A.; Valente, M. Arrhythmogenic Right Ventricular Cardiomyopathy: Dysplasia, dystrophy, or myocarditis? Circulation 1996, 94, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Lehnart, S.E.; Ackerman, M.J.; Benson, D.W.; Brugada, R.; Clancy, C.E.; Donahue, J.K.; George, A.L.; Grant, A.O.; Groft, S.C.; January, C.T.; et al. Inherited Arrhythmias. Circulation 2007, 116, 2325–2345. [Google Scholar] [CrossRef] [Green Version]
- Cannell, M.B.; Cheng, H.; Lederer, W.J. The control of calcium release in heart muscle. Science 1995, 268, 1045–1049. [Google Scholar] [CrossRef]
- Lehnart, S.; Marks, A.R. Regulation of Ryanodine Receptors in the Heart. Circ. Res. 2007, 101, 746–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehrens, X.H.; Lehnart, S.E.; Huang, F.; Vest, J.A.; Reiken, S.R.; Mohler, P.J.; Sun, J.; Guatimosim, S.; Song, L.-S.; Rosemblit, N.; et al. FKBP12.6 Deficiency and Defective Calcium Release Channel (Ryanodine Receptor) Function Linked to Exercise-Induced Sudden Cardiac Death. Cell 2003, 113, 829–840. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Velasco, M.; Rueda, A.; Rizzi, N.; Benitah, J.-P.; Colombi, B.; Napolitano, C.; Priori, S.G.; Richard, S.; Gómez, A.M. Increased Ca2+ Sensitivity of the Ryanodine Receptor Mutant RyR2 R4496C Underlies Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. Res. 2009, 104, 201–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, K.; Tanaka, H.; Mani, H.; Nakagami, T.; Takamatsu, T. Burst Emergence of Intracellular Ca 2+ Waves Evokes Arrhythmogenic Oscillatory Depolarization via the Na+ –Ca2+ Exchanger. Circ. Res. 2008, 103, 509–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myles, R.C.; Wang, L.; Kang, C.; Bers, D.; Ripplinger, C.M. Local β-Adrenergic Stimulation Overcomes Source-Sink Mismatch to Generate Focal Arrhythmia. Circ. Res. 2012, 110, 1454–1464. [Google Scholar] [CrossRef] [Green Version]
- Zaglia, T.; Pianca, N.; Borile, G.; Da Broi, F.; Richter, C.; Campione, M.; Lehnart, S.E.; Luther, S.; Corrado, D.; Miquerol, L.; et al. Optogenetic determination of the myocardial requirements for extrasystoles by cell type-specific targeting of ChannelRhodopsin-2. Proc. Natl. Acad. Sci. USA 2015, 112, E4495–E4504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halbach, M.; Pillekamp, F.; Brockmeier, K.; Hescheler, J.; Müller-Ehmsen, J.; Reppel, M. Ventricular Slices of Adult Mouse Hearts—A new Multicellular In Vitro Model for Electrophysiological Studies. Cell. Physiol. Biochem. 2006, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Habeler, W.; Peschanski, M.; Monville, C. Organotypic heart slices for cell transplantation and physiological studies. Organogenesis 2009, 5, 62–66. [Google Scholar] [CrossRef] [Green Version]
- Bussek, A.; Wettwer, E.; Christ, T.; Lohmann, H.; Camelliti, P.; Ravens, U. Tissue Slices from Adult Mammalian Hearts as a Model for Pharmacological Drug Testing. Cell. Physiol. Biochem. 2009, 24, 527–536. [Google Scholar] [CrossRef] [Green Version]
- De Boer, T.P.; Camelliti, P.; Ravens, U.; Kohl, P. Myocardial tissue slices: Organotypic pseudo-2D models for cardiac research & development. Futur. Cardiol. 2009, 5, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Lee, P.; Mirams, G.R.; Sarathchandra, P.; Borg, T.K.; Gavaghan, D.J.; Kohl, P.; Bollensdorff, C. Cardiac tissue slices: Preparation, handling, and successful optical mapping. Am. J. Physiol. Circ. Physiol. 2015, 308, H1112–H1125. [Google Scholar] [CrossRef]
- Helmchen, F.; Denk, W. Deep tissue two-photon microscopy. Nat. Methods 2005, 2, 932–940. [Google Scholar] [CrossRef]
- Rubart, M.; Wang, E.; Dunn, K.W.; Field, L.J. Two-photon molecular excitation imaging of Ca2+transients in Langendorff-perfused mouse hearts. Am. J. Physiol. Physiol. 2003, 284, C1654–C1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ripplinger, C.M. The best thing since sliced bread? Optical mapping of transverse cardiac slices in the mouse heart. J. Physiol. 2018, 596, 3825–3826. [Google Scholar] [CrossRef] [PubMed]
- Habeler, W.; Pouillot, S.; Plancheron, A.; Puceat, M.; Peschanski, M.; Monville, C. An in vitro beating heart model for long-term assessment of experimental therapeutics. Cardiovasc. Res. 2008, 81, 253–259. [Google Scholar] [CrossRef] [Green Version]
- Barclay, C.J. Modelling diffusive O2 supply to isolated preparations of mammalian skeletal and cardiac muscle. J. Muscle Res. Cell Motil. 2005, 26, 225–235. [Google Scholar] [CrossRef]
- Cordeiro, J.M.; Malone, J.E.; Di Diego, J.M.; Scornik, F.S.; Aistrup, G.L.; Antzelevitch, C.; Wasserstrom, J.A. Cellular and subcellular alternans in the canine left ventricle. Am. J. Physiol. Circ. Physiol. 2007, 293, H3506–H3516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Santulli, G.; Guo, X.; Gao, M.; Chen, B.-X.; Marks, A.R. Imaging atrial arrhythmic intracellular calcium in intact heart. J. Mol. Cell. Cardiol. 2013, 64, 120–123. [Google Scholar] [CrossRef] [Green Version]
- Plummer, B.N.; Cutler, M.J.; Wan, X.; Laurita, K.R. Spontaneous calcium oscillations during diastole in the whole heart: The influence of ryanodine reception function and gap junction coupling. Am. J. Physiol. Circ. Physiol. 2011, 300, H1822–H1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, Q.; Belevych, A.E.; Radwański, P.B.; Liu, B.; Kalyanasundaram, A.; Knollmann, B.C.; Fedorov, V.V.; Györke, S. Alternating membrane potential/calcium interplay underlies repetitive focal activity in a genetic model of calcium-dependent atrial arrhythmias. J. Physiol. 2015, 593, 1443–1458. [Google Scholar] [CrossRef]
- Kornyeyev, D.; Petrosky, A.D.; Zepeda, B.; Ferreiro, M.; Knollmann, B.; Escobar, A.L. Calsequestrin 2 deletion shortens the refractoriness of Ca2+ release and reduces rate-dependent Ca2+-alternans in intact mouse hearts. J. Mol. Cell. Cardiol. 2012, 52, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Brunello, L.; Slabaugh, J.L.; Radwański, P.B.; Ho, H.-T.; Belevych, A.E.; Lou, Q.; Chen, H.; Napolitano, C.; Lodola, F.; Priori, S.G.; et al. Decreased RyR2 refractoriness determines myocardial synchronization of aberrant Ca2+ release in a genetic model of arrhythmia. Proc. Natl. Acad. Sci. USA 2013, 110, 10312–10317. [Google Scholar] [CrossRef] [Green Version]
- Cerrone, M.; Noujaim, S.F.; Tolkacheva, E.G.; Talkachou, A.; O’Connell, R.; Berenfeld, O.; Anumonwo, J.; Pandit, S.V.; Vikstrom, K.; Napolitano, C.; et al. Arrhythmogenic Mechanisms in a Mouse Model of Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. Res. 2007, 101, 1039–1048. [Google Scholar] [CrossRef] [Green Version]
- Wleklinski, M.J.; Kannankeril, P.J.; Knollmann, B.C. Molecular and tissue mechanisms of catecholaminergic polymorphic ventricular tachycardia. J. Physiol. 2020, 598, 2817–2834. [Google Scholar] [CrossRef]
- Kistamas, K.; Veress, R.; Horváth, B.; Bányász, T.; Nánási, P.P.; Eisner, D.A. Calcium Handling Defects and Cardiac Arrhythmia Syndromes. Front. Pharmacol. 2020, 11, 72. [Google Scholar] [CrossRef]
- Sato, D.; Bartos, D.C.; Ginsburg, K.S.; Bers, D.M. Depolarization of Cardiac Membrane Potential Synchronizes Calcium Sparks and Waves in Tissue. Biophys. J. 2014, 107, 1313–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borile, G.; De Mauro, C.; Urbani, A.; Alfieri, D.; Pavone, F.S.; Mongillo, M. Multispot multiphoton Ca2+ imaging in acute myocardial slices. J. Biomed. Opt. 2014, 20, 51016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, J.N.; Nivala, M.; Garfinkel, A.; Qu, Z. Alternans and Arrhythmias. Circ. Res. 2011, 108, 98–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aistrup, G.L.; Kelly, J.E.; Kapur, S.; Kowalczyk, M.; Sysman-Wolpin, I.; Kadish, A.H.; Wasserstrom, J.A. Pacing-induced Heterogeneities in Intracellular Ca2+Signaling, Cardiac Alternans, and Ventricular Arrhythmias in Intact Rat Heart. Circ. Res. 2006, 99, E65–E73. [Google Scholar] [CrossRef] [Green Version]
- Laurita, K.R.; Rosenbaum, D.S. Cellular mechanisms of arrhythmogenic cardiac alternans. Prog. Biophys. Mol. Biol. 2008, 97, 332–347. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Lacalle, E.; Cantalapiedra, I.R.; Peñaranda, A.; Cinca, J.; Hove-Madsen, L.; Echebarria, B. Dependency of Calcium Alternans on Ryanodine Receptor Refractoriness. PLoS ONE 2013, 8, e55042. [Google Scholar] [CrossRef] [Green Version]
- Knollmann, B.C.; Chopra, N.; Hlaing, T.; Akin, B.; Yang, T.; Ettensohn, K.; Knollmann, B.E.; Horton, K.D.; Weissman, N.J.; Holinstat, I.; et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J. Clin. Investig. 2006, 116, 2510–2520. [Google Scholar] [CrossRef] [Green Version]
- Prando, V.; Da Broi, F.; Franzoso, M.; Plazzo, A.P.; Pianca, N.; Francolini, M.; Basso, C.; Kay, M.; Zaglia, T.; Mongillo, M. Dynamics of neuroeffector coupling at cardiac sympathetic synapses. J. Physiol. 2018, 596, 2055–2075. [Google Scholar] [CrossRef]
- Zaglia, T.; Mongillo, M. Cardiac sympathetic innervation, from a different point of (re)view. J. Physiol. 2017, 595, 3919–3930. [Google Scholar] [CrossRef] [PubMed]
- Pianca, N.; Di Bona, A.; Lazzeri, E.; Costantini, I.; Franzoso, M.; Prando, V.; Armani, A.; Rizzo, S.; Fedrigo, M.; Angelini, A.; et al. Cardiac sympathetic innervation network shapes the myocardium by locally controlling cardiomyocyte size through the cellular proteolytic machinery. J. Physiol. 2019, 597, 3639–3656. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Eisner, D.A.; O’Neill, S.C. Do calcium waves propagate between cells and synchronize alternating calcium release in rat ventricular myocytes? J. Physiol. 2012, 590, 6353–6361. [Google Scholar] [CrossRef]
- Bezzerides, V.J.; Caballero, A.; Wang, S.; Ai, Y.; Hylind, R.J.; Lu, F.; Heims-Waldron, D.A.; Chambers, K.D.; Zhang, D.; Abrams, D.J.; et al. Gene Therapy for Catecholaminergic Polymorphic Ventricular Tachycardia by Inhibition of Ca 2+ /Calmodulin-Dependent Kinase II. Circulation 2019, 140, 405–419. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borile, G.; Zaglia, T.; E. Lehnart, S.; Mongillo, M. Multiphoton Imaging of Ca2+ Instability in Acute Myocardial Slices from a RyR2R2474S Murine Model of Catecholaminergic Polymorphic Ventricular Tachycardia. J. Clin. Med. 2021, 10, 2821. https://doi.org/10.3390/jcm10132821
Borile G, Zaglia T, E. Lehnart S, Mongillo M. Multiphoton Imaging of Ca2+ Instability in Acute Myocardial Slices from a RyR2R2474S Murine Model of Catecholaminergic Polymorphic Ventricular Tachycardia. Journal of Clinical Medicine. 2021; 10(13):2821. https://doi.org/10.3390/jcm10132821
Chicago/Turabian StyleBorile, Giulia, Tania Zaglia, Stephan E. Lehnart, and Marco Mongillo. 2021. "Multiphoton Imaging of Ca2+ Instability in Acute Myocardial Slices from a RyR2R2474S Murine Model of Catecholaminergic Polymorphic Ventricular Tachycardia" Journal of Clinical Medicine 10, no. 13: 2821. https://doi.org/10.3390/jcm10132821