Abstract
Introduction: Thyroid peroxidase (TPO) deficiency is the most common enzymatic defect causing congenital hypothyroidism (CH). We aimed to characterize the long-term outcome of patients with TPO deficiency. Methods: Clinical and genetic data were collected retrospectively. Results: Thirty-three patients with primary CH caused by TPO deficiency were enrolled. The follow-up period was up to 43 years. Over time, 20 patients (61%) developed MNG. Eight patients (24%) underwent thyroidectomy: one of them had minimal invasive follicular thyroid carcinoma. No association was found between elevated lifetime TSH levels and the development of goiter over the years. Conclusions: This cohort represents the largest long-term follow up of patients with TPO deficiency. Our results indicate that elevated TSH alone cannot explain the high rate of goiter occurrence in patients with TPO deficiency, suggesting additional factors in goiter development. The high rate of MNG development and the risk for thyroid carcinoma indicate a need for long-term follow up with annual ultrasound scans.
1. Introduction
The incidence of congenital hypothyroidism (CH) is estimated at 1:2000–3000 births [1]. In 2020, the European Society for Pediatric Endocrinology updated guidelines for diagnosis and management for CH [2]. Thyroid dysgenesis accounts for 75–80% of the cases, and thyroid hormone synthesis disorders (dyshormonogenesis) account for 10–15% of them [3]. Thyroid dyshormonogenesis is commonly inherited in an autosomal recessive pattern and is more prevalent in highly consanguineous populations. Thyroid peroxidase (TPO) deficiency is the most common enzymatic defect, accounting for about 50% of the cases [4]. TPO catalyzes both the iodination and coupling of tyrosyl residues of thyroglobulin with a strict requirement for hydrogen peroxide, which acts as an electron acceptor [5]. A mutation in TPO was first reported in 1990 [6] (OMIM # 274500); since then, over 60 different mutations have been identified, mainly missense mutations (http://portal.biobaseinternational.com/hgmd/pro/, accessed on 18 June 2009) [5].
TPO mutations are present in primary CH with a normal-size or goitrous thyroid gland. The development of a goiter occurs either in utero, at birth, or during life. It has been postulated that the development of a goiter over the years can be attributed to elevated thyroid-stimulating hormone (TSH), resulting in hyperplasia and hypertrophy of the thyrocytes. TSH acts through stimulation of the TSH receptor in two pathways, one regulating thyroid growth, and the other regulating transcription of the genes encoding Na+/I− symporter, thyroglobulin, TPO, and pendrin to produce thyroid hormones. Elevated serum TSH levels are associated with increased risk of thyroid cancer [7]. In the past, TPO enzyme defects were diagnosed mainly by perchlorate discharge test. However, molecular sequencing of the TPO gene has rendered this test irrelevant. Several studies have described the molecular findings of patients with TPO mutations [1,4,8,9,10,11,12], but only a few them have reported the long-term clinical outcome [13]. Here, we report retrospectively on the long-term follow up (up to 43 years) of 33 patients with TPO mutations and evaluate the effect of TSH serum levels on goiter development.
2. Patients and Methods
2.1. Patients
From a cohort of 278 subjects with CH being followed at our institute, 41 (15%) were diagnosed with dyshormonogenesis. Among them, 33 patients (80%) aged 4 to 43 years had TPO enzyme deficiency and were enrolled in the study. The diagnosis of TPO deficiency was made clinically by the findings of primary CH, elevated serum thyroglobulin levels, and a normally placed thyroid gland demonstrated by 99TC scan or by ultrasound (US) imaging. I131-uptake and a perchlorate discharge test were performed in some of the patients prior to the availability of molecular sequencing. Final diagnosis was made by sequencing the TPO gene. In the case of a known mutation in the family, sequencing of the specific mutation was performed. In five subjects, the diagnosis was based on clinical characteristics or the presence of at least one additional affected member in the family. Patients were followed up every 6 months and levothyroxine (LT4) therapy doses were adjusted to maintain TSH and free T4 (FT4) levels within the normal ranges. When receiving abnormal results, L-T4 dose was adjusted and repeat tests were performed after 6 weeks from L-T4 dose change. The presence of a goiter was determined by physical examination of the thyroid gland by the physician and was confirmed by US imaging. Data were collected retrospectively from these patients’ medical files and included: clinical, biochemical, imaging, and molecular findings from birth up until the time of the study.
2.2. Biochemical Analyses
Thyroid hormone levels at birth were obtained from the national neonatal screening records. Laboratory thyroid function at birth, at annual follow up, and at the time of the study were collected retrospectively from the medical files. Thyroid hormones levels were obtained at 0800 am prior to administering LT4 therapy. To assess the effect of elevated TSH on goiter development, we used three different parameters: (i) mean FT4 serum levels, (ii) median lifetime TSH serum levels, and (iii) percentage of lifetime that TSH and FT4 were in the normal range (normal TSH, referred to as TSH below 5 mIU/L; normal FT4, referred to as FT4 between 10–20 pmol/L).
Blood samples were collected by heel puncture 48–72 h after birth. The Israeli CH screening program is based on total T4 (TT4) level followed by confirmatory TSH test, such that when the TT4 level is below the 10th percentile for age, TSH is measured. TSH values >20 mIU/L are considered indicative of primary CH.
2.3. Hormone Analyses
TSH and FT4 were measured by direct automated chemiluminescent immunoradiometric assay using the ADVIA Centaur immunoassay system (Bayer Corporation, Tarrytown, NY, USA). TSH reference values provided by our laboratory were 0.4–4.2 mIU/L and FT4 normal reference was 10–20 pmol/L.
2.4. Imaging
The size and location of the thyroid gland was demonstrated by 99TC scan at the age of 2–3 years, after withdrawing from LT4 therapy for 3 weeks. In some patients, a repeat 99TC scan was performed due to the development of MNG or the presence of a thyroid nodule. Patients underwent US imaging yearly to detect goiter development. Computerized scans (CT) were performed in cases when tracheal pressure was suspected by US scan or prior to surgery.
2.5. Molecular Analysis
After signing the appropriate informed consent, peripheral blood samples were collected for DNA analysis. Genomic DNA was extracted from peripheral mononuclear cells using the Blood Amp Kit (QIAGEN Inc., Valencia, CA, USA), according to the manufacturers’ protocol. Sequencing of the TPO gene was performed. In the case of a known mutation in the family, sequencing of the specific mutation identified in the family was performed.
2.6. Statistical Analysis
Statistical analyses were performed with SAS v9.4 statistical software package (Cary, NC, USA). Categorical variables were presented as prevalence and percent, and continuous variables as mean, median, standard deviation (SD), and ranges. Study groups were compared by Kruskal–Wallis test (continuous variables) and Chi-square or Fisher’s Exact test (categorical variables). Risk factors for goiter development were analyzed by univariate and multivariate logistic regression analyses (age at time of the study, mean FT4 and median TSH, and percentage of lifetime abnormal results of TSH and FT4). Nonparametric regression (Loess) was used to analyze the association between serum TSH levels and goiter development. Significance value was set at p < 0.05.
3. Results
Thirty-three patients were enrolled in the study. Their characteristics are presented in Table 1. All were of Arab-Muslim origin, with a high rate of consanguinity, belonging to 7 extended families (15 core families). All subjects were living in the same region in the northeast of Israel in seven neighboring villages. CH was diagnosed by national neonatal screening in 25 patients (75%), of which 7 (23%) were diagnosed clinically prior to obtaining newborn screening results. Two patients were born before the implementation of national newborn screening in Israel, and screening results were not available for 6 patients (Table 1).

Table 1.
Patient characteristics.
At diagnosis, the presenting symptoms were prolonged jaundice, macroglossia, and umbilical hernia (Table 2). At 1 year of age, 10 children (30%) had delayed psychomotor development (Table 2). At the time of the study, 9 patients (27%) had neurological sequelae; among them, 7 patients had mental retardation and 4 patients had sensorineural hearing impairment.
Table 2.
Clinical characteristics of all patients with TPO enzyme deficiency.
Congenital goiter was reported in 4 patients (Table 2), but the goiter in these cases resolved after initiation of LT4. Over the years, 20 patients developed MNG (Table 2). Most of the patients had a huge goiter as shown in Figure 1, and all had at least twice the size of a normal gland. Among them, 8 patients (40%) underwent thyroidectomy due to either local tracheal pressure or histological findings by fine needle aspiration suspecting thyroid carcinoma.
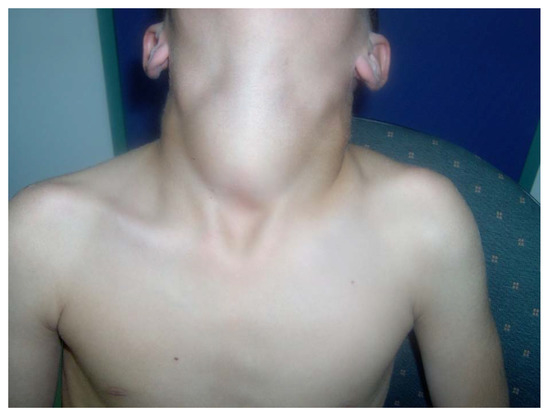
Figure 1.
Massive MNG in a 15-year-old male with TPO mutation. (Written informed consent was obtained from the patient for publication of this case report).
3.1. Imaging Results
All patients had a normally placed thyroid gland as shown by 99TC scan, and in 3 patients (12.5%), a hypertrophic gland was demonstrated at the age of 2–3 years. A second scan was performed in 10 patients and 60% showed an enlarged MNG with either cold or warm nodules. Eight patients (24%) underwent total thyroidectomy (Table 3). All patients had benign follicular adenoma or hypertrophy apart from case 14, who was diagnosed with follicular carcinoma (Table 3).
Table 3.
Summary of imaging and pathology of patients that underwent thyroidectomy.
3.2. Genetic Results
Three different mutations were identified, with either homozygous or compound heterozygous inheritance. Nineteen patients were homozygous for c.1618C>T p.Arg540Ter, 4 patients were homozygous for c.1477G>A, p.Gly493Ser, and 4 patients were compound heterozygous for both mutations. Two siblings were homozygous for the c.875C>T p.Ser292Phe mutation. When examining the presence of goiter in each mutation, we have found that 13 patients (68%) were homozygous for the c.1618C>T p.Arg540Ter mutation, 4 patients (100%) were homozygous for c.1477G>A, p.Gly493Ser, and 2 patients (100%) were homozygous for the c.875C>T p.Ser292Phe mutation. No compound heterozygous patients developed a goiter. No association was shown between the type of mutation and goiter development.
3.3. Effect of TSH Values on Development of MNG
Comparing patients with and without a goiter for mean FT4 and median TSH levels revealed no differences between the two groups (Table 4). Patients without a goiter had a higher percentage lifetime TSH level (TSH above 5 mIU/L) than those with a goiter. TSH levels were high in both groups, implicating low compliance to therapy, though in most patients, FT4 levels were in normal range during the study (normal reference was 10–20 pmol/L.) However, patients with a goiter were significantly older than those without a goiter (Table 4). No associations were found between the TSH parameters and the need for thyroidectomy.
Table 4.
Comparison of thyroid function parameters between patients that developed goiter and patients without goiter.
4. Discussion
We report on the clinical outcome of 33 patients with TPO deficiency for an extensive period of up to 43 years. We show that 61% of our patients developed MNG over time. The development of MNG was not associated with higher TSH values, indicating that elevated TSH levels have only a minor impact on goiter development. Among the 8 patients that underwent thyroidectomy, 1 patient had minimally invasive follicular carcinoma and the other 7 had either follicular adenoma or hyperplasia.
Among patients diagnosed with dyshormonogenesis, 33 (80%) had TPO deficiency. The incidence of TPO deficiency in our group was higher than that described previously. In Japan, the incidence is as low as 13%, Portugal 23% [11,12], and in Slovakia, Serbia, and the Czech Republic, 45% [4]. The high occurrence of TPO mutations in our cohort was attributed to the high rate of consanguinity in our population (80%).
The clinical presentation of TPO mutations at referral was similar to that of CH from other etiologies. Most patients (80%) presented with signs of hypothyroidism in the first days of life prior to obtaining the neonatal screening report. The main signs were prolonged jaundice, macroglossia, umbilical hernia, and coarse facial features indicating that hypothyroidism may have already existed in utero.
Congenital goiter was observed in 4 infants (12%). This finding is in contrast to a French cohort in which congenital goiter was reported in 60% of patients [14]. The high rate of congenital goiter in the French study might be attributed to the fact that the 99TC scan was performed immediately after birth, whereas in our study, the presence of goiter relied on physical examination, while the 99TC scan was performed at 2–3 years of age.
Three different mutations in the TPO gene were identified. The c.1618C>T p.Arg540Ter mutation was reported previously in populations from Japan and the Netherlands [12,15], c.1477G>A p.Gly493Ser was reported in patients from China and Portugal [9,11], and the c.875C>T p.Ser292Phe mutation was reported in patients from Tunis [10]. In family members without TPO mutations, there was no predisposition to developing a goiter.
Most of our patients (61%) developed a goiter over time, at an average age of 8.6 years. Goiter was determined by physical examination and followed by US imaging yearly once diagnosed [16]. Thyroid antibodies including anti-TPO, and anti-thyroglobulin was negative. Eight patients underwent thyroidectomy. In 5 of them, local pressure on the trachea, proven by CT scan, was the indication for surgery, whereas the other 3 patients had histological findings in fine needle aspiration of suspected malignancy. The pathological findings revealed histological characteristics of dyshormonogenesis [17]. An exception was patient 14, who was diagnosed with minimally invasive follicular carcinoma. This case was previously reported by our group [18] and it joins few other reports of follicular carcinoma in patients with TPO mutations [19,20]. Mice implanted with a thyroid gland that continuously produced TSH showed a high percentage of malignant transformations [21].
A high TSH level has been postulated to promote thyroid hypertrophy and hyperplasia, and increases the risk for thyroid malignancy [18,22]. We evaluated the association of MNG development and lifetime TSH levels in our cohort. The lifetime TSH levels were above the normal range in the entire cohort, indicating nonadherence to LT4 therapy in both groups. However, a comparison of patients with and without a goiter did not reveal higher lifelong TSH levels in patients with MNG. These results contradict the common assumption that goiter development is mainly dependent on serum TSH levels. Moreover, no association was found between a specific mutation and the development of goiter. We assume that goiter development in patients with TPO mutations is a result of a combination of genetic and environmental factors. Iodine is the main environmental factor interacting with TPO, and therefore, it has been speculated that iodine levels could modify the phenotype of patients affected with TPO mutations [5]. With regard to iodine deficiency as a parameter that may induced goiter development in our cohort, there is sufficient iodine in the diet in the Northern region of Israel. Interestingly, a previous study has shown increased risk of differentiated thyroid carcinoma in European populations with certain TPO gene variants [23].
Seven patients had a diagnosis of mental retardation, 3 of them severe. We assume that the severe neurological sequelae in these patients can be attributed mainly to a delay in diagnosis and initiation of therapy, and not to the etiology of TPO mutation. Four patients had sensorineural hearing impairment. A high rate of hearing impairment in CH has been shown recently by our group [24]. Furthermore, TPO mutations have been reported to be associated with sensorineural deafness in humans [25] as well as in animal models [26], indicating that the TPO enzyme itself may have a role in the development of the auditory system.
Finally, we should note this study’s limitations: (i) older patients in our cohort had delayed initiation of supplemental therapy, and (ii) guidelines for CH management have changed over the years [2,27].
In summary, we show a high rate of MNG in patients with TPO mutations, some of whom required thyroidectomy. No association was found between goiter development and elevated TSH, indicating that in addition to high TSH values, goiter development is attributed to other genetic and environmental factors. One patient developed follicular thyroid carcinoma, whereas all the others had hyperplastic adenomatous goiter consistent with dyshormonogenesis.
5. Conclusions
This study describes the long-term clinical outcome of a large cohort of patients with TPO mutations, emphasizing the development of MNG over the years. The high rate of MNG development and the risk for thyroid carcinoma indicates the need for long-term follow up and annual US scans in these patients.
Author Contributions
L.T., G.E.-A., O.A., S.A. and Y.T.-R. analyzed and interpreted the clinical and biochemical aspects of the patients’ data. M.K. interpreted the molecular genetic data. L.T. and Y.T.-R. contributed to writing the manuscript and designing the study. All authors have read and agreed to the published version of the manuscript.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Ethics Committee of Ha’emek Medical Center ID no. 0138-18-EMC.
Informed Consent Statement
Informed consent was not required by the Helsinki committee due to this being a retrospective study.
Acknowledgments
We thank the families for participating in the study, Camille Vainstein for professional English editing, and Naama Shwartz for statistical analysis.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cangul, H.; Aycan, Z.; Olivera-Nappa, A.; Saglam, H.; Schoenmakers, N.A.; Boelaert, K.; Cetinkaya, S.; Tarim, O.; Bober, E.; Darendeliller, F.; et al. Thyroid dyshormonogenesis is mainly caused by TPO mutations in consanguineous community. Clin. Endocrinol. 2013, 79, 275–281. [Google Scholar]
- Van Trotsenburg, P.; Stoupa, A.; Léger, J.; Rohrer, T.; Peters, C.; Fugazzola, L.; Cassio, A.; Heinrich, C.; Beauloye, V.; Pohlenz, J.; et al. Congenital Hypothyroidism: A 2020–2021 Consensus Guidelines Update—An ENDO-European Reference Network Initiative Endorsed by the European Society for Pediatric Endocrinology and the European Society for Endocrinology. Thyroid 2021, 31, 387–419. [Google Scholar]
- Rastogi, M.V.; LaFranchi, S.H. Congenital hypothyroidism: Orphanet. J. Rare Dis. J. Contin. Educ. Top. Issues 2012, 14, 33. [Google Scholar]
- Avbelj, M.; Tahirović, H.; Debeljak, M.; Kusekova, M.; Toromanovic, A.; Krzisnik, C.; Battelino, T. High prevalence of thyroid peroxidase gene mutations in patients with thyroid dyshormonogenesis. Eur. J. Endocrinol. 2007, 156, 511–519. [Google Scholar]
- Ris-Stalpers, C.; Bikker, H. Genetics and phenomics of hypothyroidism and goiter due to TPO mutations. Mol. Cell. Endocrinol. 2010, 322, 38–43. [Google Scholar]
- Abramowicz, M.; Targovnik, H.M.; Varela, V.; Cochaux, P.; Krawiec, L.; A Pisarev, M.; Propato, F.V.; Juvenal, G.; Chester, H.A.; Vassart, G. Identification of a mutation in the coding sequence of the human thyroid peroxidase gene causing congenital goiter. J. Clin. Investig. 1992, 90, 1200–1204. [Google Scholar]
- Rowe, C.W.; Paul, J.W.; Gedye, C.; Tolosa, J.M.; Bendinelli, C.; McGrath, S.; Smith, R. Targeting the TSH receptor in thyroid cancer. Endocr.-Relat. Cancer 2017, 24, R191–R202. [Google Scholar]
- Nascimento, A.C.; Guedes, D.R.; Santos, C.S.; Knobel, M.; Rubio, I.G.S.; Medeiros-Neto, G. Thyroperoxidase gene mutations in con-genital goitrous hypothyroidism with total and partial iodide organification defect. Thyroid 2003, 13, 1145–1451. [Google Scholar]
- Wu, J.-Y.; Shu, S.-G.; Yang, C.-F.; Lee, C.-C.; Tsai, F.-J. Mutation analysis of thyroid peroxidase gene in Chinese patients with total iodide organification defect: Identification of five novel mutations. J. Endocrinol. 2002, 172, 627–635. [Google Scholar]
- Bougacha-Elleuch, N.; Charfi, N.; Miled, N.; Bouhajja, H.; Belguith, N.; Mnif, M.; Jaurge, P.; Chikrouhou, N.; Ayadi, H.; Hachicha, M.; et al. Segregation of S292F TPO gene mutation in three large Tunisian families with thyroid dyshormonogenesis: Evidence of a founder effect. Eur. J. Pediatr. 2015, 174, 1491–1501. [Google Scholar]
- Rodrigues, C.D.F.; Jorge, P.; Soares, J.P.; Santos, I.; Salomão, R.; Madeira, M.; Osorió, R.V.; Santos, R. Mutation screening of the thyroid peroxidase gene in a cohort of 55 Portuguese patients with congenital hypothyroidism. Eur. J. Endocrinol. 2005, 152, 193–198. [Google Scholar]
- Narumi, S.; Muroya, K.; Asakura, Y.; Aachi, M.; Hasegawa, T. Molecular Basis of Thyroid Dyshormonogenesis: Genetic Screening in Population-Based Japanese Patients. Clin. Endocrinol. Metab. 2011, 96, E1838–E1842. [Google Scholar]
- Chiesa, A.; Rivolta, C.M.; Targovnik, H.M.; Gruñeiro-Papendieck, L. Clinical, biochemical, and molecular findings in Argen-tinean patients with goitrous congenital hypothyroidism. Endocrine 2010, 38, 377–385. [Google Scholar]
- Cavarzere, P.; Castanet, M.; Polak, M.; Raux-Demay, M.-C.; Cabrol, S.; Carel, J.C.; Léger, J.; Czernichow, P. Clinical description of infants with congenital hypothyroidism and iodide organification defects. Horm. Res. 2008, 70, 240–248. [Google Scholar]
- Bakker, B.; Bikker, H.; Vulsma, T.; de Randamie, J.S.; Wiedijk, B.M.; de Vijlder, J.J.M. Two decades of screening for congenital hypo-thyroidism in The Netherlands: TPO gene mutations in total iodide organification defects (an update). J. Clin. Endocrinol. Metab. 2000, 85, 3708–3712. [Google Scholar]
- Tritou, I.; Vakaki, M.; Sfakiotaki, R.; Kalaitzaki, K.; Raissaki, M. Pediatric thyroid ultrasound: A radiologist’s checklist. Pediatr. Radiol. 2020, 50, 563–574. [Google Scholar]
- Ghossein, R.A.; Rosai, J.; Heffess, C. Dyshormonogenetic goiter: A clinicopathologic study of 56 cases. Endocr. Pathol. 1997, 8, 283–292. [Google Scholar]
- Chertok Shacham, E.; Ishay, A.; Irit, E.; Pohlenz, J.; Tenenbaum-Rakover, Y. Minimally invasive follicular thyroid carcinoma developed in dyshormonogenetic multinodular goiter due to thyroid peroxidase gene mutation. Thyroid 2012, 22, 542–546. [Google Scholar]
- Yashiro, T.; Ito, K.; Akiba, M.; Kanaji, Y.; Obara, T.; Fujimoto, Y.; Hirayama, A.; Nakajima, H. Papillary carcinoma of the thyroid arising from dyshormonogenetic goiter. Endocrinologia Japonica 1987, 34, 955–964. [Google Scholar]
- Zhu, H.; Peng, Y.-G.; Ma, S.-G.; Liu, H. TPO gene mutations associated with thyroid carcinoma: Case report and literature review. Cancer Biomark. 2015, 15, 909–913. [Google Scholar]
- Green, C.D.; Dalton, A.J.; Morris, H.P. Malignant thyroid tumors occurring in the mouse after prolonged hormonal imbalance during the ingestion of thiouracil. J. Clin Endocrinol. Metab. 1951, 11, 1281–1295. [Google Scholar]
- Sriphrapradang, C.; Thewjitcharoen, Y.; Chanprasertyothin, S.; Nakasatien, S.; Himathongkam, T.; Trachoo, O. A novel mutation in thyroid peroxidase gene causing congenital goitrous hypothyroidism in a German-Thai Patient. J. Clin. Res. Pediatr. Endocrinol. 2016, 8, 241–245. [Google Scholar]
- Cipollini, M.; Pastor, S.; Gemignani, F.; Castell, J.; Garritano, S.; Bonotti, A.; Biarnes, J.; Figlioli, G.; Romei, C.; Marcos, R.; et al. TPO genetic variants and risk of differentiated thyroid carcinoma in two European populations. Int. J. Cancer 2013, 133, 2843–2851. [Google Scholar]
- Almagor, T.; Rath, S.; Nachtigal, D.; Sharroni, Z.; Elias-Assad, G.; Hess, O.; Havazelet, G.; Zehavi, Y.; Spiegel, R.; Bercovich, D.; et al. High Prevalence of Hearing Impairment in Primary Congenital Hypothyroidism. Eur. Thyroid J. 2020, 10, 215–221. [Google Scholar] [CrossRef]
- Pfarr, N.; Borck, G.; Turk, A.; Napiontek, U.; Keilmann, A.; Müller-Forell, W.; Kopp, P.; Pohlenz, J. Goitrous congenital hypothyroidism and hearing impairment associated with mutations in the TPO and SLC26A4/PDS genes. J. Clin. Endocrinol. Metab. 2006, 91, 2678–2681. [Google Scholar]
- Johnson, K.R.; Gagnon, L.H.; Longo-Guess, C.M.; Harris, B.S.; Chang, B. Hearing impairment in hypothyroid dwarf mice caused by mutations of the thyroid peroxidase gene. J. Assoc. Res. Otolaryngol. 2013, 15, 45–55. [Google Scholar]
- Léger, J.; Olivieri, A.; Donaldson, M.; Torresani, T.; Krude, H.; van Vliet, G.; Polak, M.; Butler, G. European Society for Paediatric Endocrinology consensus guidelines on screening, diagnosis, and management of congenital hypothyroidism. Horm. Res. Paediatr. 2014, 81, 80–103. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).