
Preclinical In Vivo Modeling of Pediatric Sarcoma—Promises and Limitations


1
Hopp Children’s Cancer Center, Heidelberg (KiTZ), 69120 Heidelberg, Germany
2
Pediatric Soft Tissue Sarcoma Research Group, German Cancer Research Center (DKFZ), 69120 Heidelberg, Germany
3
Faculty of Biosciences, Heidelberg University, 69120 Heidelberg, Germany
4
Department of General, Visceral and Transplantation Surgery, Division of Pediatric Surgery, University Hospital Heidelberg, 69120 Heidelberg, Germany
5
Institute of Pathology, University of Heidelberg, 69120 Heidelberg, Germany
*
Author to whom correspondence should be addressed.
J. Clin. Med. 2021, 10(8), 1578; https://doi.org/10.3390/jcm10081578
Submission received: 27 February 2021
/
Revised: 5 April 2021
/
Accepted: 6 April 2021
/
Published: 8 April 2021
(This article belongs to the Special Issue The Current Treatment of Childhood Cancer)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Pediatric sarcomas are an extremely heterogeneous group of genetically distinct diseases. Despite the increasing knowledge on their molecular makeup in recent years, true therapeutic advancements are largely lacking and prognosis often remains dim, particularly for relapsed and metastasized patients. Since this is largely due to the lack of suitable model systems as a prerequisite to develop and assess novel therapeutics, we here review the available approaches to model sarcoma in vivo. We focused on genetically engineered and patient-derived mouse models, compared strengths and weaknesses, and finally explored possibilities and limitations to utilize these models to advance both biological understanding as well as clinical diagnosis and therapy.
1. Introduction
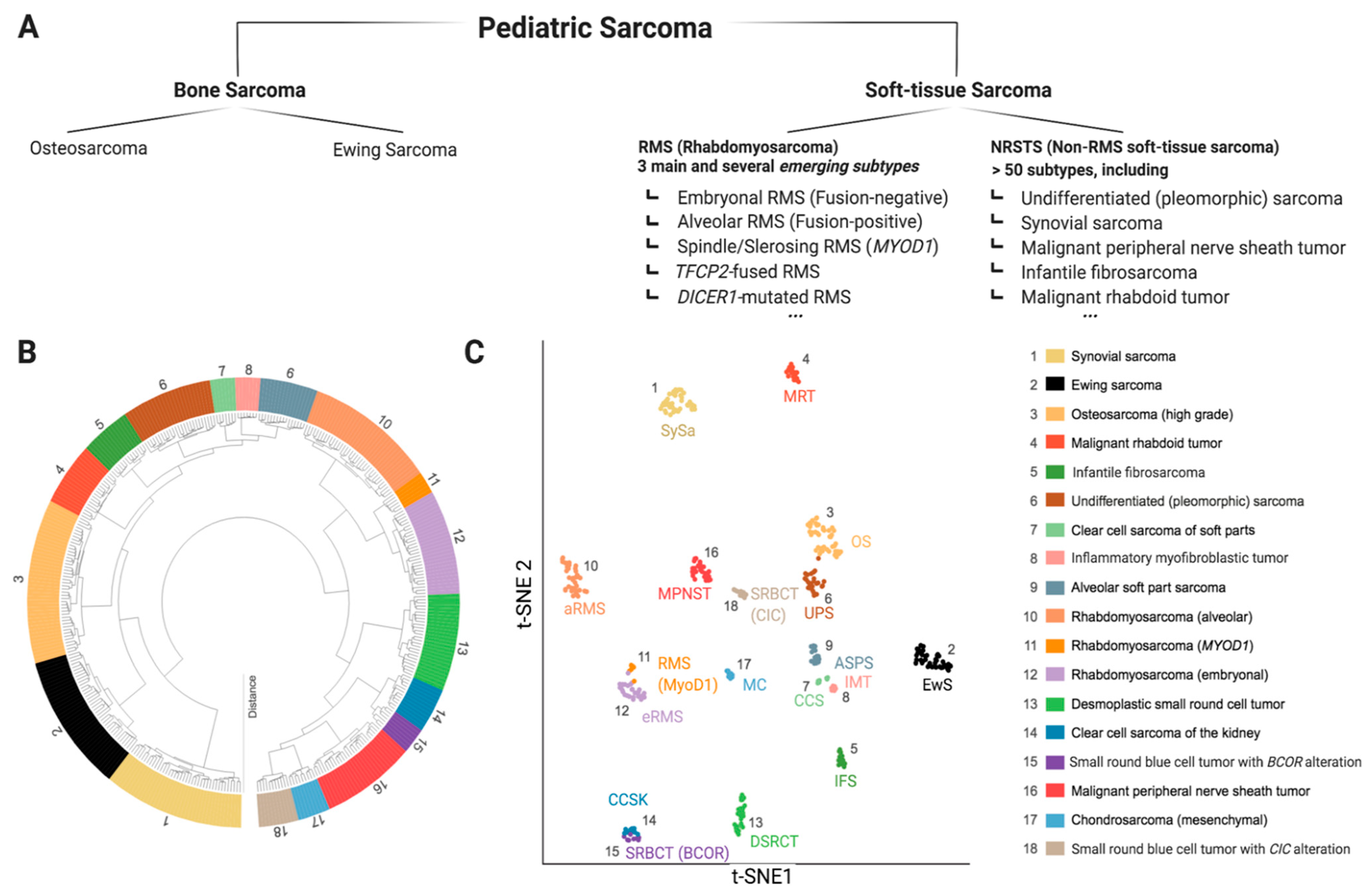
Sarcomas are mesenchymal malignancies accounting for about 15% of cancers in children and adolescents, making them the third most common group of childhood cancers, following blood malignancies and brain tumors [1]. While the last decades have seen vast improvements in pediatric cancer care with overall improved prognosis, this does not hold true for sarcomas, which are often prone to metastasis and relapse, typically accompanied by dismal prognosis [2]. Research efforts to improve this situation are complicated by the extremely diverse intrinsic nature of pediatric sarcoma with more than 60 genetically distinct entities [3]. While some pediatric sarcoma types may show widespread genomic instability (e.g., osteosarcoma (OS)), many are genetically rather simple, characterized by pathognomonic fusion oncogenes (e.g., SSX-SS18 in Synovial Sarcoma (SySa)) [4,5,6]. Ongoing molecular profiling efforts will likely lead to further sub-classification as we learn more about specific genetic and epigenetic alterations and underlying biology [7]. Figure 1 provides a snapshot of the most common pediatric sarcoma types according to the recently (2020) updated World Health Organization (WHO) classification of soft tissue and bone tumors (Figure 1a) [8,9], as well as an unbiased molecular clustering of the most common tumor entities based on DNA methylation data (Figure 1b) [10]. The development of novel therapeutic agents heavily relies on preclinical testing in disease specific models. Given the rarity of each individual pediatric sarcoma subtype, appropriate model systems are naturally scarce, and the selection of suitable models is challenging. This represents a major problem for pediatric cancer research and has significantly contributed to the lack of meaningful therapeutic improvements in pediatric sarcomas [11]. Therefore, in this review, we aim to outline the pros and cons of major in vivo modeling approaches applicable for pediatric sarcoma. We review existing models applied for specific sarcoma entities and discuss relevant points to consider for meaningful future model utilization. Due to the vast array of known malignancies, the scope of this review is limited to 18 clinically particularly relevant sarcoma entities.
2. In Vivo Modeling Approaches Applicable for Pediatric Sarcoma
Analogous to other solid tumors, four main general approaches of in vivo cancer modeling can be distinguished for pediatric sarcoma (Figure 2) [12,13]. Two of these entail the engraftment of human cancer tissue into immunocompromised mice—cell-line-derived xenograft models (CDXs) and patient-derived xenograft models (PDXs)—while the other two induce de novo tumorigenesis in immunocompetent wild type mice—environmentally-induced models (EIMMs) and genetically-engineered mouse models (GEMMs). Figure 3 highlights the major advantages and disadvantages of these approaches. Independent of the specific approach, one should consider that different mouse strains, much like humans, possess an inherent and strain-specific risk of spontaneously developing different cancer entities over their lifespans [14,15]. While these can, in some cases, also serve as useful models of human cancer, they should by no means be mistaken for specifically engrafted human or induced murine tumor tissue [16].
CDXs are the most commonly used, but least representative model when aiming to recapitulate the original disease. EIMMs are extremely powerful, but since pediatric sarcomas are usually not driven by environmental factors, they are not as relevant for childhood sarcoma. PDXs and GEMMs however, are highly representative of their human counterpart, therapeutically predictive and complementary to each other in nature.
2.1. Genetically-Engineered Mouse Models (GEMMs)
GEM models follow the principle of activating or inactivating specific cancer-associated genes in immunocompetent mice to study their impact on tumorigenesis in vivo. While early GEMM-approaches were restricted to costly embryonic stem cell alterations and extensive crossing of chimeric offspring to yield hetero- and homozygous animals for a given gene of interest [17], later work developed transgenic mouse lines expressing Cre under a variety of tissue-specific promoters, allowing conditional gene activation (typically LoxP-Stop-LoxP-Promoter-gene) or inactivation (typically LoxP-gene-LoxP) via crossing of mice [18,19]. However, germ-line models conveying constitutive gene regulation often exhibit embryonic lethality and developmental defects due to the important developmental role of many oncogenes and tumor suppressors genes [20,21,22,23,24]. This problem was overcome by the introduction of tamoxifen-inducible Cre lines, which express Cre in a Tamoxifen-inducible fashion from the endogenous Rosa26- or other more tissue-specific promoters, allowing time- and space-dependent gene regulation pre- and postnatally [25]. Profound advances in model technology entail the application of somatic gene editing approaches via lenti- and adenovirus-delivery or in vivo electroporation [26,27] and their constant optimization for in vivo use [28]. Importantly, Huang et al. could show that both CreLoxP-mediated recombination as well as somatic clustered regularly interspaced short palindromic (CRISPR)/Cas9-mediated gene editing do not lead to differences in the molecular makeup or biological behavior of induced tumors [29]. A holistic review on GEMMs in cancer research is provided by Kersten et al. [30].
2.2. Patient-Derived Xenografts (PDXs)
In PDX models, primary patient tumor material is transplanted into immunocompromised mice. Although PDXs cannot recapitulate the early steps of tumorigenesis in an intact immune microenvironment like GEMMs, their standout strength is the recapitulation of heterogeneity of their human counterparts with each PDX reflecting an individual patient [31]. This makes them a particularly valuable resource enabling the correlation of intensive molecular profiling with preclinical therapy response data to direct rational clinical trial design and select the correct patient cohorts for targeted treatments [32,33,34]. While most PDXs are established by subcutaneous (s.c.) rather than orthotopic engraftment due to easier handling and tumor surveillance, the orthotopic microenvironment may be beneficial for tumor take rates and for preserving tumor biology, as comprehensively analyzed by Stewart et al. [35]. From a technical point of view, one has to consider that tumor take rates vary greatly between about 20% and 60%, with mostly of successful engrafted tumors corresponding to aggressive/relapsed/metastasized cases. Further, stable PDX-establishment typically requires four passages in vivo, making the entire process from first engraftment to final establishment lengthy and cost-intensive, but nonetheless worthy [34,36,37,38]. Maybe most importantly, both GEMM and PDX models are regarded highly predictive for clinical therapy response [30,33,38,39].
3. Established In Vivo Models of Pediatric Sarcoma
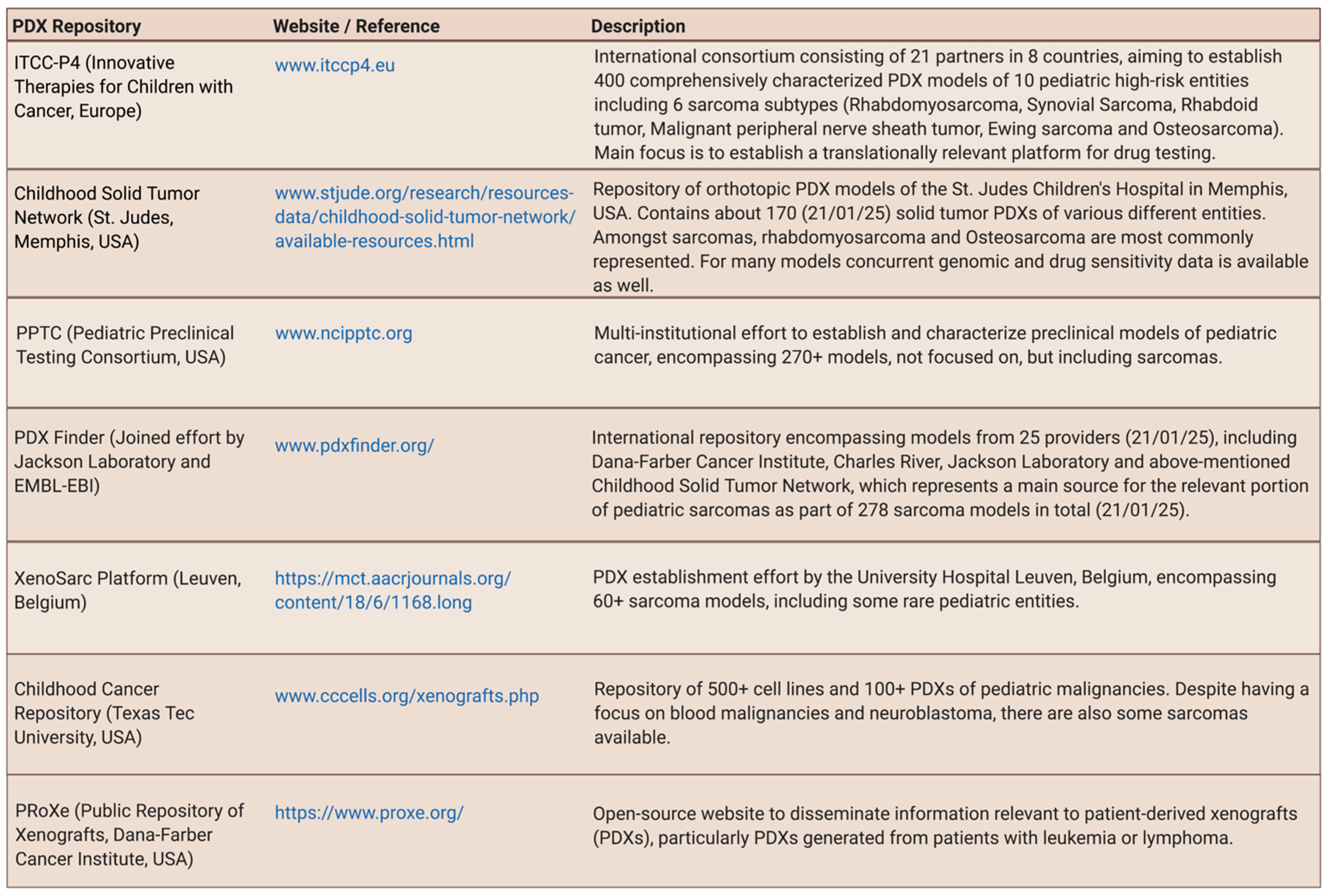
To find relevant PDX repositories entailing pediatric sarcomas (Figure 4) and to identify established GEM-modeling approaches for 18 relevant pediatric sarcoma entities (Supplementary Figure S1), a systematic literature search was performed. Additionally, the most up-to-date previous sarcoma mouse model reviews by Dodd et al. (2010) [19], Post (2012) [39], Seitz et al. (2012) [40], Brossier et al. (2012, further focused on Malignant Peripheral Nerve Sheath Tumor (MPNST) and related entities) [41], Jacques et al. (2018, further focused on bone sarcoma) [42], and O’Brien [43], Zanola et al. [44], and Yohe et al. [45] (2012/2012/2019, further focused on rhabdomyosarcoma (RMS)), were carefully considered.
3.1. Existing Cell-Line-Derived Xenograft Models (CDXs) for Pediatric Sarcoma
As extensively review by Gengenbacher and Singhal et al., the current mainstay of in vivo cancer modeling are engraftments of long-established cell lines from often cultured over many passages, using high amounts of fetal calf serum (FCS) in vitro [13]. This also holds true for sarcoma with over 70% of articles entailing sarcoma mouse models in high-ranking journals in 2016 applied CDX models both for basic and translational research while utilization of PDXs and GEMMs was under 10% [13]. This is often due to broad availability and experience as well as ease of use. Common “work horses” of sarcoma cell lines also used for in vivo engraftment are Rh30 (alveolar rhabdomyosarcoma—aRMS), A204 (embryonal rhabdomyosarcoma—eRMS), RD (RT), HS-SY-II (SySa), TC71 (Ewing Sarcoma—EwS) and KHOS (osteosarcoma—OS) among many others. Many of these models, however, do not faithfully represent the original tumor and are highly adapted to two-dimensional (2D) culture conditions. TC71, for example, one of the most commonly used EwS cell lines, harbors a BRAF mutation, which is rarely found in EwS patients and can influence therapy response [46,47]. Hinson et al. provide a holistic review on commonly used RMS cell lines and necessary precautions to consider, which can also be applied to other sarcoma entities [48].
3.2. Existing Patient-Derived Xenograft Models (PDXs) for Pediatric Sarcoma
PDX models of various entities, including sarcoma, are undisputedly highly valuable tools towards a deeper understanding of cancer biology and treatment advances [33]. The current key challenge, however, is their availability with many fragmented efforts to collect and transplant patient samples into mice, scattered over the world [49]. For sarcoma, this is particularly challenging due to the rarity of specific sub-types and the logistic challenges to obtain these samples, particularly in countries with decentralized pediatric cancer care [32,35,38,50]. Figure 4 provides a list of PDX repositories entailing pediatric sarcoma. The Memorial Sloan Kettering Cancer Center’s Department of Pediatrics will also soon be releasing their collection of pediatric PDX models including several sarcoma entities such as desmoplastic small round cell tumor (DSRCT) [51].
Some models can also be retrieved amidst larger repositories, mostly entailing models for common adulthood cancers. While many of these repositories have large amounts of models and accompanying data (e.g., https://www.europdx.eu/, last accessed 21 April 2021, with 1500+ models) and try to bridge the efforts of academic institutions and contract research organizations (e.g., https://repositive.io/, last accessed 21 April 2021, with 8000+ models), PDXs of pediatric malignancies remain largely underrepresented or not present at all. Furthermore, since many do not allow filtering available models by age group, it appears that they are often not poised to encourage deposition of pediatric PDXs.
Most repositories also supply comprehensive data sets on the molecular characterization of PDXs. This is particularly important to increase their value as preclinical testing tools. To this end, the recently proposed so-called “PDX models minimal information standard” (PDX-MI), defining a basic standard of PDX model description, could be of great value to help researchers to pick the right models for their respective question [52].
3.3. Existing Environmentally-Induced Mouse Models (EIMMs) for Pediatric Sarcoma
Environmentally-induced sarcoma models are mostly relevant for adulthood sarcoma since sarcoma in children and adolescents is typically the result of distinct genetic events rather than accumulation of genetic alterations as a result of environmental factors. Zanola et al. and Kemp et al. provide reviews entailing several EIMM systems, relevant for adulthood cancers [44,53]. Nonetheless, intramuscular (i.m.) injection of both cardiotoxin (CTX) and barium chloride to induce muscle damage and subsequent muscle regeneration with a more activated state of satellite cells (major stem cell pool for muscle regeneration), have been successfully used to induce a regenerative environment with increased susceptibility towards undifferentiated pleomorphic sarcoma (UPS) and RMS in several GEM modeling approaches (see GEMMs section) [54,55,56,57]. Interestingly, genetic models of muscular dystrophy also seem to provide a micromilieu and cellular state, which clearly facilitates sarcomagenesis with a remarkable specificity towards eRMS, which even correlates with severity of muscle dystrophy (see GEMM section) [54,58,59,60].
Radiotherapy and chemotherapy can also be regarded as external cues of mutagenic nature that children being treated for cancer frequently face on an everyday basis [61]. Since close to 10% of pediatric cancer is likely based on predisposing germ line variants, many of which can increase the susceptibility to mutagenic cues (e.g., radiotherapy in neurofibromatosis), the relevance of this could be largely underestimated [62]. To this end, Lee et al. presented a very informative comparison of murine sarcoma induction by either radiation or local injection of the mutagen 3-methylcholanthrene (MCA) in a wild type or Tp53-null background as well as a genetic model of Kras overexpression and Tp53 knockout. These comparisons revealed distinct mutagenic patterns and different levels of genomic stability, depending on the causative event [63].
3.4. Existing Genetically-Engineered Mouse Models (GEMMs) of Pediatric Sarcoma
Both the cell of origin, which is often not entirely known for many sarcomas, and mutational profile likely determine sarcoma biology and appearance [18]. While many sarcomas are determined by pathognomonic driver oncogenes, such as PAX3-FOXO1 in aRMS, UPS, and eRMS are not as clearly genetically defined. Additionally, the same genetic alterations (e.g., oncogenic RAS mutation plus TP53 inactivation) can lead to both UPS, pleomorphic RMS and eRMS, and can be seen as a disease spectrum of varying divergence in cell of origin and mutational profile [18]. Details on existing GEMMs of the 18 focus entities researched for the scope of this review is provided in Figure S1.
3.4.1. Undifferentiated Pleomorphic Sarcoma (UPS)
UPS refers to aggressive undifferentiated soft tissue and bone sarcomas, which lack an identifiable line of differentiation. While UPS are more common in adults, they may also occur in children [64]. The early work of genetic cancer modeling in mice focused on the tumor spectrum of mice deficient for major tumor suppressors such as Tp53, including specific mutations mimicking Li–Fraumeni syndrome [65]. While this does not induce specific tumor entities, but rather a plethora of different cancers with increased penetrance compared to unaffected mice, the most commonly occurring neoplasms are lymphomas and sarcomas. Sarcomas typically possess UPS morphology, more rarely also OS-, RMS-, and angiosarcoma appearance [17]. Sarcoma penetrance varied from about 10 to 50% when mice were surveilled over their entire lifespan [66,67]. If additional tumor suppressors, such as Pten, are knocked out, the efficiency increases dramatically (100% penetrance/10 weeks median latency) [68]. Furthermore, oncogenic Ras could be identified as one of the strongest oncogenes, requiring co-occurring tumor suppressor silencing (e.g., Tp53 or Rb1) to avoid apoptosis and senescence [27,69,70]. While the introduction of TP53 hotspot mutations are even more efficient in tumorigenesis than Tp53 loss and lead to spontaneous metastasis in about 13% of cases [70], mutant Ras can also cooperate with Cdkn2a inactivation to induce UPS with similar efficiency [26]. Applying Myf7-and MyoD-CreER lines, Blum et al. identified Myf7-positive muscle progenitors as a cell of origin for both UPS (62%) and RMS (38%, 63% of which were graded eRMS), while MyoD-positive progenitors only led to UPS (70% of which showed myogenic features) [55]. This is believed to correlate with activation status of satellite cells as the muscle regeneration stem cell pool, which could also be induced by i.m. injection of cardiotoxin to induce muscle damage and regeneration.
3.4.2. Embryonal/Fusion-Negative Rhabdomyosarcoma (eRMS) and Pleomorphic RMS
Embryonal rhabdomyosarcoma is the most common RMS subtype and genetically more diverse than aRMS and other sarcomas. This is reflected by the plethora of different eRMS models induced by different oncogenes and tumor suppressors over the last 25 years [71]:
- •
- Sonic Hedgehog signaling: interestingly, one of the first identified RMS GEMMs with embryonal morphology was incidentally found in a Ptch-inactivated mouse model of Gorlin syndrome, an autosomal dominant syndrome predisposing towards basal cell carcinoma, medulloblastoma, and RMS. This model developed eRMS with an incidence of 9% and 1% in CD-1 and C57BL/6 mice respectively, also highlighting the relevance of mouse strain differences for studying tumorigenesis [72]. Since then, several papers were built upon this work by dissecting the major components of Sonic Hedgehog (SHH) signaling and their influence on tumorigenesis. A tamoxifen-inducible model from Mao et al. showed that expression of a constitutively active form of the cellular signal transducer Smoothened (Smo), called SmoM2, can drive eRMS on a Ptch−/+-background with 100% penetrance within five weeks [73]. Releasing Sufu (Suppressor of fused) inhibition on the Gli effector proteins (Sufu−/+) can also drive eRMS on a Tp53−/− background while Sufu−/− is embryonically lethal [23]. Thus, as further worked out by the inducible model of Ptch-inactivation by Zibat et al., Sufu mutations appear more efficient in sarcomagenesis than Ptch mutations [74]. Interestingly, the remaining wild type allele of Ptch−/+-mice was also silenced in the course of eRMS-tumorigenesis. Hatley et al. showed that SmoM2 can even induce eRMS when expressed in the adipocytic lineage (aP2-Cre) with about 80% or up to 100% penetrance when cooperating with loss of Cdkn2a [75]. Rubin et al. utilized various Cre-drivers to investigate conditional Ptch-and Rb1-inactivation on a Tp53-null background and found a tumor spectrum of eRMS, UPS, and partly OS with satellite cells predisposed towards UPS, and maturing myoblasts towards eRMS development [18]. Finally, Fleming et al. recently showed that expression of Gli2A, a constitutively active form of SHH effector protein Gli2, via PCP2-Cre leads to small round cell tumors with Ewing-like features with nearly 100% penetrance and a median latency of about 8 weeks [24]. Pairing with SmoM2-expressing mice was embryonically lethal.
- •
- eRMS in models of muscle dystrophy and regeneration: Chamberlain et al. made the incidental observation that about 6% of muscular dystrophy X-linked (MDX) mice, which model Duchenne muscular dystrophy by harboring a spontaneous point mutation in exon 23 of the dystrophin gene, develop eRMS late in life [58]. Fernandez et al. validated this finding for MDX mice (9% eRMS penetrance late in life) and further found that mice deficient for of Alpha-Sarcoglycan (Sgca−/−), mutated in limb girdle muscular dystrophy (LGMD) can also lead to eRMS occurrence late in life (4% penetrance). Tumors exhibit Mdm2 and P53 amplification with cancer-associated P53 missense mutations. Camboni et al. bred MDX mice to homozygous or heterozygous Tp53 knockout mice and found 60%/26 weeks and 90%/17 weeks of penetrance and median latency, respectively [54]. Efficiency of tumorigenesis could further be increased by inducing muscle damage and regeneration by intramuscular CTX injection (100% penetrance/13 weeks median latency). This led to the idea that the regenerative cellular/microenvironmental state induced by muscle repair sensitizes towards sarcomagenesis. Recently, Boscolo et al. applied a more severe model of Duchenne muscular dystrophy (MDX/MtR mice) and combined it with injection of Barium chloride to induce muscle damage and regeneration [60]. Strikingly, muscle stem cells acquired an RMS-like gene signature before transformation, leading to very efficient eRMS-tumorigenesis (100%/17 weeks median latency). Van Mater et al. applied a dual recombinase system for a UPS model driven by KRASG12D and/or Tp53 inactivation and found that muscle injury can to some degree substitute for KRASG12D and leads to UPS with chromosomal gains encompassing Yap1 and Met [76]. Collectively, the work of Rubin, Boscolo, Tremblay, Van Mater and Blum et al. clearly show a connection between muscle regeneration, activation status of satellite cells and the susceptibility of sarcomagenesis towards eRMS and UPS [18,55,56,60,76].
- •
- Hippo signaling: Tremblay et al. found that this paradigm also holds true for eRMS driven by a constitutively active mutant of YAP1(S127A). Strikingly, YAP1 (Yes-associated protein 1) hyperactivity only induced sarcoma in activated satellite cells after i.m. injection of CTX or barium chloride, but not in quiescence [56]. Slemmons et al. further found cooperation between activated YAP1 and oncogenic Ras in eRMS [57].
- •
- •
- Hepatocyte-growth-factor-receptor (Hgfr)/c-Met-signaling: Takayama et al. showed that overexpression of hepatocyte growth factor/scatter factor (HGF/HF) leads to induction of various malignancies, including RMS (7% penetrance) via autocrine c-Met signaling [79]. Sharp et al., further found that Cdkn2a inactivation strongly increases the efficiency of eRMS induction upon c-Met-activation (90% penetrance/14 weeks median latency) [80].
- •
- P53 cooperation: as outlined above, various signaling pathways cooperate with Tp53-inactivation for sarcomagenesis. Comiskey et al. recently developed a model highlighting this relationship, featuring MDM2-ALT1 (splice variant 1 of murine double minute 2), which is frequently expressed in eRMS (70%) and aRMS (85%) [81]. When expressed via Sox2-Cre it promotes eRMS-induction in a Tp53−/+ (100% penetrance/20 weeks median latency, 50% of tumors are eRMS), but not Tp53−/−-background (100% penetrance/27 weeks median latency, mostly lymphoma, no eRMS. Further models include et a double knockout model of Fos/Tp53, developing eRMS in facial and orbital regions by Fleischmann et al. [82] and a cardiac model of RMS (not further specified) by Köbbert et al., who inactivated both Tp53 and Rb1 via microinjection of embryos with SV40-Tumor antigen (TAg) under the Sm22alpha promoter (active in both smooth muscle and embryonic cardiac muscle) [83].
3.4.3. Alveolar/Fusion-Positive Rhabdomyosarcoma (aRMS)
The aRMS constitute the second most common RMS subtype and usually occurs in adolescents and young adults (peak incidence at 10–25 years of age) [84]. aRMS exhibit skeletal muscle differentiation and specific molecular alterations (either a PAX3-FOXO1 or a PAX7-FOXO1 gene fusion) are detected in the majority of cases [85]. Despite this clear molecular definition, the cell of origin of aRMS is not entirely clear, complicating mouse modeling development [86]. Lagutina et al. and others found that constitutively expressing the Pax3-Foxo1 fusion from the endogenous Pax3 locus leads to developmental defects, but not tumorigenesis [87]. Heterozygous and chimeric mice showed developmental muscle defects and died perinatally from cardiac/respiratory failure. Interestingly, Pax3-Foxo1 expression from exogenous PGK-, MyoD-and rat-beta-actin promoter did not yield any phenotype.
Keller et al. later validated the developmental role of Pax3-Foxo1 upon embryonic expression and further applied a conditional model using Pax7-Cre to knock-in Pax3-Foxo1 into the endogenous Pax3 locus [88]. This expression, starting in terminally differentiating muscle cells, gave rise to aRMS, although with extremely low penetrance (1 of 228 mice, about a year after birth) [89]. Additionally, inducing haploinsufficiency for Pax3 by a second conditional allele did not accelerate tumorigenesis. Inactivation of Cdkn2a and even more so of Tp53 however, increased efficiency markedly to about 30–40% when carried out on both alleles [89]. This model was further characterized molecularly by Nishijo et al. who found a preserved gene expression signature between human and murine aRMS and observed that spontaneous metastasis, albeit occurring at very low frequency, was selected for high expression of the Pax3-Foxo1-fusion [90].
A follow-up study by Abraham et al. from the Keller laboratory further utilized this conditional Pax3-Foxo1 model, using four different Cre lines [91]: MCre (Pre-and postnatal hypaxial lineage of Pax3 that includes postnatal satellite cells), Myf5-Cre (Pre- and postnatal lineage of Myf5 that includes quiescent and activated satellite cells and early myoblasts), Myf6-CreER (Pre-and postnatal lineage of Myf6 that includes maturing myoblasts) and Pax7-CreER (Postnatal lineage of Pax7 that includes quiescent and activated satellite cells) [18]. While tumors of MCre (40% penetrance/median 29 weeks) and Myf6-CreER (100% penetrance/median 15 weeks) showed typical aRMS morphology, Pax7-CreER had spindle/pleomorphic appearance, reminiscent of fusion-negative RMS and prolonged tumor-free-survival (65% penetrance/median 48 weeks) [91]. Strikingly, reporter gene- and fusion gene expression was also lower in Pax7-CreER tumors, indicating lower transcription from the Pax3 locus in these mice and conveying divergent therapy response to histone deacetylase (HDAC) inhibitor entinostat. Myf5-Cre mice were embryonically lethal with only one mouse surviving and developing aRMS amidst increased anaplasia after 10.5 weeks:
- •
- Hippo signaling: Oristian et al. further applied the Myf6-Cre-driven conditional Pax3-Foxo1/Cdkn2aFlox/Flox/Pax3Pf/Pf model of aRMS and added Stk3Flox/Flox and Stk4Flox/Flox to activate Hippo signaling [92]. This resulted in increased tumorigenesis (88%/median 16 weeks) vs. 27%/median 26 weeks) and an increased number of tumors per animal, highlighting the role of Hippo signaling in aRMS.
Unfortunately, so far, none of the described RMS models could shed light on the age discrepancy between eRMS and aRMS patients.
3.4.4. Spindle Cell/Sclerosing RMS with MYOD1 Hotspot Mutation
MYOD1-mutant RMS represents a distinct subtype of spindle cell and sclerosing RMS. While the recurrent hotspot mutation of this biologically distinct RMS is known and appears to result in a particularly aggressive clinical course, no GEM modeling attempts could be identified in the literature to date [93]. Only a single extensively characterized patient-derived cell line model was identified [94].
3.4.5. Osteosarcoma (OS)
Osteosarcoma, a bone sarcoma characterized by a complex karyotype, can occur in children/young adults or later in life (about 60 years of age). OS is associated with genetic predisposition, in particular to Li-Fraumeni and retinoblastoma syndromes and most OS exhibit mutations/deletions of TP53 and/or RB [95,96,97]. Accordingly, several murine models relying of Tp53 inactivation have been applied to study osteosarcoma. Despite the fact that Tp53 null mice are prone to several malignances, it has been reported that 4% develop osteosarcomas (OS showing longer latency than other malignancies) with a higher frequency of OS (25%) in Tp53 heterozygous mice [17,65]. Consistent with a key role for p53 in osteosarcoma, mice harboring the p53R172H gain of function mutant knock-in develop osteosarcoma able to metastasize to other organs [67]. On the other hand, mice heterozygous for Rb deletions are not predisposed to OS, while mice homozygously deleted for Rb die at birth [98,99]. The development of several cell lineage-specific Cre expressing lines, allowed to develop many additional and improved GEMMs where Tp53 and/or Rb are inactivated in the mesenchymal/osteogenic linage; therefore, more faithfully resembling the human disease. Inactivation of Tp53 alone or together with Rb in Prx-1 positive cells (mesenchymal/skeletal progenitors) Osx, Col1A1, or Og2 positive cells (pre-osteoclasts and osteoclasts) generates OS with high penetrance often leading to metastatic disease [100,101,102,103,104,105,106]. For a detailed summary of genetically engineered mouse models for OS, see Figure S1, and recent reviews provided by Guijarro et al. [107] and Uluçkan et al. [108].
3.4.6. Ewing Sarcoma (EwS)
EwS is a small round cell sarcoma most commonly arising in the bone of children and young adults (most cases < 20 years of age). Extraskeletal manifestation of EwS occurs in about 12–20% of affected patients [109]. Ewing sarcoma’s pathognomonic driving oncogene EWSR1-ETS (typically FLI1) is functioning as a neo-transcription factor as well as an epigenetic regulator by inducing de novo enhancers at GGAA microsatellites [109]. Unfortunately, all 16 alternative attempts in 6 independent laboratories to create a transgenic Ewing sarcoma mouse model failed to date—comprehensively presented in a joined manuscript by Minas et al., 2017 [22]. Most attempts did not lead to any tumorigenesis at all despite using various tissue-specific promoters to target the potential cells of origin in EwS in different stages of development: Mesenchymal stem cells (MSCs), neural crest stem cells and embryonic osteochondrogenic progenitors [110]. Developmental EWSR1-FLI1 expression typically led to embryonic lethality while conditional expression in later stages led to various developmental defects, such as muscle degeneration. A modeling approach with successful tumorigenesis applied EWSR1-FLI1 transduction into bone marrow derived MSCs followed by intravenous (i.v.) injection into sub-lethally irradiated mice. However, it did not result in EwS, but fibrosarcoma located in the lung.
Possible explanations for these marked difficulties in model generation could lie in promoter leakiness, the lack of potential co-factors, but also in distinct biological differences between mice and humans, such as divergent splice acceptor sites, low CD99 homology, and unequal GGAA microsatellite architecture, which is not well conserved between species [111]. Particularly, the important epigenetic regulator function of EWSR1-FLI1 depends on an appropriate number of chromatin-accessible GGAA microsatellites in proximity to relevant genes to allow transformation without inducing apoptosis and might not be appropriate in mice. The fact that in vitro transformation of murine cells, such as osteochondrogenic progenitors, is possible and to some extent resembles human EwS, suggests that creating an endogenous EwS GEMM could be conceivable [112,113].
3.4.7. Synovial Sarcoma (SySa)
SySa represents a spindle cell sarcoma with variable epithelial differentiation, harboring a pathognomonic SS18-SSX1/2/4 gene fusion. Haldar et al. showed that when SS18-SSX2 expression is induced in Myf5-expressing myoblasts, 100% of mice develop synovial sarcoma-like tumors [114]. Importantly, the induction of SS18-SSX2 expression through Hprt-Cre, Pax3-Cre, or Pax7-Cre resulted in embryonic lethality, while SS18-SSX2 activation in Myf6-expressing myocytes or myofibers resulted in myopathy but no tumors. Therefore, this fusion is able to induce tumorigenesis in mice when expressed at the right time, in the right cell population (permissive cellular background) [114,115,116]. In the Myf5-Cre linage, tumors formed with 100% penetrance, presented both biphasic and monophasic histology and expressed a gene signature that partially overlapped with that of human synovial sarcoma [114]. Locally induced expression of SS18-SSX1 or SS18-SSX2 using TATCre injection also yields tumors, however with longer latency than Myf5-Cre mice. Exome sequencing identified no recurrent secondary mutations in tumors of either genotype (SS18-SSX1/2) further highlighting the idea that the fusion alone is able to drive the disease in a specific permissive background [117]. Using this localized induced model Barrott et al. showed that Pten silencing dramatically accelerated and enhances sarcomagenesis without compromising synovial sarcoma characteristics and additionally leading to spontaneous lung metastasis [118]. The same laboratory further showed that co-expression of a stabilized form of β-catenin greatly enhances synovial sarcomagenesis by enabling a stem-cell phenotype in synovial sarcoma cells, blocking epithelial differentiation and driving invasion [119].
3.4.8. Malignant Peripheral Nerve Sheath Tumor (MPNST)
Sarcomas rarely follow the benign-to-malignant multistep progression course, prototypical for many of the “big killers” in adult oncology (e.g., colorectal carcinoma). MPNST, however, can partly be regarded an exception to this rule as it often develops from plexiform or dermal neurofibromas, which represent benign lesions with homo- or heterozygous deletion of the tumor suppressor neurofibromin (NF1), acting inhibitory to Ras-signaling through its GTPase activity [120]. This can mostly be attributed to the fact that a large proportion of MPNSTs arises in neurofibromatosis patients, an autosomal dominant disease caused by inactivating NF1-mutations, but also to the fact that the unusually large NF1 gene is among the most frequently mutated genes of the human genome [121]. Additional genetic hits, such as TP53-or Cdkn2a-inactivation induce malignant progression of neurofibromas. In mice, while Nf1-plus Tp53 deletion in Schwann cells leads to MPNST, inactivation in more mesenchymal progenitors or muscle cells leads to Nf1-inactivated RMS or UPS [122]. NF1 and P53 are also exemplary of the developmental importance of many tumor suppressor genes and oncogenes. The first Nf1/Tp53 knockout models, described in 1999 by both Cichowski et al. and Vogel et al., observed embryonic lethality upon inactivation of these genes in embryonic development [20,21]. Later conditional knockout models using Cre lines specific to different tissue-specific promoters established the Schwann cell lineage as the cellular origin of MPNSTs (comprehensively reviewed Brossier et al., 2012) [41]. Since then, Dodd et al. showed that local injection of an adenovirus, delivering Cre into the sciatic nerve of Tp53-wild type mice (Nf1Flox/Flox; Ink4a/ArfFlox/Flox) also induces MPNST, while intramuscular injection leads to RMS/UPS [122]. Huang et al. validated that MPNST induction can also be obtained via lentiviral delivery of a CRISPR/Cas9 construct, targeting Nf1 and Tp53, when injected into the sciatic nerve of wild type mice [29].
3.4.9. Infantile Fibrosarcoma (IFS)
IFS represents a primitive sarcoma of fibroblastic differentiation in many of which a characteristic ETV6-NTRK3 fusion is identified [123]. While there have not been any comprehensive GEM models described to date, both ETV6-NTRK3 as well as the non-canonical fusion gene EML4-NTRK3 have been shown to be able to transform murine NIH3T3 fibroblasts, which successfully engrafted s.c. in severe combined immunodeficiency disease (SCID) and NOD SCID gamma (NSG) immunocompromised mice to result in tumors with IFS-like histomorphology [124,125].
3.4.10. Malignant Rhabdoid Tumors (MRT)
Malignant rhabdoid tumors (MRT) are aggressive, poorly differentiated pediatric cancers, characterized by the presence of germline/somatic biallelic inactivating mutations or deletions of the SMARCB1 (INI1, SNF5, or BAF47) gene, which is a component of the SWItch/Sucrose Non-Fermentable (SWI/SNF or BAF) chromatin-remodeling complex. Tumors can arise in the soft-tissue or in the kidney and less commonly in the central nervous system (referred to as atypical teratoid rhabdoid tumor; AT/RT). In mice, Smarcb1 heterozygous loss predisposes to soft-tissue sarcoma tumors consistent with human MRT but with low penetrance (approximately 12%) [126,127,128]. However, homozygous or heterozygous deletion of Tp53 (but not of Cdkn2a or Rb1) in Smarcb1 heterozygous mice accelerates tumor onset and penetrance [129,130]. Conditional inactivation of Smarcb1 in mice (Smarcb1Flox/Flox, Mx-Cre plus polyI/polyC treatment) results in rapid cancer susceptibility, with all animals developing tumors at a median age of 11 weeks [131]. These lesions exhibit many features of rhabdoid tumors, such as rhabdoid cells and complete absence of Smarcb1 expression.
3.4.11. Clear Cell Sarcoma of Soft Tissue (CCS)
CCS is an aggressive neoplasm that usually arises in the deep soft tissue of young adults. The genetic hallmark of CCS is t(12;22)(q13;q12) leading to an EWSR1-ATF1 gene fusion [132]. Straessler et al. published a model for conditional and Tamoxifen-inducible EWSR1-ATF1 expression under the endogenous Rosa26 promoter [133]. Temporal regulation of the fusion gene expression was required due to embryotoxicity of EWSR1-ATF1. Conditionally expressing human cDNA of EWSR1-ATF1 without any accompanying alterations led to highly efficient tumorigenesis of CCS-like tumors with 100% penetrance that resemble human CCS morphologically, immunohistochemically and by genome-wide expression profiling [133].
3.4.12. Alveolar Soft Part Sarcoma (ASPS)
ASPS is a deadly soft tissue malignancy, which consistently demonstrates a t(X;17)(p11.2;q25) translocation that produces the ASPSCR1-TFE3 fusion gene [134]. Expression of human cDNA in a temporal fashion (Rosa26-CreER) leads to ASPS-like tumors that resemble the human disease in terms of histology and expression patterns. Mouse tumors demonstrate angiogenic gene expression and are restricted to the tissue compartments highest in lactate, suggesting a role for lactate in alveolar soft part sarcomagenesis [135].
3.4.13. Rare Sarcomas without Current GEM Models
- •
- Clear cell sarcoma of the kidney (CCSK): CCSK is a rare neoplasm, typically arises in the kidney of infants and young children. CCSK has a dismal prognosis, often showing late relapses [136]. Recently, an internal tandem duplication of exon 15 of BCL-6 corepressor (BCOR) was identified as the major oncogenic event in CCSK [137].
- •
- Small round blue cell tumor with BCOR alteration (SRBCT-BCOR): SRBCTs represent a heterogenous group of tumors, from which SRBCT-BCOR was only recently defined as a stand-alone entity. SRBCT-BCOR typically harbors BCOR-related gene fusions (e.g., BCOR-CCNB3) or an internal tandem duplication within Exon 15 of BCOR [138,139]. SRBCT-BCOR are rare neoplasms, mostly arising in infants and young children, showing a striking male predominance [140].
- •
- Small round blue cell tumor with CIC alteration (SRBCT-CIC): similarly, to SRBCT-BCOR, SRBCT-CIC was recently identified as a distinct subtype of SRBCT [141]. In most cases a CIC-DUX4 gene fusion is identified [142]. SRBCT-CIC may arise in children and older adults; however, most cases are observed in young adults (25–35 years of age) [143].
- •
- •
- •
- Inflammatory myofibroblastic tumor (IMT): IMT is a myofibroblastic neoplasms arising in various locations, which usually shows a benign clinical course. However, few patients will present with local recurrence and/or distant metastasis [148]. In IMT, gene rearrangements affecting receptor tyrosine kinase genes (most often involving ALK) are typically identified [149].
The lack of models for many entities are a consequence of limited knowledge in regards to the tumor cell of origin, high heterogeneity of cellular backgrounds, rapid emergence of new molecular sub-types, and the need for more flexible models that allow the testing of various genetic alterations in different cellular backgrounds.
3.5. Non-Murine Animal Models for Pediatric Sarcomas
Mice as modeling organisms enable a great trade-off between appropriate resemblance of their human counterpart, and thereby translatability, as well as decently short generation times and experimental practicality. However, all aforementioned modeling approaches in mice can, in principle, also be applied in other animal species. Particularly, zebra fish represent a suitable modeling organism with largely unlocked potential in pediatric solid cancer research. While zebra fish models might not be as translatable as mouse models due to their non-mammal nature, shorter generation times, higher scalability, lower costs, extracorporeal embryonic development, and skin transparency (allowing live cell imaging) render them a powerful and complementary modeling tool for pediatric tumors [150]. Good examples of such a genetically-engineered zebrafish models for pediatric sarcoma can be found in the study of Parant et al. on MPNST [151] as well as the recent review of Casey et al. on sarcoma zebrafish models for pediatric cancers [152]. For rhabdomyosarcoma an eRMS model expressing KRASG12D in muscle satellites cells via Rag2 promoter by Langenau et al. [153,154] as well as an aRMS model, systemically expressing the PAX3-FOXO1 fusion by Kendall et al. [155] were developed to date. These models have already proven to be a valuable tool to deepen the understanding of RMS tumorigenesis [156]. Galindo et al. further showed that expression of PAX3-FOXO1 in syncytial muscle fibers of Drosophila can drive sarcomagenesis and is further supported by constitutive RAS expression [157]. Apart from GEMMs, zebra fish can also serve as host organisms for engraftment models, such as PDX. While this was previously hampered by the typical 32° living conditions of zebra fish and time-restricted to the first four weeks of life when the fish’s immune system is not yet fully developed, the recently reported prkdc−/−, il2rga−/− line represents a 37°-adapted immuno-compromised zebra fish line, allowing for engraftment of several cancer types, including eRMS and drug-response monitoring via live cell imaging [158].
Canine models have also been important for sarcoma research especially for osteosarcoma. Spontaneous OS is quite common in large dogs and highly resembles human OS in terms of gene expression profiles and histological analysis [159,160]. From a genetic perspective, OS in dogs is also characterized by complex karyotypes with variable structural and numerical chromosomal aberrations and involves many of the genes important for human OS pathogenesis including TP53, RB, and PTEN [161,162,163]. Because osteosarcoma naturally occurs with high frequency in dogs and shares many biological and clinical similarities with osteosarcoma in humans, canine OS models have provided means to understand the disease at different levels. Most importantly, they provided an opportunity to evaluate new treatment options, and indeed the development of treatment strategies in dogs and humans has mutually benefited both species [164]. Although canines have been instrumental for OS research, it is worth noting that OS in dogs occurs exclusively in old age, not entirely mimicking the human disease that peaks in adolescence [107].
4. Applications of Pediatric Sarcoma Mouse Models
Most and foremost, model generation is no end in itself. Both biological and translational advances require purposeful utilization of the right model system for the respective research question. While CDXs are still the by far most commonly used model due to broad availability and ease of use, either PDX- or GEM-models are typically the most suitable model for both basic and translational research questions (Figure 5). In general, GEM models are ideal to deepen our understanding of basic sarcoma biology while PDX models are of particularly value for preclinical testing, adequately representing patient heterogeneity. While many cell-based immunotherapies can also be tested in conventional PDX models, immunotherapies requiring endogenous immune cells can either be tested in GEMMs or humanized PDX models, the latter being very costly and technically challenging thus largely limiting their use [165,166] (Figure 6b). Both, PDX and GEM models are suitable for local therapy advancement and imaging studies, a rather underrepresented field of research, given the importance of complete resection for clinical outcome [68,167].
GEMMs of different genetic makeup can also be used to assess the fraction of tumor cells with tumorigenic potential, following the notion that some tumors rely on a small fraction of cells to drive overall cell renewal and tumor growth [168]. Following this cancer stem cell idea, Buchstaller et al., for example, compared the tumorigenic potential of two engrafted MPNST GEMMs and found that transplanted MPNST cells from Nf1+/− Ink4a/Arf−/− tumors encompassed a 10-fold higher fraction of cells with cancer-initiating potential than MPNST cells from Nf1+/− and Tp53+/− tumors [169].
Given these advantages of PDXs and GEMMs, CDXs possess one natural prime advantage GEMMs and PDXs are often lacking: they entail a corresponding in vitro system, allowing for variable functional characterization and are often very well characterized. This strength paired with the high practicality of their use makes them a highly valuable tool for present and future sarcoma research.
Considerations for Preclinical Testing
A major consideration for model utilization is how to design meaningful and translatable preclinical therapy trials. This is particularly important for pediatric sarcoma since the rarity of individual subtypes combined with the incredibly diverse array of subsets complicates rational clinical trial design. To this end, Langenau et al. provided a comprehensive and contemporary review, highlighting 10 key points to consider when designing preclinical treatment trials [170]. The key concept is to apply the same principles and guidelines used in clinical phase I, II, and III trials to the preclinical setting in a similar stepwise approach by conducting preclinical phase I, II and III trials alike [170]. This systematic approach is equally feasible for combinatorial agent testing in vivo and elucidated that some synergistic effects can be mediated by the in vivo environment and are not picked up in vitro [171]. A prerequisite of sublime importance for successful in vivo trials is the appropriate selection of promising treatment agents, based on comprehensive molecular and drug-screening in vitro data [49]. Equally important is the selection of a set of appropriate and well characterized model systems, adequately representing patient heterogeneity, including relevant patient subsets based on molecular characteristics serving as biomarkers, possibly informing about treatment response [172,173]. Connecting molecular model characterization and drug response data is an essential avenue in moving towards precision oncology in pediatrics [172]. Recent reports applying this concept by conducting single-mouse-design studies highlight the feasibility and translational relevance of this approach [33,174,175]. Approaches to use PDX models as avatars for individual patients that are parallelly being treated in the clinic are possible in principle, but typically hampered by time-consuming model establishment and variable engraftment rates [176]. Nimmervoll et al. further introduced the concept of a mouse clinic, aiming to more closely resemble the multimodal clinical therapy, including chemotherapy, radiotherapy, and local resection for testing the application of new targeted treatments [177]. While this elaborate approach will likely be to too complex and resource-intensive for general use, one should carefully consider the combination of new targeted treatments and immunotherapies with mainstays of current therapy, including local resection and radiation. A more feasible approach to deepen the insight of therapy trials is the use of molecular barcoding of engrafted cells to reveal therapy-induced clonal selection processes [178,179,180] (Figure 6c). Useful examples on how to present preclinical therapy response data can be found in the review from Gengenbacher et al. [13].
5. Future Directions
Ideally, the toolbox of preclinical sarcoma models should sufficiently represent the immense intertumoral heterogeneity of different sarcoma subtypes. The major limitation towards establishing enough of such highly predictive sarcoma models remains the scarcity of available patient material for research of these rare diseases. As recently outlined by Painter et al. in “The Angiosarcoma Project” as a part of the “Count me in” initiative by the Broad Institute, a more patient-centered research approach, empowering patients to directly share their experience, samples and data proved to be very successful in overcoming the logistic difficulties of non-centralized treatment of rare cancers and could potentially serve as a blueprint for pediatric solid tumors to help affected children and their families to engage in meaningful research to better future therapies [181]. More information on the patient-centered “Count me in” initiative, which has recently expanded to OS, can be found here: https://joincountmein.org/, last accessed 21 April 2021.
For existing models, a key challenge will be to make the scarcely used, but particularly representative and predictive GEMM- and PDX-models more available to both academic and industrial research in order to increase relevance and translational validity of obtained results [13]. This is often complicated by the legal frameworks accompanying the exchange of models, particularly for PDXs, since they entail genomic patient information. Increased model availability would make a broadly accepted standard for model description even more important (e.g., PDX-MI/PDX models minimal information standard) [52].
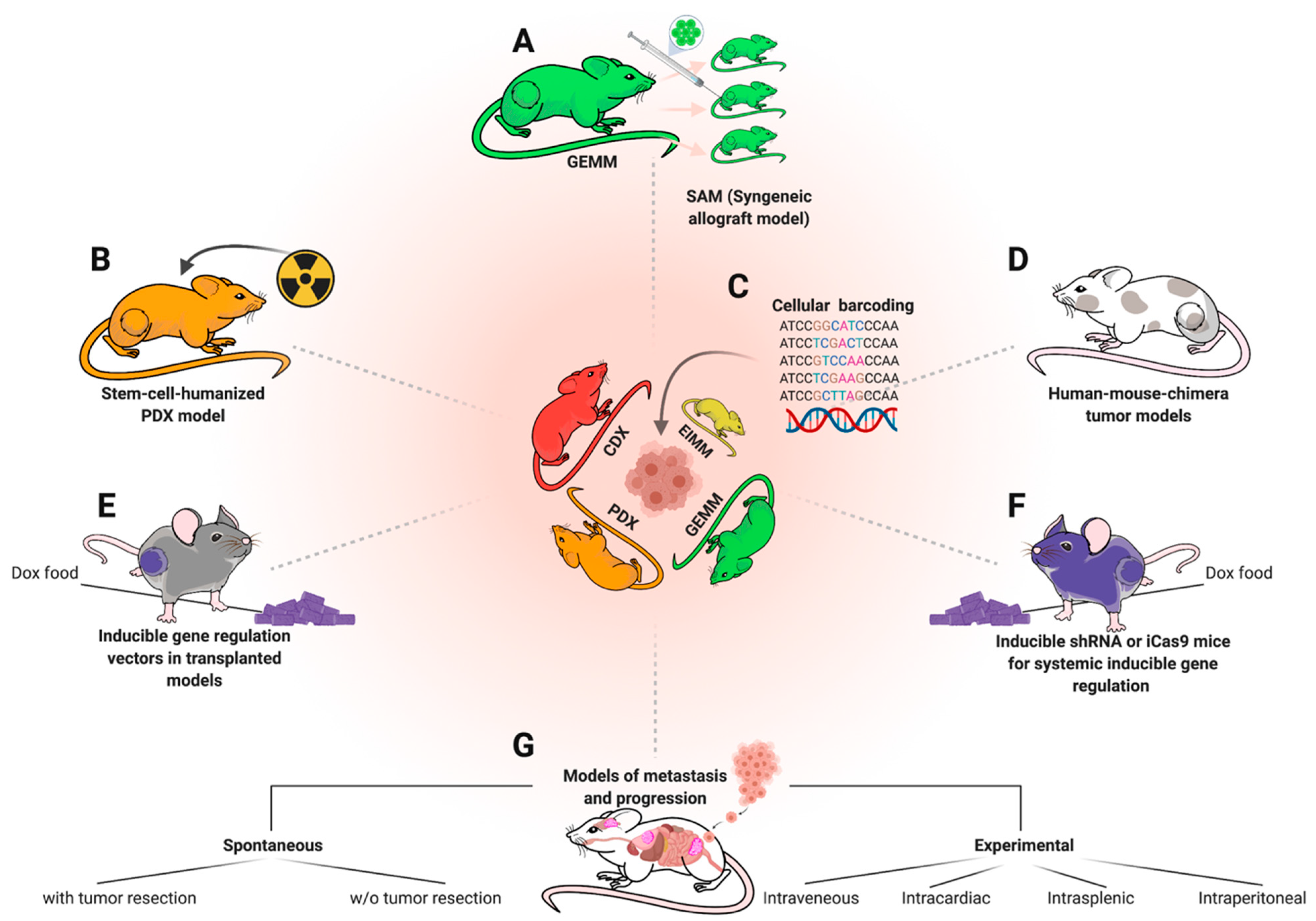
For GEMMs, more efforts should be taken towards establishing syngeneic transplantation models, allowing biobanking such as in PDX models, but with immunocompetent background. Especially immunotherapeutic approaches for children with sarcoma would immensely benefit from this approach. Importantly, once a GEMM-derived syngeneic transplantation model is established, GEM-tissue becomes expandable and the thus now scalable models can be made available beyond the host institution while costs drop from high to moderate [12] (Figure 6a). Ideally, syngeneic engraftment models (SAMs) should be accompanied by matched FCS-free 3D spheroid-/tumoroid-cultures for in vitro screening and functional studies [182,183]. This is particularly important since vast potential for future treatment advances might lie in rational combination therapies, which even more so require high-throughput in vitro screening before validation in vivo [45,61]. An additional largely unexplored potential lies in revisiting previously developed GEMMs among human samples, with up-to-date molecular profiling techniques, as exemplified by Mito et al. [184]. To this end, the recently released 285k methylation array for broad classification or the single-cell methylation analysis for mouse samples might help to deepen the knowledge on cellular origins and epigenetic determinants of various sarcoma subtypes [110,185]. To make best use of existing models, open-source platforms to access generated molecular data and microarrays to simultaneously stain for specific targets among many existing models, can be of great value to select appropriate models for particular studies [49]. Figure 6 highlights a selection of different modeling approaches that could be of additional or complementary value to various established models. For, so far, elusive GEMMs, such as EwS, which seem to require a human-specific molecular background, human–mouse chimera models could provide a solution (Figure 6d). For instance, Cohen et al. successfully developed such a model for neuroblastoma (NB) recently, by introducing NB-specific alterations on a doxycycline(Dox)-inducible vector into human stem cells (in this case hNCCs/human stem-cell derived neural crest cells) before injecting them into early mouse embryos [186]. NB-specific gene expression could later be regulated by dox administration in chimeric offspring. Another, already commonly used approach to regulate gene expression in vivo via alimental Dox are engraftment models transduced with vectors carrying a Dox-inducible RNA interference (e.g., TRE-shRNA) or inducible nuclease (e.g., Cas9-sgRNA) cassette to inducible knockdown or knockout a gene of interest [187] (Figure 6e). Such approaches are also feasible on a systemic level for to investigate the systemic toxicities due to genetic targeting of a specific gene [188,189] (Figure 6f). Genetic dropout screens via RNAi or CRISPR are also no longer limited to the in vitro setting, but can also be carried out in vivo [190].
This is also possible in models of metastasis and progression [191] (Figure 6g), a phenomenon of utmost importance for sarcoma patients, but understudied via mouse modeling [62]. Many mouse models do not metastasize spontaneously before mice have to be sacrificed due to primary tumor burden [192,193]. Thus, the most translationally relevant modeling approach for metastasis modeling is local tumor resection, typically via limb resection, and holds much promise to further elucidate mechanisms of metastasis for the various pediatric sarcoma entities, as well as possible therapeutic vulnerabilities [194,195]. At the same time, it enables utilization of PDX- and GEM models for improvement of local resection, e.g., via molecular imaging or photodynamic therapy, which, despite promising ongoing efforts, is still a rather unexplored field of pediatric sarcoma research with much unexplored potential to improve clinical outcomes [195,196,197,198,199,200].
6. Conclusions
Mouse models for pediatric sarcoma are an indispensable tool to deepen our understanding of this incredibly diverse group of diseases. Among them, genetically-engineered and patient-derived xenograft models are the most representative and predictive model types for meaningful basic and translationally relevant research. Recent technological advances, such as somatic GEM modeling, inducible in vivo gene regulation, cellular barcoding of engraftment models and first and foremost strong collaborative and international efforts to establish representative model repositories have the power to adequately represent at least major, high risk entities of the diverse landscape of pediatric sarcoma. If utilized correctly for preclinical testing, these models have the potential to transform the future clinical treatment of childhood and adolescence sarcoma.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/jcm10081578/s1, Figure S1: Existing GEM models for pediatric sarcoma of 18 focus entities.
Author Contributions
Literature curation, R.I. and A.B.; data analysis for DNA methylation clustering, F.K.F.K.; writing—original draft preparation, R.I.; writing—review and editing, R.I., F.K.F.K. and A.B.; supervision, A.B.; funding acquisition, A.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the H2020 European research Council (ERC) program, grant number 805338 (PedSarc), and the German Research Cancer Center.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created in this study. Data sharing is not applicable to this article.
Acknowledgments
We thank Nicolas Gengenbacher, Mahak Singhal, Hellmut Augustin, et al. for sharing details on sarcoma modeling from 2016 [13]. We further thank Christian Kölsche et al. for allowing to utilize DNA methylation data from the sarcoma classifier [10] and Katharina Hartwig for help with graphic design. Illustrations and figures were made with BioRender and Affinity Designer. We apologize to all authors whose related valuable work was not discussed due to space restrains.
Conflicts of Interest
The authors declare no conflict of interest.
Glossary
| Major in vivo modeling approaches | |
| CDX | Cell-line-derived xenograft |
| EIMM | Environmentally-induced mouse model |
| GEMM | Genetically-engineered mouse model |
| PDOX | Patient-derived orthotopic xenograft |
| PDX | Patient-derived xenograft |
| Focus sarcoma entities of this review with abbreviations and typical drivers | |
| aRMS | Alveolar Rhabdomyosarcoma (PAX3/7-FOXO1) |
| ASPS | Alveolar soft part sarcoma (ASPSCR1-TFE3) |
| CCS | Clear cell sarcoma (of soft tissue) (EWSR1-ATF1) |
| CCSK | Clear cell sarcoma of the kidney (BCOR ITDs, YWAHE-NUTM2) |
| DSRCT | Desmoplastic small round cell tumor (EWSR1-WT1) |
| eRMS | Embryonal Rhabdomyosarcoma (KRAS and TP53 alterations among various others) |
| EwS | Ewing Sarcoma (EWSR1-FLI1, EWSR1-ERG, STAG2) |
| IFS | Infantile fibrosarcoma (ETV6-NTRK3) |
| IMT | Inflammatory myofibroblastic tumor (ALK fusions, among others) |
| MC | Mesenchymal chondrosarcoma (HEY1-NCOA2) |
| MPNST | Malignant peripheral nerve sheath tumor (NF1 and TP53 deletions) |
| MRT | Malignant rhabdoid tumor (SMARCB1) |
| MYOD1-RMS | MYOD-mutated (typically spindle cell) rhabdomyosarcoma (MYOD-hotspot mutation among others) |
| OS | Osteosarcoma (TP53, SNVs) |
| SRBCT | Small round blue cell tumors, previously called Ewing-like tumors, subgroups harboring BCOR- and CIC rearrangements |
| SySa | Synovial Sarcoma (SS18-SSX1/2/4) |
| UPS | Undifferentiated pleomorphic sarcoma, previously called Malignant fibrous histiocytoma (KRAS and TP53 among various others) |
References
- Allen-Rhoades, W.; Whittle, S.B.; Rainusso, N. Pediatric Solid Tumors in Children and Adolescents: An Overview. Pediatr. Rev. 2018, 39, 444–453. [Google Scholar] [CrossRef]
- Sandler, G.; Yokoi, A.; Hayes-Jordan, A. An update in the management of pediatric sarcoma. Curr. Opin. Pediatr. 2019, 31, 368–377. [Google Scholar] [CrossRef]
- Cao, J.; An, Q.; Wang, L. Pediatric sarcomas. Oncol. Lett. 2018, 15, 1397–1402. [Google Scholar] [CrossRef]
- Nakano, K.; Takahashi, S. Translocation-Related Sarcomas. Int. J. Mol. Sci. 2018, 19, 3784. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, M.; Felisiak-Golabek, A.; Luiña Contreras, A.; Glod, J.; Kaplan, R.N.; Killian, J.K.; Lasota, J. New fusion sarcomas: Histopathology and clinical significance of selected entities. Hum. Pathol. 2019, 86, 57–65. [Google Scholar] [CrossRef]
- Laetsch, T.W.; Roy, A.; Xu, L.; Black, J.O.; Coffin, C.M.; Chi, Y.Y.; Tian, J.; Spunt, S.L.; Hawkins, D.S.; Bridge, J.A.; et al. Undifferentiated sarcomas in children harbor clinically relevant oncogenic fusions and gene copy-number alterations: A report from the children’s oncology group. Clin. Cancer Res. 2018, 24, 3888–3897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shern, J.F.; Chen, L.; Chmielecki, J.; Wei, J.S.; Patidar, R.; Rosenberg, M.; Ambrogio, L.; Auclair, D.; Wang, J.; Song, Y.K.; et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014, 4, 216–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO Classification of Tumours Editorial Board (Ed.) World Health Organization Classification of Soft Tissue and Bone Tumours, 5th ed.; IARC Press: Lyon, France, 2020. [Google Scholar]
- Anderson, W.J.; Doyle, L.A. Updates from the 2020 World Health Organization Classification of Soft Tissue and Bone Tumours. Histopathology 2021, his.14265. [Google Scholar] [CrossRef]
- Koelsche, C.; Schrimpf, D.; Stichel, D.; Sill, M.; Sahm, F.; Reuss, D.E.; Blattner, M.; Worst, B.; Heilig, C.E.; Beck, K.; et al. Sarcoma classification by DNA methylation profiling. Nat. Commun. 2021, 12, 498. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.W.; Banito, A.; Grünewald, T.G.P.; Haber, M.; Jäger, N.; Kool, M.; Milde, T.; Molenaar, J.J.; Nabbi, A.; Pugh, T.J.; et al. Molecular characteristics and therapeutic vulnerabilities across paediatric solid tumours. Nat. Rev. Cancer 2019, 19, 420–438. [Google Scholar] [CrossRef]
- Day, C.-P.; Merlino, G.; Van Dyke, T. Preclinical mouse cancer models: A maze of opportunities and challenges. Cell 2015, 163, 39–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gengenbacher, N.; Singhal, M.; Augustin, H.G. Preclinical mouse solid tumour models: Status quo, challenges and perspectives. Nat. Rev. Cancer 2017, 17, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Hunter, K.W. Mouse models of cancer: Does the strain matter? Nat. Rev. Cancer 2012, 12, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Puccini, J.; Dorstyn, L.; Kumar, S. Genetic background and tumour susceptibility in mouse models. Cell Death Differ. 2013, 20, 964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyer, A.M.; Yu, J.; Sinnwell, J.P.; Dockter, T.J.; Suman, V.J.; Weinshilboum, R.M.; Boughey, J.C.; Goetz, M.P.; Visscher, D.W.; Wang, L. Spontaneous murine tumors in the development of patient-derived xenografts: A potential pitfall. Oncotarget 2019, 10, 3924–3930. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Rubin, B.P.; Nishijo, K.; Chen, H.I.H.; Yi, X.; Schuetze, D.P.; Pal, R.; Prajapati, S.I.; Abraham, J.; Arenkiel, B.R.; Chen, Q.R.; et al. Evidence for an Unanticipated Relationship between Undifferentiated Pleomorphic Sarcoma and Embryonal Rhabdomyosarcoma. Cancer Cell 2011, 19, 177–191. [Google Scholar] [CrossRef] [Green Version]
- Dodd, R.D.; Mito, J.K.; Kirsch, D.G. Animal models of soft-tissue sarcoma. DMM Dis. Model. Mech. 2010, 3, 557–566. [Google Scholar] [CrossRef] [Green Version]
- Vogel, K.S.; Klesse, L.J.; Velasco-Miguel, S.; Meyers, K.; Rushing, E.J.; Parada, L.F. Mouse tumor model for neurofibromatosis type 1. Science 1999, 286, 2176–2179. [Google Scholar] [CrossRef]
- Cichowski, K.; Shih, T.S.; Schmitt, E.; Santiago, S.; Reilly, K.; McLaughlin, M.E.; Bronson, R.T.; Jacks, T. Mouse models of tumor development in neurofibromatosis type 1. Science 1999, 286, 2172–2176. [Google Scholar] [CrossRef]
- Minas, T.Z.; Surdez, D.; Javaheri, T.; Tanaka, M.; Howarth, M.; Kang, H.-J.; Han, J.; Han, Z.-Y.; Sax, B.; Kream, B.E.; et al. Combined experience of six independent laboratories attempting to create an Ewing sarcoma mouse model. Oncotarget 2017, 8, 34141–34163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Kawagoe, R.; Sasai, K.; Li, Y.; Russell, H.R.; Curran, T.; McKinnon, P.J. Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene 2007, 26, 6442–6447. [Google Scholar] [CrossRef] [Green Version]
- Fleming, J.T.; Brignola, E.; Chen, L.; Guo, Y.; Zhao, S.; Wang, Q.; Li, B.; Correa, H.; Ermilov, A.N.; Dlugosz, A.A.; et al. Insight into the etiology of undifferentiated soft tissue sarcomas from a novel mouse model. Mol. Cancer Res. 2019, 17, 1024–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hameyer, D.; Loonstra, A.; Eshkind, L.; Schmitt, S.; Antunes, C.; Groen, A.; Bindels, E.; Jonkers, J.; Krimpenfort, P.; Meuwissen, R.; et al. Toxicity of ligand-dependent Cre recombinases and generation of a conditional Cre deleter mouse allowing mosaic recombination in peripheral tissues. Physiol. Genom. 2007, 31, 32–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirsch, D.G.; Dinulescu, D.M.; Miller, J.B.; Grimm, J.; Santiago, P.M.; Young, N.P.; Nielsen, G.P.; Quade, B.J.; Chaber, C.J.; Schultz, C.P.; et al. A spatially and temporally restricted mouse model of soft tissue sarcoma. Nat. Med. 2007, 13, 992–997. [Google Scholar] [CrossRef]
- Tsumura, H.; Yoshida, T.; Saito, H.; Imanaka-Yoshida, K.; Suzuki, N. Cooperation of oncogenic K-ras and p53 deficiency in pleomorphic rhabdomyosarcoma development in adult mice. Oncogene 2006, 25, 7673–7679. [Google Scholar] [CrossRef]
- Maddalo, D.; Manchado, E.; Concepcion, C.P.; Bonetti, C.; Vidigal, J.A.; Han, Y.C.; Ogrodowski, P.; Crippa, A.; Rekhtman, N.; De Stanchina, E.; et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature 2014, 516, 423–428. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Chen, M.; Whitley, M.J.; Kuo, H.-C.C.; Xu, E.S.; Walens, A.; Mowery, Y.M.; Van Mater, D.; Eward, W.C.; Cardona, D.M.; et al. Generation and comparison of CRISPR-Cas9 and Cre-mediated genetically engineered mouse models of sarcoma. Nat. Commun. 2017, 8, 15999. [Google Scholar] [CrossRef]
- Kersten, K.; de Visser, K.E.; van Miltenburg, M.H.; Jonkers, J. Genetically engineered mouse models in oncology research and cancer medicine. EMBO Mol. Med. 2017, 9, 137–153. [Google Scholar] [CrossRef]
- Risbridger, G.P.; Lawrence, M.G.; Taylor, R.A. PDX: Moving Beyond Drug Screening to Versatile Models for Research Discovery. J. Endocr. Soc. 2020, 4, bvaa132. [Google Scholar] [CrossRef]
- Rokita, J.L.; Rathi, K.S.; Cardenas, M.F.; Upton, K.A.; Jayaseelan, J.; Cross, K.L.; Pfeil, J.; Egolf, L.E.; Way, G.P.; Farrel, A.; et al. Genomic Profiling of Childhood Tumor Patient-Derived Xenograft Models to Enable Rational Clinical Trial Design. Cell Rep. 2019, 29, 1675–1689.e9. [Google Scholar] [CrossRef]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Bhimani, J.; Ball, K.; Stebbing, J. Patient-derived xenograft models—the future of personalised cancer treatment. Br. J. Cancer 2020, 122, 601–602. [Google Scholar] [CrossRef]
- Stewart, E.; Federico, S.M.; Chen, X.; Shelat, A.A.; Bradley, C.; Gordon, B.; Karlstrom, A.; Twarog, N.R.; Clay, M.R.; Bahrami, A.; et al. Orthotopic patient-derived xenografts of paediatric solid tumours. Nature 2017, 549, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Surdez, D.; Landuzzi, L.; Scotlandi, K.; Manara, M.C. Ewing Sarcoma PDX Models. Methods Mol. Biol. 2021, 2226, 223–242. [Google Scholar] [CrossRef]
- Lu, W.; Chao, T.; Ruiqi, C.; Juan, S.; Zhihong, L. Patient-derived xenograft models in musculoskeletal malignancies. J. Transl. Med. 2018, 16, 107. [Google Scholar] [CrossRef] [PubMed]
- Cornillie, J.; Wozniak, A.; Li, H.; Wang, Y.; Boeckx, B.; Gebreyohannes, Y.K.; Wellens, J.; Vanleeuw, U.; Hompes, D.; Stas, M.; et al. Establishment and Characterization of Histologically and Molecularly Sfelix Soft-tissue Sarcoma Xenograft Models for Biological Studies and Preclinical Drug Testing. Mol. Cancer Ther. 2019, 18, 1168–1178. [Google Scholar] [CrossRef] [Green Version]
- Post, S.M. Mouse models of sarcomas: Critical tools in our understanding of the pathobiology. Clin. Sarcoma Res. 2012, 2, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seitz, G.; Armeanu-Ebinger, S.; Warmann, S.; Fuchs, J. Animal models of extracranial pediatric solid tumors (Review). Oncol. Lett. 2012, 4, 859–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brossier, N.M.; Carroll, S.L. Genetically engineered mouse models shed new light on the pathogenesis of neurofibromatosis type I-related neoplasms of the peripheral nervous system. Brain Res. Bull. 2012, 88, 58–71. [Google Scholar] [CrossRef] [Green Version]
- Jacques, C.; Renema, N.; Lezot, F.; Ory, B.; Walkley, C.R.; Grigoriadis, A.E.; Heymann, D. Small animal models for the study of bone sarcoma pathogenesis:characteristics, therapeutic interests and limitations. J. Bone Oncol. 2018, 12, 7–13. [Google Scholar] [CrossRef]
- O’Brien, D.; Jacob, A.G.; Qualman, S.J.; Chandler, D.S. Advances in pediatric rhabdomyosarcoma characterization and disease model development. Histol. Histopathol. 2012, 27, 13–22. [Google Scholar] [CrossRef]
- Zanola, A.; Rossi, S.; Faggi, F.; Monti, E.; Fanzani, A. Rhabdomyosarcomas: An overview on the experimental animal models. J. Cell. Mol. Med. 2012, 16, 1377–1391. [Google Scholar] [CrossRef]
- Yohe, M.E.; Heske, C.M.; Stewart, E.; Adamson, P.C.; Ahmed, N.; Antonescu, C.R.; Chen, E.; Collins, N.; Ehrlich, A.; Galindo, R.L.; et al. Insights into pediatric rhabdomyosarcoma research: Challenges and goals. Pediatr. Blood Cancer 2019, 66, 1–10. [Google Scholar] [CrossRef]
- Crompton, B.D.; Stewart, C.; Taylor-Weiner, A.; Alexe, G.; Kurek, K.C.; Calicchio, M.L.; Kiezun, A.; Carter, S.L.; Shukla, S.A.; Mehta, S.S.; et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014, 4, 1326–1341. [Google Scholar] [CrossRef] [Green Version]
- Çelik, H.; Sciandra, M.; Flashner, B.; Gelmez, E.; Kayraklloǧlu, N.; Allegakoen, D.V.; Petro, J.R.; Conn, E.J.; Hour, S.; Han, J.; et al. Clofarabine inhibits Ewing sarcoma growth through a novel molecular mechanism involving direct binding to CD99. Oncogene 2018, 37, 2181–2196. [Google Scholar] [CrossRef]
- Hinson, A.R.P.; Jones, R.; Crose, L.E.S.; Belyea, B.C.; Barr, F.G.; Linardic, C.M. Human rhabdomyosarcoma cell lines for rhabdomyosarcoma research: Utility and pitfalls. Front. Oncol. 2013, 3, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, E.; Federico, S.; Karlstrom, A.; Shelat, A.; Sablauer, A.; Pappo, A.; Dyer, M.A. The Childhood Solid Tumor Network: A new resource for the developmental biology and oncology research communities. Dev. Biol. 2016, 411, 287–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conte, N.; Mason, J.C.; Halmagyi, C.; Neuhauser, S.; Mosaku, A.; Yordanova, G.; Chatzipli, A.; Begley, D.A.; Krupke, D.M.; Parkinson, H.; et al. PDX Finder: A portal for patient-derived tumor xenograft model discovery. Nucleic Acids Res. 2019, 47, D1073–D1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, R.S.; Odintsov, I.; Liu, Z.; 2+, A.; Lui, J.-W.; Hayashi, T.; Vojnic, M.; Suehara, Y.; Delasos, L.; Mattar, M.S.; et al. Establishment of multiple novel patient-derived models of desmoplastic small round cell tumor enabling functional characterization of ERBB pathway signaling and pre-clinical evaluation of a novel targeted therapy approach. bioRxiv 2021. [Google Scholar] [CrossRef]
- Meehan, T.F.; Conte, N.; Goldstein, T.; Inghirami, G.; Murakami, M.A.; Brabetz, S.; Gu, Z.; Wiser, J.A.; Dunn, P.; Begley, D.A.; et al. PDX-MI: Minimal information for patient-derived tumor xenograft models. Cancer Res. 2017, 77, e62–e66. [Google Scholar] [CrossRef] [Green Version]
- Kemp, C.J. Animal Models of Chemical Carcinogenesis: Driving Breakthroughs in Cancer Research for 100 Years. Cold Spring Harb. Protoc. 2015, 2015, 865–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camboni, M.; Hammond, S.; Martin, L.T.; Martin, P.T. Induction of a regenerative microenvironment in skeletal muscle is sufficient to induce embryonal rhabdomyosarcoma in p53-deficient mice. J. Pathol. 2012, 226, 40–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, J.M.; Añó, L.; Li, Z.; Van Mater, D.; Bennett, B.D.; Sachdeva, M.; Lagutina, I.; Zhang, M.; Mito, J.K.; Dodd, L.G.; et al. Distinct and overlapping sarcoma subtypes initiated from muscle stem and progenitor cells. Cell Rep. 2013, 5, 933–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremblay, A.M.; Missiaglia, E.; Galli, G.G.; Hettmer, S.; Urcia, R.; Carrara, M.; Judson, R.N.; Thway, K.; Nadal, G.; Selfe, J.L.; et al. The Hippo transducer YAP1 transforms activated satellite cells and is a potent effector of embryonal rhabdomyosarcoma formation. Cancer Cell 2014, 26, 273–287. [Google Scholar] [CrossRef] [Green Version]
- Slemmons, K.K.; Crose, L.E.S.; Rudzinski, E.; Bentley, R.C.; Linardic, C.M. Role of the YAP oncoprotein in priming ras-driven rhabdomyosarcoma. PLoS ONE 2015, 10, e0140781. [Google Scholar] [CrossRef]
- Chamberlain, J.S.; Metzger, J.; Reyes, M.; Townsend, D.; Faulkner, J.A. Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J. 2007, 21, 2195–2204. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, K.; Serinagaoglu, Y.; Hammond, S.; Martin, L.T.; Martin, P.T. Mice lacking dystrophin or α sarcoglycan spontaneously develop embryonal rhabdomyosarcoma with cancer-associated p53 mutations and alternatively spliced or mutant Mdm2 transcripts. Am. J. Pathol. 2010, 176, 416–434. [Google Scholar] [CrossRef] [Green Version]
- Boscolo Sesillo, F.; Fox, D.; Sacco, A. Muscle Stem Cells Give Rise to Rhabdomyosarcomas in a Severe Mouse Model of Duchenne Muscular Dystrophy. Cell Rep. 2019, 26, 689–701.e6. [Google Scholar] [CrossRef] [Green Version]
- Grünewald, T.G.; Alonso, M.; Avnet, S.; Banito, A.; Burdach, S.; Cidre-Aranaz, F.; Di Pompo, G.; Distel, M.; Dorado-Garcia, H.; Garcia-Castro, J.; et al. Sarcoma treatment in the era of molecular medicine. EMBO Mol. Med. 2020, 12, 1–33. [Google Scholar] [CrossRef]
- Gröbner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.-L.; Mowery, Y.M.; Daniel, A.R.; Zhang, D.; Sibley, A.B.; Delaney, J.R.; Wisdom, A.J.; Qin, X.; Wang, X.; Caraballo, I.; et al. Mutational landscape in genetically engineered, carcinogen-induced, and radiation-induced mouse sarcoma. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alaggio, R.; Collini, P.; Randall, R.L.; Barnette, P.; Million, L.; Coffin, C.M. Undifferentiated High-Grade Pleomorphic Sarcomas in Children: A Clinicopathologic Study of 10 Cases and Review of Literature. Pediatr. Dev. Pathol. 2010, 13, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Jacks, T.; Remington, L.; Williams, B.O.; Schmitt, E.M.; Halachmi, S.; Bronson, R.T.; Weinberg, R.A. Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 1994, 4, 1–7. [Google Scholar] [CrossRef]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef] [Green Version]
- Buchakjian, M.R.; Merritt, N.M.; Moose, D.L.; Dupuy, A.J.; Tanas, M.R.; Henry, M.D. A Trp53fl/flPtenfl/fl mouse model of undifferentiated pleomorphic sarcoma mediated by adeno-Cre injection and in vivo bioluminescence imaging. PLoS ONE 2017, 12, e0183469. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, T.; Venier, R.; Dickson, B.C.; Kabaroff, L.; Alkema, M.; Chen, L.; Shern, J.F.; Yohe, M.E.; Khan, J.; Gladdy, R.A. Kras activation in p53-deficient myoblasts results in high-grade sarcoma formation with impaired myogenic differentiation. Oncotarget 2015, 6, 14220–14232. [Google Scholar] [CrossRef] [Green Version]
- Doyle, B.; Morton, J.P.; Delaney, D.W.; Ridgway, R.A.; Wilkins, J.A.; Sansom, O.J. p53 Mutation and loss have different effects on tumourigenesis in a novel mouse model of pleomorphic rhabdomyosarcoma. J. Pathol. 2010, 222, 129–137. [Google Scholar] [CrossRef]
- Skapek, S.X.; Ferrari, A.; Gupta, A.A.; Lupo, P.J.; Butler, E.; Shipley, J.; Barr, F.G.; Hawkins, D.S. Rhabdomyosarcoma. Nat. Rev. Dis. Prim. 2019, 5. [Google Scholar] [CrossRef]
- Hahn, H.; Wojnowski, L.; Zimmer, A.M.; Hall, J.; Miller, G.; Zimmer, A. Rhabdomyosarcomas and radiation hypersensitivity in a mouse model of Gorlin syndrome. Nat. Med. 1998, 4, 619–622. [Google Scholar] [CrossRef]
- Mao, J.; Ligon, K.L.; Rakhlin, E.Y.; Thayer, S.P.; Bronson, R.T.; Rowitch, D.; McMahon, A.P. A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer Res. 2006, 66, 10171–10178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zibat, A.; Uhmann, A.; Nitzki, F.; Wijgerde, M.; Frommhold, A.; Heller, T.; Armstrong, V.; Wojnowski, L.; Quintanilla-Martinez, L.; Reifenberger, J.; et al. Time-point and dosage of gene inactivation determine the tumor spectrum in conditional Ptch knockouts. Carcinogenesis 2009, 30, 918–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabello, C.M.; Bair, W.B.; Lamore, S.D.; Ley, S.; Alexandra, S.; Azimian, S.; Wondrak, G.T. A mouse model of rhabdomyosarcoma originating from the adipocyte lineage. Cancer Cell 2012, 22, 536–546. [Google Scholar] [CrossRef]
- Van Mater, D.; Xu, E.; Reddy, A.; Añó, L.; Sachdeva, M.; Huang, W.; Williams, N.; Ma, Y.; Love, C.; Happ, L.; et al. Injury promotes sarcoma development in a genetically and temporally restricted manner. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Nanni, P.; Nicoletti, G.; De Giovanni, C.; Croci, S.; Astolfi, A.; Landuzzi, L.; Di Carlo, E.; Iezzi, M.; Musiani, P.; Lollini, P.L. Development of rhabdomyosarcoma in HER-2/neu transgenic p53 mutant mice. Cancer Res. 2003, 63, 2728–2732. [Google Scholar]
- Ianzano, M.L.; Croci, S.; Nicoletti, G.; Palladini, A.; Landuzzi, L.; Grosso, V.; Ranieri, D.; Dall’Ora, M.; Santeramo, I.; Urbini, M.; et al. Tumor suppressor genes promote rhabdomyosarcoma progression in p53 heterozygous, HER-2/neu transgenic mice. Oncotarget 2014, 5, 108–119. [Google Scholar] [CrossRef]
- Takayama, H.; Larochelle, W.J.; Sharp, R.; Otsuka, T.; Kriebel, P.; Anver, M.; Aaronson, S.A.; Merlino, G. Diverse tumorigenesis associated with aberrant development in mice overexpressing hepatocyte growth factor/scatter factor. Proc. Natl. Acad. Sci. USA 1997, 94, 701–706. [Google Scholar] [CrossRef] [Green Version]
- Sharp, R.; Recio, J.A.; Jhappan, C.; Otsuka, T.; Liu, S.; Yu, Y.; Liu, W.; Anver, M.; Navid, F.; Helman, L.J.; et al. Synergism between INK4a/ARF inactivation and aberrant HGF/SF signaling in rhabdomyosarcomagenesis. Nat. Med. 2002, 8, 1276–1280. [Google Scholar] [CrossRef]
- Comiskey, D.F.; Jacob, A.G.; Sanford, B.L.; Montes, M.; Goodwin, A.K.; Steiner, H.; Matsa, E.; Tapia-Santos, A.S.; Bebee, T.W.; Grieves, J.; et al. A novel mouse model of rhabdomyosarcoma underscores the dichotomy of MDM2-ALT1 function in vivo. Oncogene 2018, 37, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Fleischmann, A.; Jochum, W.; Eferl, R.; Witowsky, J.; Wagner, E.F. Rhabdomyosarcoma development in mice lacking Trp53 and Fos: Tumor suppression by the Fos protooncogene. Cancer Cell 2003, 4, 477–482. [Google Scholar] [CrossRef] [Green Version]
- Köbbert, C.; Möllmann, C.; Schäfers, M.; Hermann, S.; Baba, H.A.; Hoffmeier, A.; Breithardt, G.; Scheld, H.H.; Weissen-Plenz, G.; Sindermann, J.R. Transgenic model of cardiac rhabdomyosarcoma formation. J. Thorac. Cardiovasc. Surg. 2008, 136, 1178–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bompas, E.; Campion, L.; Italiano, A.; Le Cesne, A.; Chevreau, C.; Isambert, N.; Toulmonde, M.; Mir, O.; Ray-Coquard, I.; Piperno-Neumann, S.; et al. Outcome of 449 adult patients with rhabdomyosarcoma: An observational ambispective nationwide study. Cancer Med. 2018, 7, 4023–4035. [Google Scholar] [CrossRef] [PubMed]
- Galili, N.; Davis, R.J.; Fredericks, W.J.; Mukhopadhyay, S.; Rauscher, F.J.; Emanuel, B.S.; Rovera, G.; Barr, F.G. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 1993, 5, 230–235. [Google Scholar] [CrossRef]
- Charytonowicz, E.; Cordon-Cardo, C.; Matushansky, I.; Ziman, M. Alveolar rhabdomyosarcoma: Is the cell of origin a mesenchymal stem cell? Cancer Lett. 2009, 279, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Lagutina, I.; Conway, S.J.; Sublett, J.; Grosveld, G.C. Pax3-FKHR Knock-In Mice Show Developmental Aberrations but Do Not Develop Tumors. Mol. Cell. Biol. 2002, 22, 7204–7216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, C.; Hansen, M.S.; Coffin, C.M.; Capecchi, M.R. Pax3:Fkhr interferes with embryonic Pax3 and Pax7 function: Implications for alveolar rhabdomyosarcoma cell of origin. Genes Dev. 2004, 18, 2608–2613. [Google Scholar] [CrossRef] [Green Version]
- Keller, C.; Arenkiel, B.R.; Coffin, C.M.; El-Bardeesy, N.; DePinho, R.A.; Capecchi, M.R. Alveolar rhabdomyosarcomas in conditional Pax3:Fkhr mice: Cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev. 2004, 18, 2614–2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishijo, K.; Chen, Q.-R.; Zhang, L.; McCleish, A.T.; Rodriguez, A.; Cho, M.J.; Prajapati, S.I.; Gelfond, J.A.L.; Chisholm, G.B.; Michalek, J.E.; et al. Credentialing a preclinical mouse model of alveolar rhabdomyosarcoma. Cancer Res. 2009, 69, 2902–2911. [Google Scholar] [CrossRef] [Green Version]
- Abraham, J.; Nuñez-Álvarez, Y.; Hettmer, S.; Carrió, E.; Chen, H.I.H.; Nishijo, K.; Huang, E.T.; Prajapati, S.I.; Walker, R.L.; Davis, S.; et al. Lineage of origin in rhabdomyosarcoma informs pharmacological response. Genes Dev. 2014, 28, 1578–1591. [Google Scholar] [CrossRef] [Green Version]
- Oristian, K.M.; Crose, L.E.S.; Kuprasertkul, N.; Bentley, R.C.; Lin, Y.T.; Williams, N.; Kirsch, D.G.; Linardic, C.M. Loss of MST/Hippo signaling in a genetically engineered mouse model of fusion-positive rhabdomyosarcoma accelerates tumorigenesis. Cancer Res. 2018, 78, 5513–5520. [Google Scholar] [CrossRef] [Green Version]
- Agaram, N.P.; Chen, C.-L.; Zhang, L.; LaQuaglia, M.P.; Wexler, L.; Antonescu, C.R. Recurrent MYOD1 mutations in pediatric and adult sclerosing and spindle cell rhabdomyosarcomas: Evidence for a common pathogenesis. Genes. Chromosomes Cancer 2014, 53, 779–787. [Google Scholar] [CrossRef] [Green Version]
- Schleicher, S.; Grote, S.; Malenke, E.; Chan, K.C.H.; Schaller, M.; Fehrenbacher, B.; Riester, R.; Kluba, T.; Frauenfeld, L.; Boesmueller, H.; et al. Establishment and Characterization of a Sclerosing Spindle Cell Rhabdomyosarcoma Cell Line with a Complex Genomic Profile. Cells 2020, 9, 2668. [Google Scholar] [CrossRef] [PubMed]
- Overholtzer, M.; Rao, P.H.; Favis, R.; Lu, X.-Y.; Elowitz, M.B.; Barany, F.; Ladanyi, M.; Gorlick, R.; Levine, A.J. The presence of p53 mutations in human osteosarcomas correlates with high levels of genomic instability. Proc. Natl. Acad. Sci. USA 2003, 100, 11547–11552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, M.F.; Koufos, A.; Gallie, B.L.; Phillips, R.A.; Fodstad, O.; Brøgger, A.; Gedde-Dahl, T.; Cavenee, W.K. Osteosarcoma and retinoblastoma: A shared chromosomal mechanism revealing recessive predisposition. Proc. Natl. Acad. Sci. USA 1985, 82, 6216–6220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wadayama, B.; Toguchida, J.; Shimizu, T.; Ishizaki, K.; Sasaki, M.S.; Kotoura, Y.; Yamamuro, T. Mutation spectrum of the retinoblastoma gene in osteosarcomas. Cancer Res. 1994, 54, 3042–3048. [Google Scholar]
- Jacks, T.; Fazeli, A.; Schmitt, E.M.; Bronson, R.T.; Goodell, M.A.; Weinberg, R.A. Effects of an Rb mutation in the mouse. Nature 1992, 359, 295–300. [Google Scholar] [CrossRef]
- Vooijs, M.; Berns, A. Developmental defects and tumor predisposition in Rb mutant mice. Oncogene 1999, 18, 5293–5303. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.P.; Pandey, M.K.; Jin, F.; Raymond, A.K.; Akiyama, H.; Lozano, G. Targeted mutation of p53 and Rb in mesenchymal cells of the limb bud produces sarcomas in mice. Carcinogenesis 2009, 30, 1789–1795. [Google Scholar] [CrossRef] [Green Version]
- Calo, E.; Quintero-Estades, J.A.; Danielian, P.S.; Nedelcu, S.; Berman, S.D.; Lees, J.A. Rb regulates fate choice and lineage commitment in vivo. Nature 2010, 466, 1110–1114. [Google Scholar] [CrossRef] [Green Version]
- Berman, S.D.; Calo, E.; Landman, A.S.; Danielian, P.S.; Miller, E.S.; West, J.C.; Fonhoue, B.D.; Caron, A.; Bronson, R.; Bouxsein, M.L.; et al. Metastatic osteosarcoma induced by inactivation of Rb and p53 in the osteoblast lineage. Proc. Natl. Acad. Sci. USA 2008, 105, 11851–11856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walkley, C.R.; Qudsi, R.; Sankaran, V.G.; Perry, J.A.; Gostissa, M.; Roth, S.I.; Rodda, S.J.; Snay, E.; Dunning, P.; Fahey, F.H.; et al. Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes Dev. 2008, 22, 1662–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lengner, C.J.; Steinman, H.A.; Gagnon, J.; Smith, T.W.; Henderson, J.E.; Kream, B.E.; Stein, G.S.; Lian, J.B.; Jones, S.N. Osteoblast differentiation and skeletal development are regulated by Mdm2-p53 signaling. J. Cell Biol. 2006, 172, 909–921. [Google Scholar] [CrossRef]
- Molyneux, S.D.; Di Grappa, M.A.; Beristain, A.G.; McKee, T.D.; Wai, D.H.; Paderova, J.; Kashyap, M.; Hu, P.; Maiuri, T.; Narala, S.R.; et al. Prkar1a is an osteosarcoma tumor suppressor that defines a molecular subclass in mice. J. Clin. Investig. 2010, 120, 3310–3325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutsaers, A.J.; Ng, A.J.M.; Baker, E.K.; Russell, M.R.; Chalk, A.M.; Wall, M.; Liddicoat, B.J.J.; Ho, P.W.M.; Slavin, J.L.; Goradia, A.; et al. Modeling distinct osteosarcoma subtypes in vivo using Cre:lox and lineage-restricted transgenic shRNA. Bone 2013, 55, 166–178. [Google Scholar] [CrossRef]
- Guijarro, M.V.; Ghivizzani, C.S.; Gibbs, P.C. Animal models in osteosarcoma. Front. Genet. 2014, 5, 475. [Google Scholar] [CrossRef] [Green Version]
- Uluçkan, Ö.; Segaliny, A.; Botter, S.; Santiago, J.M.; Mutsaers, A.J. Preclinical mouse models of osteosarcoma. Bonekey Rep. 2015, 4, 670. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; De Álava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing sarcoma. Nat. Rev. Dis. Prim. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Sheffield, N.C.; Pierron, G.; Klughammer, J.; Datlinger, P.; Schönegger, A.; Schuster, M.; Hadler, J.; Surdez, D.; Guillemot, D.; Lapouble, E.; et al. DNA methylation heterogeneity defines a disease spectrum in Ewing sarcoma. Nat. Med. 2017, 23, 386–395. [Google Scholar] [CrossRef]
- Gangwal, K.; Sankar, S.; Hollenhorst, P.C.; Kinsey, M.; Haroldsen, S.C.; Shah, A.A.; Boucher, K.M.; Watkins, W.S.; Jorde, L.B.; Graves, B.J.; et al. Microsatellites as EWS/FLI response elements in Ewing’s sarcoma. Proc. Natl. Acad. Sci. USA 2008, 105, 10149–10154. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Yamazaki, Y.; Kanno, Y.; Igarashi, K.; Aisaki, K.I.; Kanno, J.; Nakamura, T. Ewing’s sarcoma precursors are highly enriched in embryonic osteochondrogenic progenitors. J. Clin. Investig. 2014, 124, 3061–3074. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, R.; Tanaka, M.; Tsutsumi, S.; Aburatani, H.; Yamazaki, Y.; Homme, M.; Kitagawa, Y.; Nakamura, T. EWS-FLI1 regulates a transcriptional program in cooperation with Foxq1 in mouse Ewing sarcoma. Cancer Sci. 2018, 109, 2907–2918. [Google Scholar] [CrossRef] [Green Version]
- Haldar, M.; Hancock, J.D.; Coffin, C.M.; Lessnick, S.L.; Capecchi, M.R. A Conditional Mouse Model of Synovial Sarcoma: Insights into a Myogenic Origin. Cancer Cell 2007, 11, 375–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nacev, B.A.; Jones, K.B.; Intlekofer, A.M.; Yu, J.S.E.; Allis, C.D.; Tap, W.D.; Ladanyi, M.; Nielsen, T.O. The epigenomics of sarcoma. Nat. Rev. Cancer 2020, 1, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.R.; Meltzer, P.S. Modeling synovial sarcoma: Timing is everything. Cancer Cell 2007, 11, 305–307. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.B.; Barrott, J.J.; Xie, M.; Haldar, M.; Jin, H.; Zhu, J.-F.; Monument, M.J.; Mosbruger, T.L.; Langer, E.M.; Randall, R.L.; et al. The impact of chromosomal translocation locus and fusion oncogene coding sequence in synovial sarcomagenesis. Oncogene 2016, 35, 5021–5032. [Google Scholar] [CrossRef] [Green Version]
- Barrott, J.J.; Kafchinski, L.A.; Jin, H.; Potter, J.W.; Kannan, S.D.; Kennedy, R.; Mosbruger, T.; Wang, W.-L.; Tsai, J.-W.; Araujo, D.M.; et al. Modeling synovial sarcoma metastasis in the mouse: PI3’-lipid signaling and inflammation. J. Exp. Med. 2016, 213, 2989–3005. [Google Scholar] [CrossRef] [PubMed]
- Barrott, J.J.; Illum, B.E.; Jin, H.; Zhu, J.-F.; Mosbruger, T.; Monument, M.J.; Smith-Fry, K.; Cable, M.G.; Wang, Y.; Grossmann, A.H.; et al. β-catenin stabilization enhances SS18-SSX2-driven synovial sarcomagenesis and blocks the mesenchymal to epithelial transition. Oncotarget 2015, 6, 22758–22766. [Google Scholar] [CrossRef] [Green Version]
- Rubin, J.B.; Gutmann, D.H. Neurofibromatosis type 1—A model for nervous system tumour formation? Nat. Rev. Cancer 2005, 5, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Philpott, C.; Tovell, H.; Frayling, I.M.; Cooper, D.N.; Upadhyaya, M. The NF1 somatic mutational landscape in sporadic human cancers. Hum. Genom. 2017, 11, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Dodd, R.D.; Mito, J.K.; Eward, W.C.; Chitalia, R.; Sachdeva, M.; Ma, Y.; Barretina, J.; Dodd, L.; Kirsch, D.G. NF1 deletion generates multiple subtypes of soft-tissue sarcoma that respond to MEK inhibition. Mol. Cancer Ther. 2013, 12, 1906–1917. [Google Scholar] [CrossRef] [Green Version]
- Sultan, I.; Casanova, M.; Al-Jumaily, U.; Meazza, C.; Rodriguez-Galindo, C.; Ferrari, A. Soft tissue sarcomas in the first year of life. Eur. J. Cancer 2010, 46, 2449–2456. [Google Scholar] [CrossRef] [PubMed]
- Wai, D.H.; Knezevich, S.R.; Lucas, T.; Jansen, B.; Kay, R.J.; Sorensen, P.H.B. The ETV6-NTRK3 gene fusion encodes a chimeric protein tyrosine kinase that transforms NIH3T3 cells. Oncogene 2000, 19, 906–915. [Google Scholar] [CrossRef] [Green Version]
- Tannenbaum-Dvir, S.; Glade Bender, J.L.; Church, A.J.; Janeway, K.A.; Harris, M.H.; Mansukhani, M.M.; Nagy, P.L.; Andrews, S.J.; Murty, V.V.; Kadenhe-Chiweshe, A.; et al. Characterization of a novel fusion gene EML4-NTRK3 in a case of recurrent congenital fibrosarcoma. Cold Spring Harb. Mol. Case Stud. 2015, 1, a000471. [Google Scholar] [CrossRef] [Green Version]
- Roberts, C.W.; Galusha, S.A.; McMenamin, M.E.; Fletcher, C.D.; Orkin, S.H. Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc. Natl. Acad. Sci. USA 2000, 97, 13796–13800. [Google Scholar] [CrossRef] [Green Version]
- Guidi, C.J.; Sands, A.T.; Zambrowicz, B.P.; Turner, T.K.; Demers, D.A.; Webster, W.; Smith, T.W.; Imbalzano, A.N.; Jones, S.N. Disruption of Ini1 leads to peri-implantation lethality and tumorigenesis in mice. Mol. Cell. Biol. 2001, 21, 3598–3603. [Google Scholar] [CrossRef] [Green Version]
- Klochendler-Yeivin, A.; Fiette, L.; Barra, J.; Muchardt, C.; Babinet, C.; Yaniv, M. The murine SNF5/INI1 chromatin remodeling factor is essential for embryonic development and tumor suppression. EMBO Rep. 2000, 1, 500–506. [Google Scholar] [CrossRef] [Green Version]
- DelBove, J.; Kuwahara, Y.; Mora-Blanco, E.L.; Godfrey, V.; Funkhouser, W.K.; Fletcher, C.D.M.; Van Dyke, T.; Roberts, C.W.M.; Weissman, B.E. Inactivation of SNF5 cooperates with p53 loss to accelerate tumor formation in Snf5(+/-);p53(+/-) mice. Mol. Carcinog. 2009, 48, 1139–1148. [Google Scholar] [CrossRef] [Green Version]
- Isakoff, M.S.; Sansam, C.G.; Tamayo, P.; Subramanian, A.; Evans, J.A.; Fillmore, C.M.; Wang, X.; Biegel, J.A.; Pomeroy, S.L.; Mesirov, J.P.; et al. Inactivation of the Snf5 tumor suppressor stimulates cell cycle progression and cooperates with p53 loss in oncogenic transformation. Proc. Natl. Acad. Sci. USA 2005, 102, 17745–17750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, C.W.M.; Leroux, M.M.; Fleming, M.D.; Orkin, S.H. Highly penetrant, rapid tumorigenesis through conditional inversion of the tumor suppressor gene Snf5. Cancer Cell 2002, 2, 415–425. [Google Scholar] [CrossRef] [Green Version]
- Stenman, G.; Kindblom, L.G.; Angervall, L. Reciprocal translocation t(12;22)(q13;q13) in clear-cell sarcoma of tendons and aponeuroses. Genes. Chromosomes Cancer 1992, 4, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Straessler, K.M.; Jones, K.B.; Hu, H.; Jin, H.; van de Rijn, M.; Capecchi, M.R. Modeling clear cell sarcomagenesis in the mouse: Cell of origin differentiation state impacts tumor characteristics. Cancer Cell 2013, 23, 215–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladanyi, M.; Lui, M.Y.; Antonescu, C.R.; Krause-Boehm, A.; Meindl, A.; Argani, P.; Healey, J.H.; Ueda, T.; Yoshikawa, H.; Meloni-Ehrig, A.; et al. The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene 2001, 20, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, M.L.; Jin, H.; Straessler, K.; Smith-Fry, K.; Zhu, J.-F.; Monument, M.J.; Grossmann, A.; Randall, R.L.; Capecchi, M.R.; Jones, K.B. Modeling alveolar soft part sarcomagenesis in the mouse: A role for lactate in the tumor microenvironment. Cancer Cell 2014, 26, 851–862. [Google Scholar] [CrossRef] [Green Version]
- Gooskens, S.L.; Furtwängler, R.; Spreafico, F.; van Tinteren, H.; de Kraker, J.; Vujanic, G.M.; Leuschner, I.; Coulomb-L’Herminé, A.; Godzinski, J.; Schleiermacher, G.; et al. Treatment and outcome of patients with relapsed clear cell sarcoma of the kidney: A combined SIOP and AIEOP study. Br. J. Cancer 2014, 111, 227–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, A.; Kumar, V.; Zorman, B.; Fang, E.; Haines, K.M.; Doddapaneni, H.; Hampton, O.A.; White, S.; Bavle, A.A.; Patel, N.R.; et al. Recurrent internal tandem duplications of BCOR in clear cell sarcoma of the kidney. Nat. Commun. 2015, 6, 8891. [Google Scholar] [CrossRef] [PubMed]
- Pierron, G.; Tirode, F.; Lucchesi, C.; Reynaud, S.; Ballet, S.; Cohen-Gogo, S.; Perrin, V.; Coindre, J.M.; Delattre, O. A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat. Genet. 2012, 44, 461–466. [Google Scholar] [CrossRef] [Green Version]
- Kao, Y.-C.; Sung, Y.-S.; Zhang, L.; Huang, S.-C.; Argani, P.; Chung, C.T.; Graf, N.S.; Wright, D.C.; Kellie, S.J.; Agaram, N.P.; et al. Recurrent BCOR Internal Tandem Duplication and YWHAE-NUTM2B Fusions in Soft Tissue Undifferentiated Round Cell Sarcoma of Infancy. Am. J. Surg. Pathol. 2016, 40, 1009–1020. [Google Scholar] [CrossRef] [Green Version]
- Kao, Y.-C.; Owosho, A.A.; Sung, Y.-S.; Zhang, L.; Fujisawa, Y.; Lee, J.-C.; Wexler, L.; Argani, P.; Swanson, D.; Dickson, B.C.; et al. BCOR-CCNB3 Fusion Positive Sarcomas: A Clinicopathologic and Molecular Analysis of 36 Cases With Comparison to Morphologic Spectrum and Clinical Behavior of Other Round Cell Sarcomas. Am. J. Surg. Pathol. 2018, 42, 604–615. [Google Scholar] [CrossRef]
- Specht, K.; Sung, Y.-S.; Zhang, L.; Richter, G.H.S.; Fletcher, C.D.; Antonescu, C.R. Distinct transcriptional signature and immunoprofile of CIC-DUX4 fusion-positive round cell tumors compared to EWSR1-rearranged Ewing sarcomas: Further evidence toward distinct pathologic entities. Genes. Chromosomes Cancer 2014, 53, 622–633. [Google Scholar] [CrossRef] [Green Version]
- Kawamura-Saito, M.; Yamazaki, Y.; Kaneko, K.; Kawaguchi, N.; Kanda, H.; Mukai, H.; Gotoh, T.; Motoi, T.; Fukayama, M.; Aburatani, H.; et al. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum. Mol. Genet. 2006, 15, 2125–2137. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Owosho, A.A.; Zhang, L.; Chen, S.; Deniz, K.; Huryn, J.M.; Kao, Y.C.; Huang, S.C.; Singer, S.; Tap, W.; et al. Sarcomas with CIC-rearrangements Are a Distinct Pathologic Entity with Aggressive Outcome. Am. J. Surg. Pathol. 2017, 41, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Gerald, W.L.; Miller, H.K.; Battifora, H.; Miettinen, M.; Silva, E.G.; Rosai, J. Intra-abdominal desmoplastic small round-cell tumor. Report of 19 cases of a distinctive type of high-grade polyphenotypic malignancy affecting young individuals. Am. J. Surg. Pathol. 1991, 15, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, J.R.; Tryka, A.F.; Lewis, J.M. A novel reciprocal chromosome translocation t(11;22)(p13;q12) in an intraabdominal desmoplastic small round-cell tumor. Am. J. Surg. Pathol. 1992, 16, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Frezza, A.M.; Cesari, M.; Baumhoer, D.; Biau, D.; Bielack, S.; Campanacci, D.A.; Casanova, J.; Esler, C.; Ferrari, S.; Funovics, P.T.; et al. Mesenchymal chondrosarcoma: Prognostic factors and outcome in 113 patients. A European Musculoskeletal Oncology Society study. Eur. J. Cancer 2015, 51, 374–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Motoi, T.; Khanin, R.; Olshen, A.; Mertens, F.; Bridge, J.; Dal Cin, P.; Antonescu, C.R.; Singer, S.; Hameed, M.; et al. Identification of a novel, recurrent HEY1-NCOA2 fusion in mesenchymal chondrosarcoma based on a genome-wide screen of exon-level expression data. Genes. Chromosomes Cancer 2012, 51, 127–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.-D.; Wu, T.; Wang, J.-Q.; Zhang, W.-D.; He, X.-L.; Bao, G.-Q.; Li, Y.; Gong, L.; Wang, Q. Primary inflammatory myofibroblastic tumor of the breast with rapid recurrence and metastasis: A case report. Oncol. Lett. 2013, 5, 97–100. [Google Scholar] [CrossRef]
- Mariño-Enríquez, A.; Dal Cin, P. ALK as a paradigm of oncogenic promiscuity: Different mechanisms of activation and different fusion partners drive tumors of different lineages. Cancer Genet. 2013, 206, 357–373. [Google Scholar] [CrossRef]
- Hayes, M.N.; Langenau, D.M. Discovering novel oncogenic pathways and new therapies using zebrafish models of sarcoma. Methods Cell Biol. 2017, 138, 525–561. [Google Scholar] [CrossRef]
- Parant, J.; Fogley, R.; Zon, L. Cancer and Zebrafish; Langenau, D.M., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2016; Volume 916, ISBN 978-3-319-30652-0. [Google Scholar]
- Casey, M.J.; Stewart, R.A. Pediatric Cancer Models in Zebrafish. Trends Cancer 2020, 6, 407–418. [Google Scholar] [CrossRef] [Green Version]
- Langenau, D.M.; Keefe, M.D.; Storer, N.Y.; Guyon, J.R.; Kutok, J.L.; Le, X.; Goessling, W.; Neuberg, D.S.; Kunkel, L.M.; Zon, L.I. Effects of RAS on the genesis of embryonal rhabdomyosarcoma. Genes Dev. 2007, 21, 1382–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storer, N.Y.; White, R.M.; Uong, A.; Price, E.; Nielsen, G.P.; Langenau, D.M.; Zon, L.I. Zebrafish rhabdomyosarcoma reflects the developmental stage of oncogene expression during myogenesis. Development 2013, 140, 3040–3050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendall, G.C.; Watson, S.; Xu, L.; Lavigne, C.A.; Murchison, W.; Rakheja, D.; Skapek, S.X.; Tirode, F.; Delattre, O.; Amatruda, J.F. PAX3-FOXO1 transgenic zebrafish models identify HES3 as a mediator of rhabdomyosarcoma tumorigenesis. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Tenente, I.M.; Hayes, M.N.; Ignatius, M.S.; McCarthy, K.; Yohe, M.; Sindiri, S.; Gryder, B.; Oliveira, M.L.; Ramakrishnan, A.; Tang, Q.; et al. Myogenic regulatory transcription factors regulate growth in rhabdomyosarcoma. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galindo, R.L.; Allport, J.A.; Olson, E.N. A Drosophila model of the rhabdomyosarcoma initiator PAX7-FKHR. Proc. Natl. Acad. Sci. USA 2006, 103, 13439–13444. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Brunson, D.C.; Tang, Q.; Do, D.; Iftimia, N.A.; Moore, J.C.; Hayes, M.N.; Welker, A.M.; Garcia, E.G.; Dubash, T.D.; et al. Visualizing Engrafted Human Cancer and Therapy Responses in Immunodeficient Zebrafish. Cell 2019, 177, 1903–1914.e14. [Google Scholar] [CrossRef]
- Selvarajah, G.T.; Kirpensteijn, J.; van Wolferen, M.E.; Rao, N.A.S.; Fieten, H.; Mol, J.A. Gene expression profiling of canine osteosarcoma reveals genes associated with short and long survival times. Mol. Cancer 2009, 8, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, F.; Fuchs, B.; Kaser-Hotz, B. Comparative biology of human and canine osteosarcoma. Anticancer Res. 2007, 27, 155–164. [Google Scholar]
- van Leeuwen, I.S.; Cornelisse, C.J.; Misdorp, W.; Goedegebuure, S.A.; Kirpensteijn, J.; Rutteman, G.R. P53 gene mutations in osteosarcomas in the dog. Cancer Lett. 1997, 111, 173–178. [Google Scholar] [CrossRef]
- Levine, R.A.; Forest, T.; Smith, C. Tumor suppressor PTEN is mutated in canine osteosarcoma cell lines and tumors. Vet. Pathol. 2002, 39, 372–378. [Google Scholar] [CrossRef]
- Paoloni, M.; Davis, S.; Lana, S.; Withrow, S.; Sangiorgi, L.; Picci, P.; Hewitt, S.; Triche, T.; Meltzer, P.; Khanna, C. Canine tumor cross-species genomics uncovers targets linked to osteosarcoma progression. BMC Genomics 2009, 10, 625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paoloni, M.; Khanna, C. Translation of new cancer treatments from pet dogs to humans. Nat. Rev. Cancer 2008, 8, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Chester, C.; Melero, I.; Kohrt, H. Defining the optimal murine models to investigate immune checkpoint blockers and their combination with other immunotherapies. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Lyu, Y.; Yang, Y.G.; Hu, Z. Humanized Rodent Models for Cancer Research. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Dorado Garcia, H.; Scheer, M.; Henssen, A.G. Current and Future Treatment Strategies for Rhabdomyosarcoma. Front. Oncol. 2019, 9, 1–18. [Google Scholar] [CrossRef]
- Atashzar, M.R.; Baharlou, R.; Karami, J.; Abdollahi, H.; Rezaei, R.; Pourramezan, F.; Zoljalali Moghaddam, S.H. Cancer stem cells: A review from origin to therapeutic implications. J. Cell. Physiol. 2020, 235, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Joseph, N.M.; Mosher, J.T.; Buchstaller, J.; Snider, P.; McKeever, P.E.; Lim, M.; Conway, S.J.; Parada, L.F.; Zhu, Y.; Morrison, S.J. The loss of Nf1 transiently promotes self-renewal but not tumorigenesis by neural crest stem cells. Cancer Cell 2008, 13, 129–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langenau, D.M.; Sweet-Cordero, A.; Wechsler-Reya, R.; Dyer, M.A. Preclinical Models Provide Scientific Justification and Translational Relevance for Moving Novel Therapeutics into Clinical Trials for Pediatric Cancer. Cancer Res. 2015, 75, 5176–5186. [Google Scholar] [CrossRef] [Green Version]
- Houghton, P.J.; Morton, C.L.; Gorlick, R.; Lock, R.B.; Carol, H.; Reynolds, C.P.; Kang, M.H.; Maris, J.M.; Keir, S.T.; Kolb, E.A.; et al. Stage 2 combination testing of rapamycin with cytotoxic agents by the pediatric preclinical testing program. Mol. Cancer Ther. 2010, 9, 101–112. [Google Scholar] [CrossRef] [Green Version]
- DuBois, S.G.; Corson, L.B.; Stegmaier, K.; Janeway, K.A. Ushering in the next generation of precision trials for pediatric cancer. Science 2019, 363, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Russano, M.; Napolitano, A.; Ribelli, G.; Iuliani, M.; Simonetti, S.; Citarella, F.; Pantano, F.; Dell’Aquila, E.; Anesi, C.; Silvestris, N.; et al. Liquid biopsy and tumor heterogeneity in metastatic solid tumors: The potentiality of blood samples. J. Exp. Clin. Cancer Res. 2020, 39, 95. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.; Yin, H.; Maris, J.M.; Kolb, E.A.; Gorlick, R.; Reynolds, C.P.; Kang, M.H.; Keir, S.T.; Kurmasheva, R.T.; Dvorchik, I.; et al. Evaluation of alternative in vivo drug screening methodology: A single mouse analysis. Cancer Res. 2016, 76, 5798–5809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghilu, S.; Li, Q.; Fontaine, S.D.; Santi, D.V.; Kurmasheva, R.T.; Zheng, S.; Houghton, P.J. Prospective use of the single-mouse experimental design for the evaluation of PLX038A. Cancer Chemother. Pharmacol. 2020, 85, 251–263. [Google Scholar] [CrossRef]
- Stebbing, J.; Paz, K.; Schwartz, G.K.; Wexler, L.H.; Maki, R.; Pollock, R.E.; Morris, R.; Cohen, R.; Shankar, A.; Blackman, G.; et al. Patient-derived xenografts for individualized care in advanced sarcoma. Cancer 2014, 120, 2006–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimmervoll, B.V.; Boulos, N.; Bianski, B.; Dapper, J.; DeCuypere, M.; Shelat, A.; Terranova, S.; Terhune, H.E.; Gajjar, A.; Patel, Y.T.; et al. Establishing a Preclinical Multidisciplinary Board for Brain Tumors. Clin. Cancer Res. 2018, 24, 1654–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merino, D.; Weber, T.S.; Serrano, A.; Vaillant, F.; Liu, K.; Pal, B.; Di Stefano, L.; Schreuder, J.; Lin, D.; Chen, Y.; et al. Barcoding reveals complex clonal behavior in patient-derived xenografts of metastatic triple negative breast cancer. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Seth, S.; Li, C.Y.; Ho, I.L.; Corti, D.; Loponte, S.; Sapio, L.; Del Poggetto, E.; Yen, E.Y.; Robinson, F.S.; Peoples, M.; et al. Pre-existing Functional Heterogeneity of Tumorigenic Compartment as the Origin of Chemoresistance in Pancreatic Tumors. Cell Rep. 2019, 26, 1518–1532.e9. [Google Scholar] [CrossRef] [Green Version]
- Echeverria, G.V.; Powell, E.; Seth, S.; Ge, Z.; Carugo, A.; Bristow, C.; Peoples, M.; Robinson, F.; Qiu, H.; Shao, J.; et al. High-resolution clonal mapping of multi-organ metastasis in triple negative breast cancer. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Painter, C.A.; Jain, E.; Tomson, B.N.; Dunphy, M.; Stoddard, R.E.; Thomas, B.S.; Damon, A.L.; Shah, S.; Kim, D.; Gómez Tejeda Zañudo, J.; et al. The Angiosarcoma Project: Enabling genomic and clinical discoveries in a rare cancer through patient-partnered research. Nat. Med. 2020, 26, 181–187. [Google Scholar] [CrossRef]
- Choi, S.Y.C.; Lin, D.; Gout, P.W.; Collins, C.C.; Xu, Y.; Wang, Y. Lessons from patient-derived xenografts for better in vitro modeling of human cancer. Adv. Drug Deliv. Rev. 2014, 79, 222–237. [Google Scholar] [CrossRef] [Green Version]
- Sesillo, F.B.; Sacco, A. Tumorsphere derivation and treatment from primary tumor cells isolated from mouse rhabdomyosarcomas. J. Vis. Exp. 2019, 2019. [Google Scholar] [CrossRef]
- Mito, J.K.; Riedel, R.F.; Dodd, L.; Lahat, G.; Lazar, A.J.; Dodd, R.D.; Stangenberg, L.; Eward, W.C.; Hornicek, F.J.; Yoon, S.S.; et al. Cross Species Genomic Analysis Identifies a Mouse Model as Undifferentiated Pleomorphic Sarcoma/Malignant Fibrous Histiocytoma. PLoS ONE 2009, 4, e8075. [Google Scholar] [CrossRef]
- Needhamsen, M.; Ewing, E.; Lund, H.; Gomez-Cabrero, D.; Harris, R.A.; Kular, L.; Jagodic, M. Usability of human Infinium MethylationEPIC BeadChip for mouse DNA methylation studies. BMC Bioinform. 2017, 18, 486. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.A.; Zhang, S.; Sengupta, S.; Ma, H.; Bell, G.W.; Horton, B.; Sharma, B.; George, R.E.; Spranger, S.; Jaenisch, R. Formation of Human Neuroblastoma in Mouse-Human Neural Crest Chimeras. Cell Stem Cell 2020, 26, 579–592.e6. [Google Scholar] [CrossRef] [PubMed]
- Karim, M.E.; Tha, K.K.; Othman, I.; Uddin, M.B.; Chowdhury, E.H. Therapeutic potency of nanoformulations of siRNAs and shRNAs in animal models of cancers. Pharmaceutics 2018, 10, 65. [Google Scholar] [CrossRef] [Green Version]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef] [Green Version]
- Bolden, J.E.; Tasdemir, N.; Dow, L.E.; van Es, J.H.; Wilkinson, J.E.; Zhao, Z.; Clevers, H.; Lowe, S.W. Inducible in vivo silencing of Brd4 identifies potential toxicities of sustained BET protein inhibition. Cell Rep 2014, 8, 1919–1929. [Google Scholar] [CrossRef] [Green Version]
- Maresch, R.; Mueller, S.; Veltkamp, C.; Öllinger, R.; Friedrich, M.; Heid, I.; Steiger, K.; Weber, J.; Engleitner, T.; Barenboim, M.; et al. Multiplexed pancreatic genome engineering and cancer induction by transfection-based CRISPR/Cas9 delivery in mice. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Sanjana, N.E.; Zheng, K.; Shalem, O.; Lee, K.; Shi, X.; Scott, D.A.; Song, J.; Pan, J.Q.; Weissleder, R.; et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 2015, 160, 1246–1260. [Google Scholar] [CrossRef] [Green Version]
- Khanna, C.; Hunter, K. Modeling metastasis in vivo. Carcinogenesis 2005, 26, 513–523. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Cuadrado, L.; Tracey, N.; Ma, R.; Qian, B.; Brunton, V.G. Mouse models of metastasis: Progress and prospects. Dis. Model. Mech. 2017, 10, 1061–1074. [Google Scholar] [CrossRef] [Green Version]
- Rwandamuriye, F.X.; Weston, B.J.; Johns, T.G.; Lesterhuis, W.J.; Zemek, R.M. A mouse model of incompletely resected soft tissue sarcoma for testing (Neo)adjuvant therapies. J. Vis. Exp. 2020, 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lagares-Tena, L.; García-Monclús, S.; López-Alemany, R.; Almacellas-Rabaiget, O.; Huertas-Martínez, J.; Sáinz-Jaspeado, M.; Mateo-Lozano, S.; Rodríguez-Galindo, C.; Rello-Varona, S.; Herrero-Martín, D.; et al. Caveolin-1 promotes Ewing sarcoma metastasis regulating MMP-9 expression through MAPK/ERK pathway. Oncotarget 2016, 7, 56889–56903. [Google Scholar] [CrossRef] [Green Version]
- Urla, C.; Armeanu-Ebinger, S.; Fuchs, J.; Seitz, G. Successful in vivo tumor visualization using fluorescence laparoscopy in a mouse model of disseminated alveolar rhabdomyosarcoma. Surg. Endosc. 2015, 29, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Duchi, S.; Ramos-Romero, S.; Dozza, B.; Guerra-Rebollo, M.; Cattini, L.; Ballestri, M.; Dambruoso, P.; Guerrini, A.; Sotgiu, G.; Varchi, G.; et al. Development of near-infrared photoactivable phthalocyanine-loaded nanoparticles to kill tumor cells: An improved tool for photodynamic therapy of solid cancers. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 1885–1897. [Google Scholar] [CrossRef] [PubMed]
- Martano, M.; Morello, E.; Avnet, S.; Costa, F.; Sammartano, F.; Kusuzaki, K.; Baldini, N. Photodynamic Surgery for Feline Injection-Site Sarcoma. Biomed Res. Int. 2019, 2019, 8275935. [Google Scholar] [CrossRef] [PubMed]
- Kusuzaki, K.; Takai, T.; Yoshimura, H.; Inoue, K.; Takai, S.; Baldini, N. Clinical trial of radiotherapy after intravenous injection of acridine orange for patients with cancer. Anticancer Res. 2018, 38, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Alzeibak, R.; Mishchenko, T.A.; Shilyagina, N.Y.; Balalaeva, I.V.; Vedunova, M.V.; Krysko, D.V. Targeting immunogenic cancer cell death by photodynamic therapy: Past, present and future. J. Immunother. Cancer 2021, 9, e001926. [Google Scholar] [CrossRef]
Figure 1.
The diverse landscape of pediatric sarcoma. (A) Extraction from the current WHO classification of soft-tissue and bone tumors (5th edition, 2020) with focus on pediatric sarcoma [8]. (B,C) Molecular classification by whole genome DNA methylation data, here depicted for 18 sarcoma entities relevant for childhood and adolescence. Data shown as hierarchical clustering analysis, (B) and t-distributed stochastic neighbor embedding (t-SNE), (C) data adapted from Kölsche et al., 2021 [10].
Figure 1.
The diverse landscape of pediatric sarcoma. (A) Extraction from the current WHO classification of soft-tissue and bone tumors (5th edition, 2020) with focus on pediatric sarcoma [8]. (B,C) Molecular classification by whole genome DNA methylation data, here depicted for 18 sarcoma entities relevant for childhood and adolescence. Data shown as hierarchical clustering analysis, (B) and t-distributed stochastic neighbor embedding (t-SNE), (C) data adapted from Kölsche et al., 2021 [10].

Figure 2.
Different approaches to model sarcoma in vivo. Relative sizes of mouse pictograms resemble approximate utilization of modeling approaches in current sarcoma research. Dashed line illustrates that environmentally-induced models (EIMMs) are typically not particularly relevant for childhood sarcoma.
Figure 2.
Different approaches to model sarcoma in vivo. Relative sizes of mouse pictograms resemble approximate utilization of modeling approaches in current sarcoma research. Dashed line illustrates that environmentally-induced models (EIMMs) are typically not particularly relevant for childhood sarcoma.

Figure 3.
Pros and cons of different in vivo modeling approaches. While cell-line-derived xenograft models (CDXs) and patient-derived xenograft models (PDXs) are both engraftment models, environmentally-induced models (EIMMs) and genetically-engineered mouse models (GEMMs) can be utilized to establish syngeneic engraftment models (SAMs), enabling scalability for these models, too. Pros and cons of different engraftment sites depicted at the bottom apply to all of the engraftment models, regardless of origin.
Figure 3.
Pros and cons of different in vivo modeling approaches. While cell-line-derived xenograft models (CDXs) and patient-derived xenograft models (PDXs) are both engraftment models, environmentally-induced models (EIMMs) and genetically-engineered mouse models (GEMMs) can be utilized to establish syngeneic engraftment models (SAMs), enabling scalability for these models, too. Pros and cons of different engraftment sites depicted at the bottom apply to all of the engraftment models, regardless of origin.

Figure 4.
Selection of PDX-repositories relevant for pediatric sarcoma.

Figure 5.
Utilization of sarcoma mouse models. Schematic overview of different research questions in sarcoma research. Color codes represent the generally most suitable modeling approach for the respective research field ((green) = GEMM, (orange) = PDX, (red) = CDX).
Figure 5.
Utilization of sarcoma mouse models. Schematic overview of different research questions in sarcoma research. Color codes represent the generally most suitable modeling approach for the respective research field ((green) = GEMM, (orange) = PDX, (red) = CDX).

Figure 6.
Complementary in vivo modeling approaches. The depicted methods are not specific to sarcoma, but can be applied to complement and optimize utilization of existing models and approaches. (A) Derivation of syngeneic engraftment models (SAMs) from GEMMs to increase scalability. (B) Humanization of existing PDX models for immunotherapy trials requiring an endogenous immune system, e.g., checkpoint inhibitor therapies. (C) Cellular barcoding of engraftment models to study clonal selection effects under therapy and metastasis. (D) Human mouse chimera genetic models to bypass mouse-human biology discrepancies. (E) Transduction of tumor cells from engraftment models using Doxycycline (Dox)-inducible vectors (e.g., TRE-shRNA to inducible knockdown a gene of interest) to investigate molecular dependencies in vivo. (F) Applying the same approach (E) on a systemic level to additionally study systemic toxicity in a target-gene-dependent fashion. (G) Overview of divergent metastasis modeling approaches.
Figure 6.
Complementary in vivo modeling approaches. The depicted methods are not specific to sarcoma, but can be applied to complement and optimize utilization of existing models and approaches. (A) Derivation of syngeneic engraftment models (SAMs) from GEMMs to increase scalability. (B) Humanization of existing PDX models for immunotherapy trials requiring an endogenous immune system, e.g., checkpoint inhibitor therapies. (C) Cellular barcoding of engraftment models to study clonal selection effects under therapy and metastasis. (D) Human mouse chimera genetic models to bypass mouse-human biology discrepancies. (E) Transduction of tumor cells from engraftment models using Doxycycline (Dox)-inducible vectors (e.g., TRE-shRNA to inducible knockdown a gene of interest) to investigate molecular dependencies in vivo. (F) Applying the same approach (E) on a systemic level to additionally study systemic toxicity in a target-gene-dependent fashion. (G) Overview of divergent metastasis modeling approaches.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Imle, R.; Kommoss, F.K.F.; Banito, A. Preclinical In Vivo Modeling of Pediatric Sarcoma—Promises and Limitations. J. Clin. Med. 2021, 10, 1578. https://doi.org/10.3390/jcm10081578
AMA Style
Imle R, Kommoss FKF, Banito A. Preclinical In Vivo Modeling of Pediatric Sarcoma—Promises and Limitations. Journal of Clinical Medicine. 2021; 10(8):1578. https://doi.org/10.3390/jcm10081578
Chicago/Turabian StyleImle, Roland, Felix K. F. Kommoss, and Ana Banito. 2021. "Preclinical In Vivo Modeling of Pediatric Sarcoma—Promises and Limitations" Journal of Clinical Medicine 10, no. 8: 1578. https://doi.org/10.3390/jcm10081578
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.