
Arrhythmogenic Cardiomyopathy Is a Multicellular Disease Affecting Cardiac and Bone Marrow Mesenchymal Stromal Cells

 ,
,  , ,
, ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Models
2.2. Isolation of Mesenchymal Stromal Cells (MSCs) from Murine Hearts
2.3. Isolation of MSCs from Murine Bone Marrow (BM)
2.4. RTqPCR
2.5. Western Blotting (WB)
2.6. Viral-Assisted Dsg2 Silencing in Normal Rat Cultured BM-MSCs
2.7. Morphometric Analyses of Cultured MSCs
2.8. Transwell Migration Assay
2.9. In Vivo Competitive Homing Assay
2.10. Statistical Analysis
3. Results
3.1. Cardiac and Bone Marrow Mesenchymal Stromal Cells Express Desmosomal Proteins
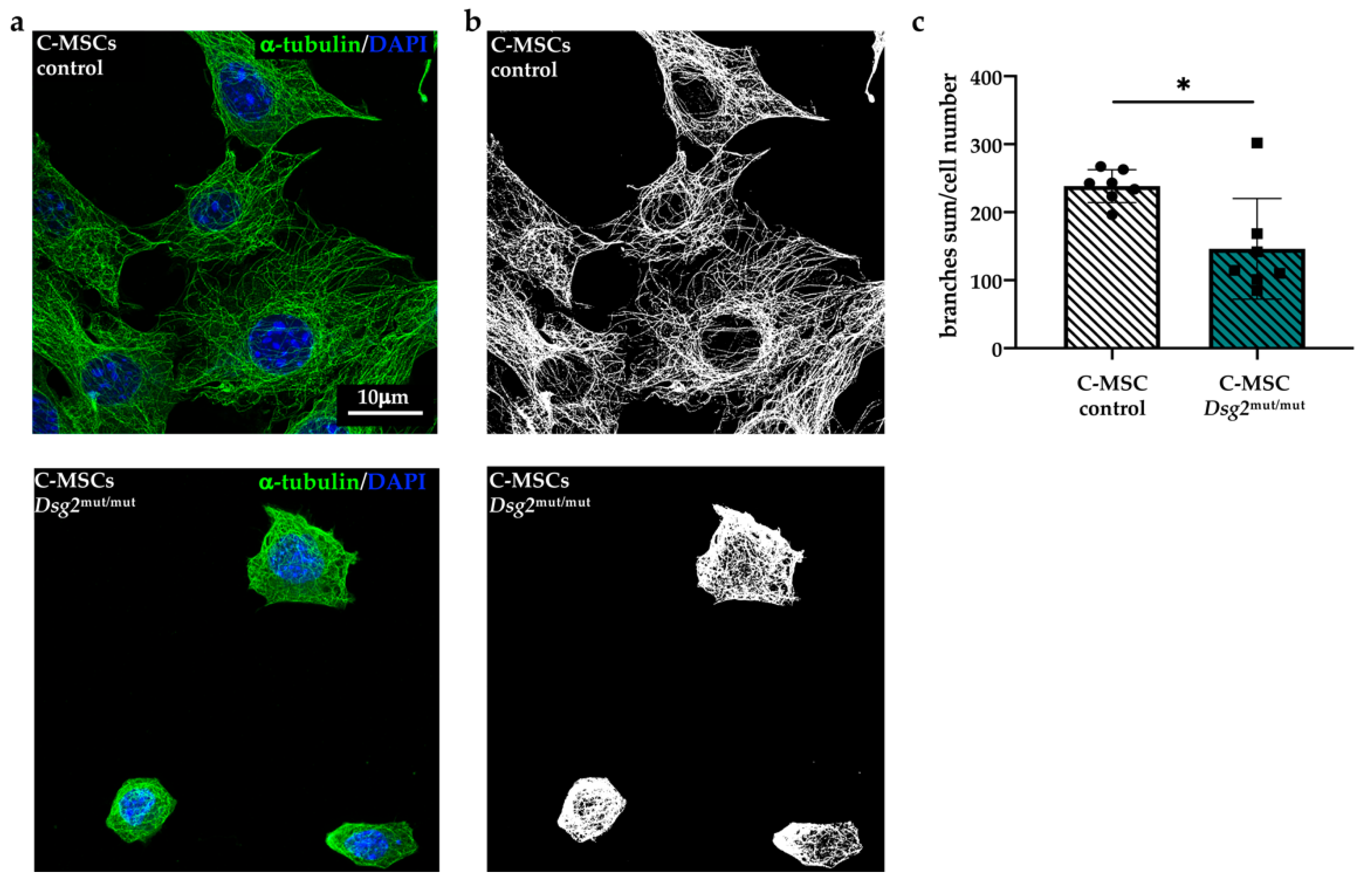
3.2. Dsg2mut/mut Cardiac and Bone Marrow Mesenchymal Stromal Cells Show Cytoskeletal Alteration
3.3. DSG2 Downregulation Affects Mesenchymal Stromal Cells Cytoskeletal Organization
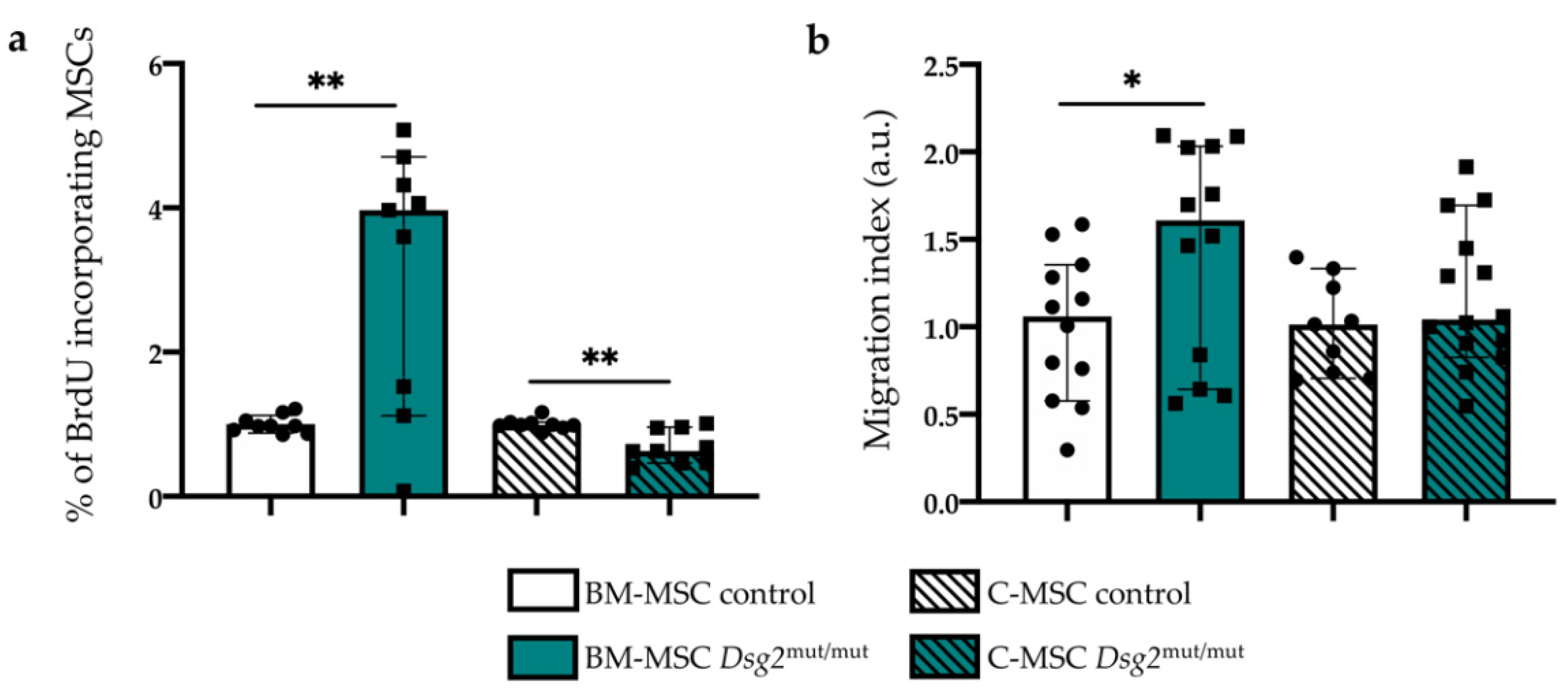
3.4. AC-linked Dsg2 Variant Affects Proliferation Rate and Migratory Capacity of Cardiac and Bone Marrow Mesenchymal Stromal Cells, In Vitro
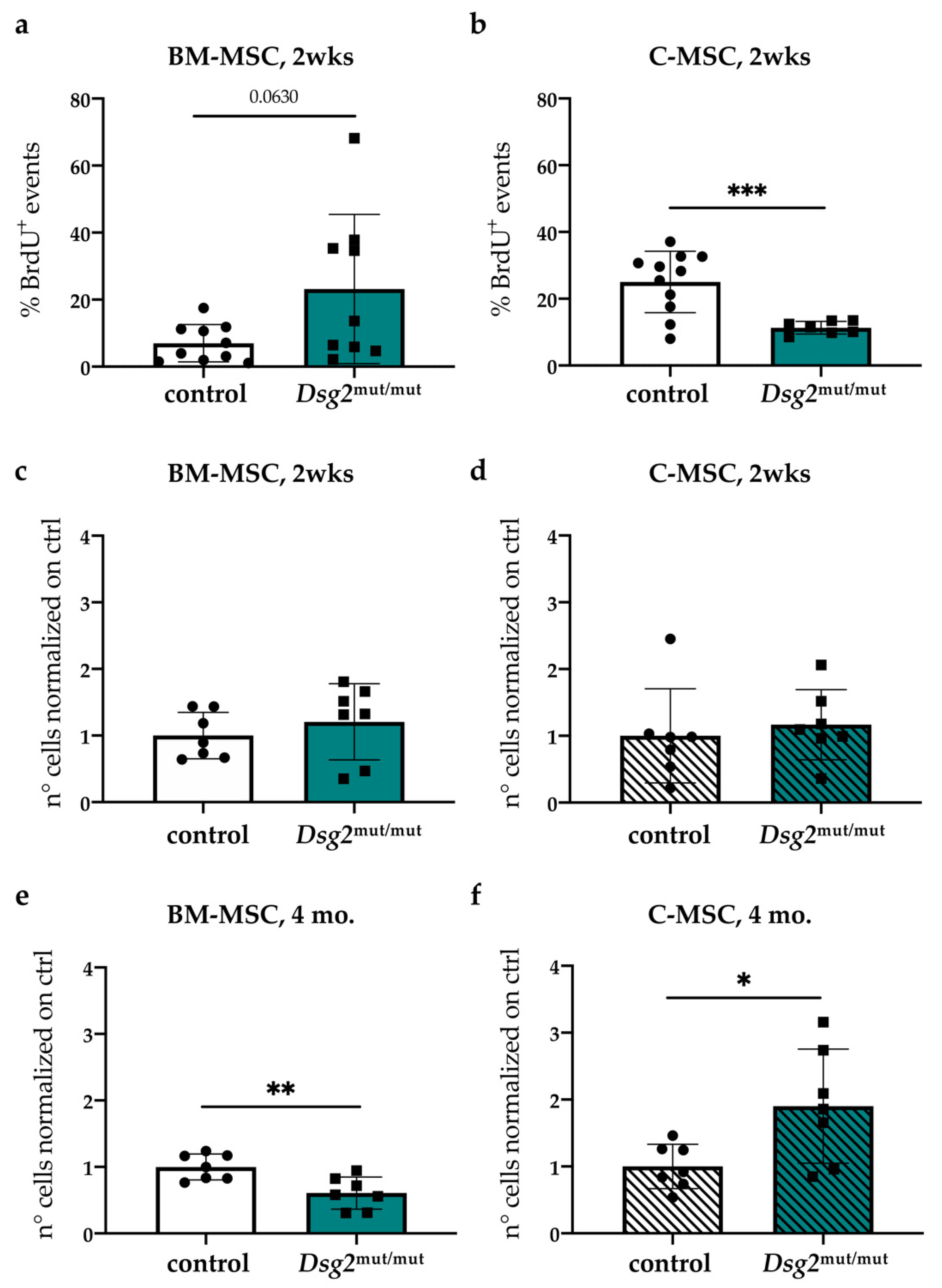
3.5. In Vivo Quantitation of Proliferative Index and Content of Cardiac vs. Bone Marrow Mesenchymal Stromal Cells, during AC Progression
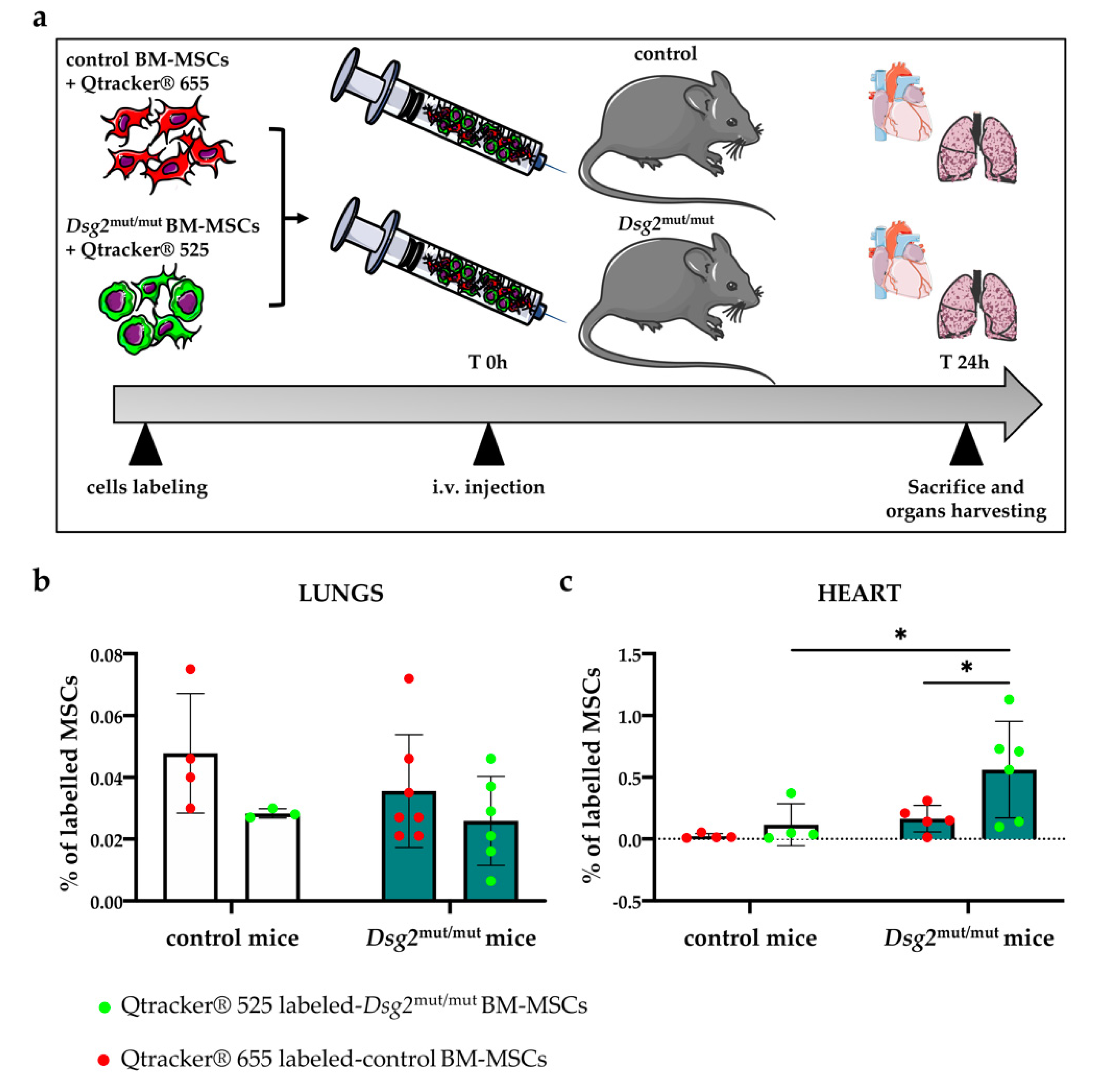
3.6. Intravenously Injected Bone Marrow-Derived Dsg2mut/mut Mesenchymal Stromal Cells Have Increased Propensity to Migrate to the AC Myocardium
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thiene, G.; Nava, A.; Corrado, D.; Rossi, L.; Pennelli, N. Right ventricular cardiomyopathy and sudden death in young people. N. Engl. J. Med. 1988, 318, 129–133. [Google Scholar] [CrossRef]
- Basso, C.; Thiene, G.; Corrado, D.; Angelini, A.; Nava, A.; Valente, M. Arrhythmogenic right ventricular cardiomyopathy: Dysplasia, dystrophy, or myocarditis? Circulation 1996, 94, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Bauce, B.; Corrado, D.; Thiene, G. Pathophysiology of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2012, 223–233. [Google Scholar] [CrossRef]
- Asimaki, A.; Saffitz, J.E. Remodeling of cell-cell junctions in arrhythmogenic cardiomyopathy. Cell Commun. Adhes. 2014, 13–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrado, D.; Basso, C.; Rizzoli, G.; Schiavon, M.; Thiene, G. Does sports activity enhance the risk of sudden death in adolescents and young adults? J. Am. Coll. Cardiol. 2003, 42, 1959–1963. [Google Scholar] [CrossRef] [PubMed]
- James, C.A.; Bhonsale, A.; Tichnell, C.; Murray, B.; Russell, S.D.; Tandri, H.; Tedford, R.J.; Judge, D.P.; Calkins, H. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J. Am. Coll. Cardiol. 2013, 62, 1290–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilichou, K.; Thiene, G.; Bauce, B.; Rigato, I.; Lazzarini, E.; Migliore, F.; Perazzolo Marra, M.; Rizzo, S.; Zorzi, A.; Daliento, L.; et al. Arrhythmogenic cardiomyopathy. Orphanet J. Rare Dis. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrado, D.; Van Tintelen, P.J.; McKenna, W.J.; Hauer, R.N.W.; Anastastakis, A.; Asimaki, A.; Basso, C.; Bauce, B.; Brunckhorst, C.; Bucciarelli-Ducci, C.; et al. Arrhythmogenic right ventricular cardiomyopathy: Evaluation of the current diagnostic criteria and differential diagnosis. Eur. Heart J. 2020, 1414–1429. [Google Scholar] [CrossRef] [Green Version]
- Corrado, D.; Perazzolo Marra, M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; De Lazzari, M.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of Arrhythmogenic Cardiomyopathy: The Padua Criteria. Int. J. Cardiol. 2020, 319, 106–114. [Google Scholar] [CrossRef]
- Sommariva, E.; Brambilla, S.; Carbucicchio, C.; Gambini, E.; Meraviglia, V.; Dello Russo, A.; Farina, F.M.; Casella, M.; Catto, V.; Pontone, G.; et al. Cardiac mesenchymal stromal cells are a source of adipocytes in arrhythmogenic cardiomyopathy. Eur. Heart J. 2016, 57, 1835–1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uccelli, A.; Moretta, L.; Pistoia, V. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 2008, 726–736. [Google Scholar] [CrossRef]
- Nombela-Arrieta, C.; Ritz, J.; Silberstein, L.E. The elusive nature and function of mesenchymal stem cells. Nat. Rev. Mol. Cell Biol. 2011, 126–131. [Google Scholar] [CrossRef] [Green Version]
- Bianco, P. “Mesenchymal” stem cells. Annu. Rev. Cell Dev. Biol. 2014, 677–704. [Google Scholar] [CrossRef]
- Chelko, S.P.; Asimaki, A.; Andersen, P.; Bedja, D.; Amat-Alarcon, N.; DeMazumder, D.; Jasti, R.; MacRae, C.A.; Leber, R.; Kleber, A.G.; et al. Central role for GSK3β in the Pathogenesis of arrhythmogenic cardiomyopathy. JCI Insight 2016, 1. [Google Scholar] [CrossRef] [Green Version]
- Pilichou, K.; Nava, A.; Basso, C.; Beffagna, G.; Bauce, B.; Lorenzon, A.; Frigo, G.; Vettori, A.; Valente, M.; Towbin, J.; et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation 2006, 113, 1171–1179. [Google Scholar] [CrossRef] [Green Version]
- Ackers-Johnson, M.; Li, P.Y.; Holmes, A.P.; O’Brien, S.M.; Pavlovic, D.; Foo, R.S. A simplified, langendorff-free method for concomitant isolation of viable cardiac myocytes and nonmyocytes from the adult mouse heart. Circ. Res. 2016, 119, 909–920. [Google Scholar] [CrossRef] [Green Version]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. the international society for cellular therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Angioni, R.; Liboni, C.; Herkenne, S.; Sánchez-Rodríguez, R.; Borile, G.; Marcuzzi, E.; Calì, B.; Muraca, M.; Viola, A. CD73+ extracellular vesicles inhibit angiogenesis through adenosine A2B receptor signalling. J. Extracell. Vesicles 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Zaglia, T.; Milan, G.; Ruhs, A.; Franzoso, M.; Bertaggia, E.; Pianca, N.; Carpi, A.; Carullo, P.; Pesce, P.; Sacerdoti, D.; et al. Atrogin-1 deficiency promotes cardiomyopathy and premature death via impaired autophagy. J. Clin. Investig. 2014, 124, 2410–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muinao, T.; Pal, M.; Boruah, H.P.D. Cytosolic and transmembrane protein extraction methods of breast and ovarian cancer cells: A comparative study. J. Biomol. Tech. 2018, 29, 71–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavares, S.; Vieira, A.F.; Taubenberger, A.V.; Araújo, M.; Martins, N.P.; Brás-Pereira, C.; Polónia, A.; Herbig, M.; Barreto, C.; Otto, O.; et al. Actin Stress fiber organization promotes cell stiffening and proliferation of pre-invasive breast cancer cells. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Giusti, B.; Margheri, F.; Rossi, L.; Lapini, I.; Magi, A.; Serratì, S.; Chillà, A.; Laurenzana, A.; Magnelli, L.; Calorini, L.; et al. Correction: Desmoglein-2-integrin beta-8 interaction regulates actin assembly in endothelial cells: Deregulation in Systemic sclerosis. PLoS ONE 2013, 8, E68117. [Google Scholar] [CrossRef] [Green Version]
- Ebert, L.M.; Tan, L.Y.; Johan, M.Z.; Min, K.K.M.; Cockshell, M.P.; Parham, K.A.; Betterman, K.L.; Szeto, P.; Boyle, S.; Silva, L.; et al. A Non-canonical role for Desmoglein-2 in endothelial cells: Implications for neoangiogenesis. Angiogenesis 2016, 19, 463–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kant, S.; Freytag, B.; Herzog, A.; Reich, A.; Merkel, R.; Hoffmann, B.; Krusche, C.A.; Leube, R.E. Desmoglein 2 mutation provokes skeletal muscle actin expression and accumulation at intercalated discs in murine hearts. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatzfeld, M.; Haffner, C.; Schulze, K.; Vinzens, U. The function of plakophilin 1 in desmosome assembly and actin filament organization. J. Cell Biol. 2000, 149, 209–222. [Google Scholar] [CrossRef]
- Bouchet, B.P.; Akhmanova, A. Microtubules in 3D cell motility. J. Cell Sci. 2017, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Qu, R.; Fan, T.; Zhu, X.; Feng, Y.; Yang, Y.; Deng, T.; Peng, Y.; Huang, W.; Ouyang, J.; et al. Cross-talk between microtubules and the linker of nucleoskeleton complex plays a critical role in the adipogenesis of human adipose-derived stem cells. Stem. Cell Res. Ther. 2018, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Biedzinski, S.; Faivre, L.; Vianay, B.; Delord, M.; Blanchoin, L.; Larghero, J.; Théry, M.; Brunet, S. Microtubules deform the nucleus and force chromatin reorganization during early differentiation of human hematopoietic stem cells. bioRxiv 2019, 763326. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.F.; Liu, G.S.; Wang, L.; Paul, C.; Wen, Z.L.; Wang, Y. Repair injured heart by regulating cardiac regenerative signals. Stem. Cells Int. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Agha, E.; Kramann, R.; Schneider, R.K.; Li, X.; Seeger, W.; Humphreys, B.D.; Bellusci, S. Mesenchymal stem cells in fibrotic disease. Cell Stem. Cell. 2017, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Fischer, U.M.; Harting, M.T.; Jimenez, F.; Monzon-Posadas, W.O.; Xue, H.; Savitz, S.I.; Laine, G.A.; Cox, C.S. Pulmonary passage is a major obstacle for intravenous stem cell delivery: The pulmonary first-pass effect. Stem. Cells Dev. 2009, 18, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Agrimi, J.; Scalco, A.; Agafonova, J.; Williams III, L.; Pansari, N.; Keceli, G.; Jun, S.; Wang, N.; Mastorci, F.; Tichnell, C.; et al. Psychosocial stress hastens disease progression and sudden death in mice with arrhythmogenic cardiomyopathy. J. Clin. Med. 2020, 9, 3804. [Google Scholar] [CrossRef] [PubMed]
- Chelko, S.P.; Asimaki, A.; Lowenthal, J.; Bueno-Beti, C.; Bedja, D.; Scalco, A.; Amat-Alarcon, N.; Andersen, P.; Judge, D.P.; Tung, L.; et al. Therapeutic modulation of the immune response in arrhythmogenic cardiomyopathy. Circulation 2019, 140, 1491–1505. [Google Scholar] [CrossRef]
- Hu, K.X.; Sun, Q.Y.; Guo, M.; Ai, H.S. The Radiation Protection and therapy effects of mesenchymal stem cells in mice with acute radiation injury. Br. J. Radiol. 2010, 83, 52–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez Perez, R.; Brauer, J.; Rühle, A.; Trinh, T.; Sisombath, S.; Wuchter, P.; Grosu, A.L.; Debus, J.; Saffrich, R.; Huber, P.E.; et al. Human mesenchymal stem cells are resistant to UV-B irradiation. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scalco, A.; Liboni, C.; Angioni, R.; Di Bona, A.; Albiero, M.; Bertoldi, N.; Fadini, G.P.; Thiene, G.; Chelko, S.P.; Basso, C.; et al. Arrhythmogenic Cardiomyopathy Is a Multicellular Disease Affecting Cardiac and Bone Marrow Mesenchymal Stromal Cells. J. Clin. Med. 2021, 10, 1871. https://doi.org/10.3390/jcm10091871
Scalco A, Liboni C, Angioni R, Di Bona A, Albiero M, Bertoldi N, Fadini GP, Thiene G, Chelko SP, Basso C, et al. Arrhythmogenic Cardiomyopathy Is a Multicellular Disease Affecting Cardiac and Bone Marrow Mesenchymal Stromal Cells. Journal of Clinical Medicine. 2021; 10(9):1871. https://doi.org/10.3390/jcm10091871
Chicago/Turabian StyleScalco, Arianna, Cristina Liboni, Roberta Angioni, Anna Di Bona, Mattia Albiero, Nicole Bertoldi, Gian Paolo Fadini, Gaetano Thiene, Stephen P. Chelko, Cristina Basso, and et al. 2021. "Arrhythmogenic Cardiomyopathy Is a Multicellular Disease Affecting Cardiac and Bone Marrow Mesenchymal Stromal Cells" Journal of Clinical Medicine 10, no. 9: 1871. https://doi.org/10.3390/jcm10091871