
Cellular and Molecular Players in the Tumor Microenvironment of Renal Cell Carcinoma
 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
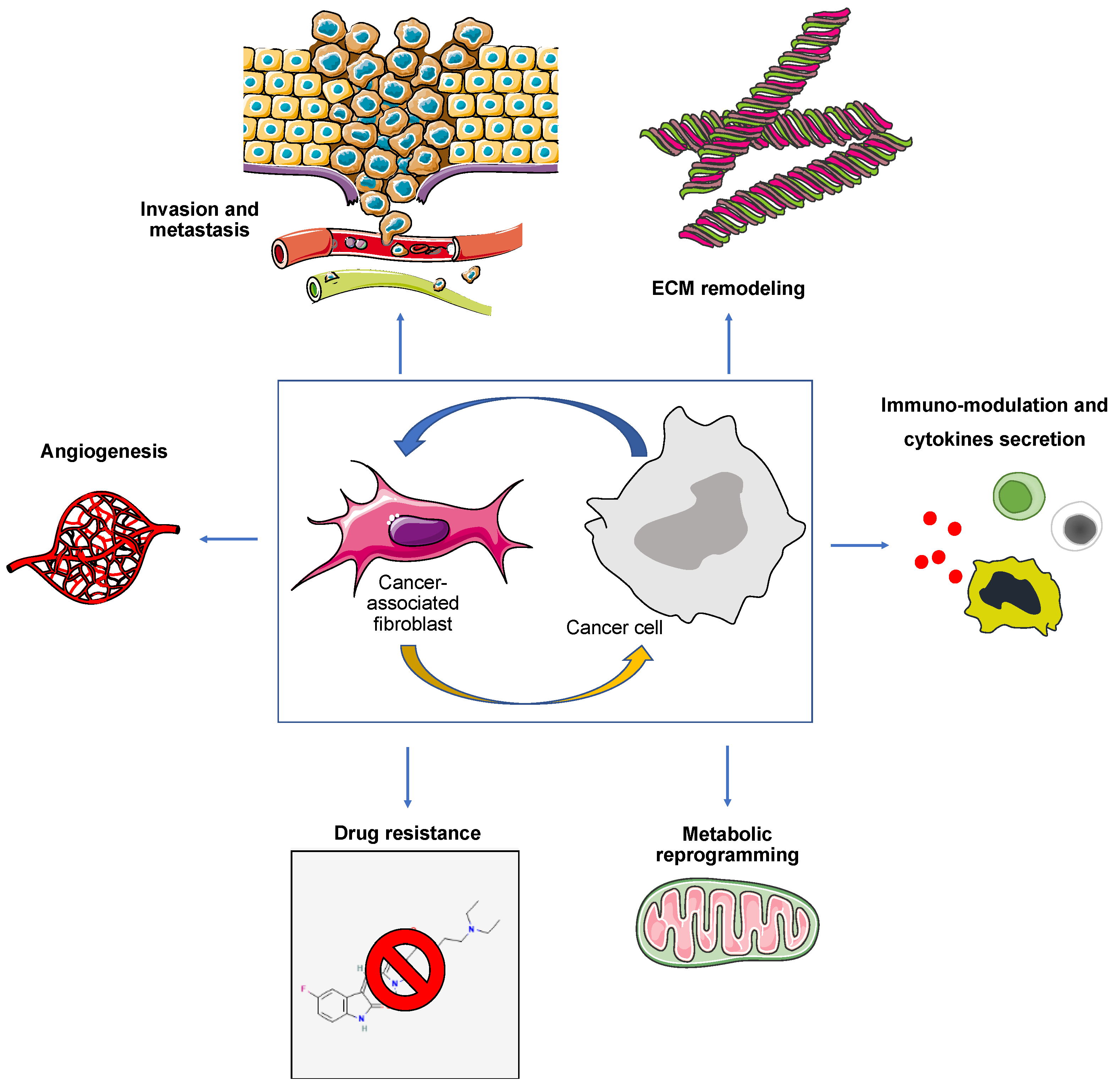
2. Tumor Microenvironment
2.1. Cancer-Associated Fibroblasts (CAFs)
2.2. Tumor Vascular Cells
2.3. Tumor-Associated Adipocytes
2.4. Tumor Immune Microenvironment
2.4.1. T Cells
2.4.2. Tumor-Associated Myeloid Cells
3. Metabolic Reprogramming and Immune Escape in the TME
4. Clinical Role of the TME and Therapeutic Implications
4.1. Angiogenesis Inhibitors
4.2. Mammalian Target of Rapamycin (mTOR) Inhibitors
4.3. Cytokine Therapy and Immune Checkpoint Inhibitors (ICIs)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Raghubar, A.M.; Roberts, M.J.; Wood, S.; Healy, H.G.; Kassianos, A.J.; Mallett, A.J. Cellular Milieu in Clear Cell Renal Cell Carcinoma. Front. Oncol. 2022, 12, 943583. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, G.; Galleggiante, V.; Rutigliano, M.; Vavallo, A.; Ditonno, P.; Battaglia, M. Isolation and Characterization of Cancer Stem Cells in Renal Cell Carcinoma. Urologia 2015, 82, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Galleggiante, V.; Rutigliano, M.; Sallustio, F.; Ribatti, D.; Ditonno, P.; Bettocchi, C.; Selvaggi, F.P.; Lucarelli, G.; Battaglia, M. CTR2 Identifies a Population of Cancer Cells with Stem Cell-like Features in Patients with Clear Cell Renal Cell Carcinoma. J. Urol. 2014, 192, 1831–1841. [Google Scholar] [CrossRef] [PubMed]
- di Meo, N.A.; Lasorsa, F.; Rutigliano, M.; Loizzo, D.; Ferro, M.; Stella, A.; Bizzoca, C.; Vincenti, L.; Pandolfo, S.D.; Autorino, R.; et al. Renal Cell Carcinoma as a Metabolic Disease: An Update on Main Pathways, Potential Biomarkers, and Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 14360. [Google Scholar] [CrossRef]
- di Meo, N.A.; Lasorsa, F.; Rutigliano, M.; Milella, M.; Ferro, M.; Battaglia, M.; Ditonno, P.; Lucarelli, G. The Dark Side of Lipid Metabolism in Prostate and Renal Carcinoma: Novel Insights into Molecular Diagnostic and Biomarker Discovery. Expert. Rev. Mol. Diagn. 2023, 23, 297–313. [Google Scholar] [CrossRef]
- Lucarelli, G.; Loizzo, D.; Franzin, R.; Battaglia, S.; Ferro, M.; Cantiello, F.; Castellano, G.; Bettocchi, C.; Ditonno, P.; Battaglia, M. Metabolomic Insights into Pathophysiological Mechanisms and Biomarker Discovery in Clear Cell Renal Cell Carcinoma. Expert. Rev. Mol. Diagn. 2019, 19, 397–407. [Google Scholar] [CrossRef]
- Lucarelli, G.; Ferro, M.; Battaglia, M. Multi-Omics Approach Reveals the Secrets of Metabolism of Clear Cell-Renal Cell Carcinoma. Transl. Androl. Urol. 2016, 5, 801–803. [Google Scholar] [CrossRef] [Green Version]
- Lucarelli, G.; Galleggiante, V.; Rutigliano, M.; Sanguedolce, F.; Cagiano, S.; Bufo, P.; Lastilla, G.; Maiorano, E.; Ribatti, D.; Giglio, A.; et al. Metabolomic Profile of Glycolysis and the Pentose Phosphate Pathway Identifies the Central Role of Glucose-6-Phosphate Dehydrogenase in Clear Cell-Renal Cell Carcinoma. Oncotarget 2015, 6, 13371–13386. [Google Scholar] [CrossRef] [Green Version]
- Ragone, R.; Sallustio, F.; Piccinonna, S.; Rutigliano, M.; Vanessa, G.; Palazzo, S.; Lucarelli, G.; Ditonno, P.; Battaglia, M.; Fanizzi, F.P.; et al. Renal Cell Carcinoma: A Study through NMR-Based Metabolomics Combined with Transcriptomics. Diseases 2016, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, C.; Meregalli, C.; Bombelli, S.; Di Stefano, V.; Salerno, F.; Torsello, B.; De Marco, S.; Bovo, G.; Cifola, I.; Mangano, E.; et al. The Glucose and Lipid Metabolism Reprogramming Is Grade-Dependent in Clear Cell Renal Cell Carcinoma Primary Cultures and Is Targetable to Modulate Cell Viability and Proliferation. Oncotarget 2017, 8, 113502–113515. [Google Scholar] [CrossRef] [Green Version]
- Lucarelli, G.; Rutigliano, M.; Sallustio, F.; Ribatti, D.; Giglio, A.; Lepore Signorile, M.; Grossi, V.; Sanese, P.; Napoli, A.; Maiorano, E.; et al. Integrated Multi-Omics Characterization Reveals a Distinctive Metabolic Signature and the Role of NDUFA4L2 in Promoting Angiogenesis, Chemoresistance, and Mitochondrial Dysfunction in Clear Cell Renal Cell Carcinoma. Aging 2018, 10, 3957–3985. [Google Scholar] [CrossRef] [PubMed]
- De Marco, S.; Torsello, B.; Minutiello, E.; Morabito, I.; Grasselli, C.; Bombelli, S.; Zucchini, N.; Lucarelli, G.; Strada, G.; Perego, R.A.; et al. The Cross-Talk between Abl2 Tyrosine Kinase and TGFβ1 Signalling Modulates the Invasion of Clear Cell Renal Cell Carcinoma Cells. FEBS Lett. 2023, 597, 1098–1113. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, G.; Ferro, M.; Loizzo, D.; Bianchi, C.; Terracciano, D.; Cantiello, F.; Bell, L.N.; Battaglia, S.; Porta, C.; Gernone, A.; et al. Integration of Lipidomics and Transcriptomics Reveals Reprogramming of the Lipid Metabolism and Composition in Clear Cell Renal Cell Carcinoma. Metabolites 2020, 10, 509. [Google Scholar] [CrossRef]
- Bombelli, S.; Torsello, B.; De Marco, S.; Lucarelli, G.; Cifola, I.; Grasselli, C.; Strada, G.; Bovo, G.; Perego, R.A.; Bianchi, C. 36-KDa Annexin A3 Isoform Negatively Modulates Lipid Storage in Clear Cell Renal Cell Carcinoma Cells. Am. J. Pathol. 2020, 190, 2317–2326. [Google Scholar] [CrossRef]
- Battaglia, M.; Lucarelli, G. The Role of Renal Surgery in the Era of Targeted Therapy: The Urologist’s Perspective. Urologia 2015, 82, 137–138. [Google Scholar] [CrossRef] [PubMed]
- Pandolfo, S.D.; Beksac, A.T.; Derweesh, I.; Celia, A.; Schiavina, R.; Bianchi, L.; Costa, G.; Carbonara, U.; Loizzo, D.; Lucarelli, G.; et al. Percutaneous Ablation vs Robot-Assisted Partial Nephrectomy for Completely Endophytic Renal Masses: A Multicenter Trifecta Analysis with a Minimum 3-Year Follow-Up. J. Endourol. 2023, 37, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Pandolfo, S.D.; Loizzo, D.; Beksac, A.T.; Derweesh, I.; Celia, A.; Bianchi, L.; Elbich, J.; Costa, G.; Carbonara, U.; Lucarelli, G.; et al. Percutaneous Thermal Ablation for CT1 Renal Mass in Solitary Kidney: A Multicenter Trifecta Comparative Analysis versus Robot-Assisted Partial Nephrectomy. Eur. J. Surg. Oncol. 2023, 49, 486–490. [Google Scholar] [CrossRef]
- Vartolomei, L.; Cotruș, A.; Stanciu, C.; Delcea, C.; Tozzi, M.; Lievore, E.; Crocetto, F.; Del Giudice, F.; Lucarelli, G.; Muto, M.; et al. Quality of Life and Psychological Distress among Patients with Small Renal Masses. J. Clin. Med. 2022, 11, 3944. [Google Scholar] [CrossRef]
- Monti, M.; Lunardini, S.; Magli, I.A.; Campi, R.; Primiceri, G.; Berardinelli, F.; Amparore, D.; Terracciano, D.; Lucarelli, G.; Schips, L.; et al. Micro-RNAs Predict Response to Systemic Treatments in Metastatic Renal Cell Carcinoma Patients: Results from a Systematic Review of the Literature. Biomedicines 2022, 10, 1287. [Google Scholar] [CrossRef]
- Papale, M.; Vocino, G.; Lucarelli, G.; Rutigliano, M.; Gigante, M.; Rocchetti, M.T.; Pesce, F.; Sanguedolce, F.; Bufo, P.; Battaglia, M.; et al. Urinary RKIP/p-RKIP Is a Potential Diagnostic and Prognostic Marker of Clear Cell Renal Cell Carcinoma. Oncotarget 2017, 8, 40412–40424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gigante, M.; Lucarelli, G.; Divella, C.; Netti, G.S.; Pontrelli, P.; Cafiero, C.; Grandaliano, G.; Castellano, G.; Rutigliano, M.; Stallone, G.; et al. Soluble Serum AKlotho Is a Potential Predictive Marker of Disease Progression in Clear Cell Renal Cell Carcinoma. Medicine 2015, 94, e1917. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, G.; Rutigliano, M.; Sanguedolce, F.; Galleggiante, V.; Giglio, A.; Cagiano, S.; Bufo, P.; Maiorano, E.; Ribatti, D.; Ranieri, E.; et al. Increased Expression of the Autocrine Motility Factor Is Associated With Poor Prognosis in Patients with Clear Cell-Renal Cell Carcinoma. Medicine 2015, 94, e2117. [Google Scholar] [CrossRef]
- Lucarelli, G.; Ditonno, P.; Bettocchi, C.; Vavallo, A.; Rutigliano, M.; Galleggiante, V.; Larocca, A.M.V.; Castellano, G.; Gesualdo, L.; Grandaliano, G.; et al. Diagnostic and Prognostic Role of Preoperative Circulating CA 15-3, CA 125, and Beta-2 Microglobulin in Renal Cell Carcinoma. Dis. Markers 2014, 2014, 689795. [Google Scholar] [CrossRef] [PubMed]
- Ferro, M.; Musi, G.; Marchioni, M.; Maggi, M.; Veccia, A.; Del Giudice, F.; Barone, B.; Crocetto, F.; Lasorsa, F.; Antonelli, A.; et al. Radiogenomics in Renal Cancer Management-Current Evidence and Future Prospects. Int. J. Mol. Sci. 2023, 24, 4615. [Google Scholar] [CrossRef] [PubMed]
- Ferro, M.; Crocetto, F.; Barone, B.; Del Giudice, F.; Maggi, M.; Lucarelli, G.; Busetto, G.M.; Autorino, R.; Marchioni, M.; Cantiello, F.; et al. Artificial Intelligence and Radiomics in Evaluation of Kidney Lesions: A Comprehensive Literature Review. Ther. Adv. Urol. 2023, 15, 17562872231164804. [Google Scholar] [CrossRef] [PubMed]
- Tataru, O.S.; Marchioni, M.; Crocetto, F.; Barone, B.; Lucarelli, G.; Del Giudice, F.; Busetto, G.M.; Veccia, A.; Lo Giudice, A.; Russo, G.I.; et al. Molecular Imaging Diagnosis of Renal Cancer Using 99mTc-Sestamibi SPECT/CT and Girentuximab PET-CT-Current Evidence and Future Development of Novel Techniques. Diagnostics 2023, 13, 593. [Google Scholar] [CrossRef]
- Vuong, L.; Kotecha, R.R.; Voss, M.H.; Hakimi, A.A. Tumor Microenvironment Dynamics in Clear-Cell Renal Cell Carcinoma. Cancer Discov. 2019, 9, 1349–1357. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.; Tang, F.; Huang, Y.; He, C.; Chen, C.; Zhao, J.; Wu, W.; He, Z. The Tumour Microenvironment and Metabolism in Renal Cell Carcinoma Targeted or Immune Therapy. J. Cell. Physiol. 2021, 236, 1616–1627. [Google Scholar] [CrossRef]
- de Visser, K.E.; Joyce, J.A. The Evolving Tumor Microenvironment: From Cancer Initiation to Metastatic Outgrowth. Cancer Cell. 2023, 41, 374–403. [Google Scholar] [CrossRef]
- Heidegger, I.; Pircher, A.; Pichler, R. Targeting the Tumor Microenvironment in Renal Cell Cancer Biology and Therapy. Front. Oncol. 2019, 9, 490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calon, A.; Tauriello, D.V.F.; Batlle, E. TGF-Beta in CAF-Mediated Tumor Growth and Metastasis. Semin. Cancer Biol. 2014, 25, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Fearon, D.T. The Carcinoma-Associated Fibroblast Expressing Fibroblast Activation Protein and Escape from Immune Surveillance. Cancer Immunol. Res. 2014, 2, 187–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grout, J.A.; Sirven, P.; Leader, A.M.; Maskey, S.; Hector, E.; Puisieux, I.; Steffan, F.; Cheng, E.; Tung, N.; Maurin, M.; et al. Spatial Positioning and Matrix Programs of Cancer-Associated Fibroblasts Promote T-Cell Exclusion in Human Lung Tumors. Cancer Discov. 2022, 12, 2606–2625. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.-L.; Xiong, L.-B.; Zhou, Z.-H.; Ning, K.; Li, Z.; Wu, Z.-S.; Deng, M.-H.; Wei, W.-S.; Wang, N.; Zou, X.-P.; et al. Single-Cell Transcriptomics Reveals a Low CD8+ T Cell Infiltrating State Mediated by Fibroblasts in Recurrent Renal Cell Carcinoma. J. Immunother. Cancer 2022, 10, e004206. [Google Scholar] [CrossRef] [PubMed]
- Fiori, M.E.; Di Franco, S.; Villanova, L.; Bianca, P.; Stassi, G.; De Maria, R. Cancer-Associated Fibroblasts as Abettors of Tumor Progression at the Crossroads of EMT and Therapy Resistance. Mol. Cancer 2019, 18, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef]
- Aubert, S.; Fauquette, V.; Hémon, B.; Lepoivre, R.; Briez, N.; Bernard, D.; Van Seuningen, I.; Leroy, X.; Perrais, M. MUC1, a New Hypoxia Inducible Factor Target Gene, Is an Actor in Clear Renal Cell Carcinoma Tumor Progression. Cancer Res. 2009, 69, 5707–5715. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Wong, S.; Borrelli, A.; Chung, M.A. Down-Regulation of MUC1 in Cancer Cells Inhibits Cell Migration by Promoting E-Cadherin/Catenin Complex Formation. Biochem. Biophys. Res. Commun. 2007, 362, 740–746. [Google Scholar] [CrossRef]
- Piva, F.; Giulietti, M.; Santoni, M.; Occhipinti, G.; Scarpelli, M.; Lopez-Beltran, A.; Cheng, L.; Principato, G.; Montironi, R. Epithelial to Mesenchymal Transition in Renal Cell Carcinoma: Implications for Cancer Therapy. Mol. Diagn. Ther. 2016, 20, 111–117. [Google Scholar] [CrossRef]
- Sun, R.; Kong, X.; Qiu, X.; Huang, C.; Wong, P.-P. The Emerging Roles of Pericytes in Modulating Tumor Microenvironment. Front. Cell. Dev. Biol. 2021, 9, 676342. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.A.; Chang, S.-H.; Shih, S.-C.; Dvorak, A.M.; Dvorak, H.F. Heterogeneity of the Tumor Vasculature. Semin. Thromb. Hemost. 2010, 36, 321–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comandone, A.; Vana, F.; Comandone, T.; Tucci, M. Antiangiogenic Therapy in Clear Cell Renal Carcinoma (CCRC): Pharmacological Basis and Clinical Results. Cancers 2021, 13, 5896. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, C.; Xie, H.; Fan, Y.; Yang, Z.; Ma, J.; He, D.; Li, L. Infiltrating Mast Cells Promote Renal Cell Carcinoma Angiogenesis by Modulating PI3K →︀ AKT →︀ GSK3β →︀ AM Signaling. Oncogene 2017, 36, 2879–2888. [Google Scholar] [CrossRef] [PubMed]
- Akino, T.; Hida, K.; Hida, Y.; Tsuchiya, K.; Freedman, D.; Muraki, C.; Ohga, N.; Matsuda, K.; Akiyama, K.; Harabayashi, T.; et al. Cytogenetic Abnormalities of Tumor-Associated Endothelial Cells in Human Malignant Tumors. Am. J. Pathol. 2009, 175, 2657–2667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Z.; Li, C.; Fan, J.; He, D.; Li, L. Androgen Receptor (AR) Signaling Promotes RCC Progression via Increased Endothelial Cell Proliferation and Recruitment by Modulating AKT → NF-ΚB → CXCL5 Signaling. Sci. Rep. 2016, 6, 37085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amersfoort, J.; Eelen, G.; Carmeliet, P. Immunomodulation by Endothelial Cells—Partnering up with the Immune System? Nat. Rev. Immunol. 2022, 22, 576–588. [Google Scholar] [CrossRef]
- Ma, Q.; Dieterich, L.C.; Detmar, M. Multiple Roles of Lymphatic Vessels in Tumor Progression. Curr. Opin. Immunol. 2018, 53, 7–12. [Google Scholar] [CrossRef]
- Bruna, F.A.; Romeo, L.R.; Campo-Verde-Arbocco, F.; Contador, D.; Gómez, S.; Santiano, F.; Sasso, C.V.; Zyla, L.; López-Fontana, C.; Calvo, J.C.; et al. Human Renal Adipose Tissue from Normal and Tumor Kidney: Its Influence on Renal Cell Carcinoma. Oncotarget 2019, 10, 5454–5467. [Google Scholar] [CrossRef] [Green Version]
- Horiguchi, A.; Sumitomo, M.; Asakuma, J.; Asano, T.; Zheng, R.; Asano, T.; Nanus, D.M.; Hayakawa, M. Leptin Promotes Invasiveness of Murine Renal Cancer Cells via Extracellular Signal-Regulated Kinases and Rho Dependent Pathway. J. Urol. 2006, 176, 1636–1641. [Google Scholar] [CrossRef]
- Pallegar, N.K.; Christian, S.L. Adipocytes in the Tumour Microenvironment. Adv. Exp. Med. Biol. 2020, 1234, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Gu, C.; Zhu, Y.; Luo, L.; Dong, D.; Wan, F.; Zhang, H.; Shi, G.; Sun, L.; Ye, D. ADIPOQ Polymorphism Rs182052 Is Associated with Clear Cell Renal Cell Carcinoma. Cancer Sci. 2015, 106, 687–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdu Allah, A.M.; El-Hefnway, S.M.; Alhanafy, A.M.; Zahran, A.M.; Kasem, H.E. Leptin Receptor Gene (A/G) Polymorphism Rs1137101 and Renal Cell Carcinoma. Mol. Cell. Biochem. 2018, 448, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Ferrando, M.; Bruna, F.A.; Romeo, L.R.; Contador, D.; Moya-Morales, D.L.; Santiano, F.; Zyla, L.; Gomez, S.; Lopez-Fontana, C.M.; Calvo, J.C.; et al. Renal Peritumoral Adipose Tissue Undergoes a Browning Process and Stimulates the Expression of Epithelial-Mesenchymal Transition Markers in Human Renal Cells. Sci. Rep. 2022, 12, 8687. [Google Scholar] [CrossRef]
- Olea-Flores, M.; Juárez-Cruz, J.; Mendoza-Catalán, M.; Padilla-Benavides, T.; Navarro-Tito, N. Signaling Pathways Induced by Leptin during Epithelial–Mesenchymal Transition in Breast Cancer. Int. J. Mol. Sci. 2018, 19, 3493. [Google Scholar] [CrossRef] [Green Version]
- Philip, M.; Schietinger, A. CD8+ T Cell Differentiation and Dysfunction in Cancer. Nat. Rev. Immunol. 2022, 22, 209–223. [Google Scholar] [CrossRef]
- Lasorsa, F.; di Meo, N.A.; Rutigliano, M.; Milella, M.; Ferro, M.; Pandolfo, S.D.; Crocetto, F.; Tataru, O.S.; Autorino, R.; Battaglia, M.; et al. Immune Checkpoint Inhibitors in Renal Cell Carcinoma: Molecular Basis and Rationale for Their Use in Clinical Practice. Biomedicines 2023, 11, 1071. [Google Scholar] [CrossRef]
- Pardoll, D.M. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Tseng, S.-Y.; Otsuji, M.; Gorski, K.; Huang, X.; Slansky, J.E.; Pai, S.I.; Shalabi, A.; Shin, T.; Pardoll, D.M.; Tsuchiya, H. B7-Dc, a New Dendritic Cell Molecule with Potent Costimulatory Properties for T Cells. J. Exp. Med. 2001, 193, 839–846. [Google Scholar] [CrossRef]
- Braun, D.A.; Street, K.; Burke, K.P.; Cookmeyer, D.L.; Denize, T.; Pedersen, C.B.; Gohil, S.H.; Schindler, N.; Pomerance, L.; Hirsch, L.; et al. Progressive Immune Dysfunction with Advancing Disease Stage in Renal Cell Carcinoma. Cancer Cell. 2021, 39, 632–648.e8. [Google Scholar] [CrossRef]
- Krishna, C.; DiNatale, R.G.; Kuo, F.; Srivastava, R.M.; Vuong, L.; Chowell, D.; Gupta, S.; Vanderbilt, C.; Purohit, T.A.; Liu, M.; et al. Single-Cell Sequencing Links Multiregional Immune Landscapes and Tissue-Resident T Cells in CcRCC to Tumor Topology and Therapy Efficacy. Cancer Cell. 2021, 39, 662–677.e6. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, N.A.; Becht, E.; Pagès, F.; Skliris, G.; Verkarre, V.; Vano, Y.; Mejean, A.; Saint-Aubert, N.; Lacroix, L.; Natario, I.; et al. Orchestration and Prognostic Significance of Immune Checkpoints in the Microenvironment of Primary and Metastatic Renal Cell Cancer. Clin. Cancer Res. 2015, 21, 3031–3040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giraldo, N.A.; Becht, E.; Vano, Y.; Petitprez, F.; Lacroix, L.; Validire, P.; Sanchez-Salas, R.; Ingels, A.; Oudard, S.; Moatti, A.; et al. Tumor-Infiltrating and Peripheral Blood T-Cell Immunophenotypes Predict Early Relapse in Localized Clear Cell Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 4416–4428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhary, B.; Elkord, E. Regulatory T Cells in the Tumor Microenvironment and Cancer Progression: Role and Therapeutic Targeting. Vaccines 2016, 4, 28. [Google Scholar] [CrossRef] [Green Version]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T Cells in Cancer Immunosuppression—Implications for Anticancer Therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef]
- Gigante, M.; Pontrelli, P.; Herr, W.; Gigante, M.; D’Avenia, M.; Zaza, G.; Cavalcanti, E.; Accetturo, M.; Lucarelli, G.; Carrieri, G.; et al. MiR-29b and MiR-198 Overexpression in CD8+ T Cells of Renal Cell Carcinoma Patients down-Modulates JAK3 and MCL-1 Leading to Immune Dysfunction. J. Transl. Med. 2016, 14, 84. [Google Scholar] [CrossRef] [Green Version]
- Taylor, N.A.; Vick, S.C.; Iglesia, M.D.; Brickey, W.J.; Midkiff, B.R.; McKinnon, K.P.; Reisdorf, S.; Anders, C.K.; Carey, L.A.; Parker, J.S.; et al. Treg Depletion Potentiates Checkpoint Inhibition in Claudin-Low Breast Cancer. J. Clin. Investig. 2017, 127, 3472–3483. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Zhang, X.H.-F. Tumor-Associated Neutrophils and Macrophages-Heterogenous but Not Chaotic. Front. Immunol. 2020, 11, 553967. [Google Scholar] [CrossRef]
- Sica, A.; Bronte, V. Altered Macrophage Differentiation and Immune Dysfunction in Tumor Development. J. Clin. Investig. 2007, 117, 1155–1166. [Google Scholar] [CrossRef] [Green Version]
- Loeuillard, E.; Yang, J.; Buckarma, E.; Wang, J.; Liu, Y.; Conboy, C.; Pavelko, K.D.; Li, Y.; O’Brien, D.; Wang, C.; et al. Targeting Tumor-Associated Macrophages and Granulocytic Myeloid-Derived Suppressor Cells Augments PD-1 Blockade in Cholangiocarcinoma. J. Clin. Investig. 2020, 130, 5380–5396. [Google Scholar] [CrossRef]
- Tseng, D.; Volkmer, J.-P.; Willingham, S.B.; Contreras-Trujillo, H.; Fathman, J.W.; Fernhoff, N.B.; Seita, J.; Inlay, M.A.; Weiskopf, K.; Miyanishi, M.; et al. Anti-CD47 Antibody-Mediated Phagocytosis of Cancer by Macrophages Primes an Effective Antitumor T-Cell Response. Proc. Natl. Acad. Sci. USA 2013, 110, 11103–11108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Jee, S.; Bang, S.; Son, H.; Cha, H.; Myung, J.; Sim, J.; Kim, Y.; Paik, S.; Kim, H. CD47 Expression Predicts Unfavorable Prognosis in Clear Cell Renal Cell Carcinoma after Curative Resection. Diagnostics 2022, 12, 2291. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leek, R.D.; Talks, K.L.; Pezzella, F.; Turley, H.; Campo, L.; Brown, N.S.; Bicknell, R.; Taylor, M.; Gatter, K.C.; Harris, A.L. Relation of Hypoxia-Inducible Factor-2 Alpha (HIF-2 Alpha) Expression in Tumor-Infiltrative Macrophages to Tumor Angiogenesis and the Oxidative Thymidine Phosphorylase Pathway in Human Breast Cancer. Cancer Res. 2002, 62, 1326–1329. [Google Scholar] [PubMed]
- Toge, H.; Inagaki, T.; Kojimoto, Y.; Shinka, T.; Hara, I. Angiogenesis in Renal Cell Carcinoma: The Role of Tumor-Associated Macrophages. Int. J. Urol. 2009, 16, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Komohara, Y.; Hasita, H.; Ohnishi, K.; Fujiwara, Y.; Suzu, S.; Eto, M.; Takeya, M. Macrophage Infiltration and Its Prognostic Relevance in Clear Cell Renal Cell Carcinoma. Cancer Sci. 2011, 102, 1424–1431. [Google Scholar] [CrossRef]
- Cros, J.; Sbidian, E.; Posseme, K.; Letierce, A.; Guettier, C.; Benoît, G.; Ferlicot, S. Nestin Expression on Tumour Vessels and Tumour-Infiltrating Macrophages Define a Poor Prognosis Subgroup of Pt1 Clear Cell Renal Cell Carcinoma. Virchows Arch. 2016, 469, 331–337. [Google Scholar] [CrossRef]
- Chittezhath, M.; Dhillon, M.K.; Lim, J.Y.; Laoui, D.; Shalova, I.N.; Teo, Y.L.; Chen, J.; Kamaraj, R.; Raman, L.; Lum, J.; et al. Molecular Profiling Reveals a Tumor-Promoting Phenotype of Monocytes and Macrophages in Human Cancer Progression. Immunity 2014, 41, 815–829. [Google Scholar] [CrossRef] [Green Version]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef] [Green Version]
- López-Lago, M.A.; Posner, S.; Thodima, V.J.; Molina, A.M.; Motzer, R.J.; Chaganti, R.S.K. Neutrophil Chemokines Secreted by Tumor Cells Mount a Lung Antimetastatic Response during Renal Cell Carcinoma Progression. Oncogene 2013, 32, 1752–1760. [Google Scholar] [CrossRef] [Green Version]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-Beta: “N1” versus “N2” TAN. Cancer Cell. 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, H.K.; Donskov, F.; Marcussen, N.; Nordsmark, M.; Lundbeck, F.; von der Maase, H. Presence of Intratumoral Neutrophils Is an Independent Prognostic Factor in Localized Renal Cell Carcinoma. J. Clin. Oncol. 2009, 27, 4709–4717. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Li, L.; He, D.; Xie, H.; Chen, J.; Yeh, C.-R.; Chang, L.S.-S.; Yeh, S.; Chang, C. Infiltrating Neutrophils Promote Renal Cell Carcinoma (RCC) Proliferation via Modulating Androgen Receptor (AR) → c-Myc Signals. Cancer Lett. 2015, 368, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-Derived Suppressor Cells in the Era of Increasing Myeloid Cell Diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I Production in the Tumor Microenvironment by Mature Myeloid Cells Inhibits T-Cell Receptor Expression and Antigen-Specific T-Cell Responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef] [Green Version]
- Raber, P.L.; Thevenot, P.; Sierra, R.; Wyczechowska, D.; Halle, D.; Ramirez, M.E.; Ochoa, A.C.; Fletcher, M.; Velasco, C.; Wilk, A.; et al. Subpopulations of Myeloid-Derived Suppressor Cells Impair T Cell Responses through Independent Nitric Oxide-Related Pathways. Int. J. Cancer 2014, 134, 2853–2864. [Google Scholar] [CrossRef]
- Yu, J.; Du, W.; Yan, F.; Wang, Y.; Li, H.; Cao, S.; Yu, W.; Shen, C.; Liu, J.; Ren, X. Myeloid-Derived Suppressor Cells Suppress Antitumor Immune Responses through IDO Expression and Correlate with Lymph Node Metastasis in Patients with Breast Cancer. J. Immunol. 2013, 190, 3783–3797. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ding, Y.; Guo, N.; Wang, S. MDSCs: Key Criminals of Tumor Pre-Metastatic Niche Formation. Front. Immunol. 2019, 10, 172. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Wu, J.; Ding, C.-K.; Lu, M.; Keenan, M.M.; Lin, C.-C.; Lin, C.-A.; Wang, C.C.; George, D.; Hsu, D.S.; et al. Cystine Deprivation Triggers Programmed Necrosis in VHL-Deficient Renal Cell Carcinomas. Cancer Res. 2016, 76, 1892–1903. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory Effect of Tumor Cell-Derived Lactic Acid on Human T Cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef] [Green Version]
- Watson, M.J.; Vignali, P.D.A.; Mullett, S.J.; Overacre-Delgoffe, A.E.; Peralta, R.M.; Grebinoski, S.; Menk, A.V.; Rittenhouse, N.L.; DePeaux, K.; Whetstone, R.D.; et al. Metabolic Support of Tumour-Infiltrating Regulatory T Cells by Lactic Acid. Nature 2021, 591, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.N.; Rathmell, J.C. Metabolic Programming and Immune Suppression in the Tumor Microenvironment. Cancer Cell. 2023, 41, 421–433. [Google Scholar] [CrossRef]
- Gouirand, V.; Guillaumond, F.; Vasseur, S. Influence of the Tumor Microenvironment on Cancer Cells Metabolic Reprogramming. Front. Oncol. 2018, 8, 117. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Zhao, Y.; Lopez, J.I.; Rowan, A.; Au, L.; Fendler, A.; Hazell, S.; Xu, H.; Horswell, S.; Shepherd, S.T.C.; et al. Spatial Patterns of Tumour Growth Impact Clonal Diversification in a Computational Model and the TRACERx Renal Study. Nat. Ecol. Evol. 2022, 6, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Vavallo, A.; Simone, S.; Lucarelli, G.; Rutigliano, M.; Galleggiante, V.; Grandaliano, G.; Gesualdo, L.; Campagna, M.; Cariello, M.; Ranieri, E.; et al. Pre-Existing Type 2 Diabetes Mellitus Is an Independent Risk Factor for Mortality and Progression in Patients with Renal Cell Carcinoma. Medicine 2014, 93, e183. [Google Scholar] [CrossRef]
- Bao, X.; Zhang, J.; Huang, G.; Yan, J.; Xu, C.; Dou, Z.; Sun, C.; Zhang, H. The Crosstalk between HIFs and Mitochondrial Dysfunctions in Cancer Development. Cell. Death Dis. 2021, 12, 215. [Google Scholar] [CrossRef] [PubMed]
- Ferro, F.; Servais, S.; Besson, P.; Roger, S.; Dumas, J.-F.; Brisson, L. Autophagy and Mitophagy in Cancer Metabolic Remodelling. Semin. Cell. Dev. Biol. 2020, 98, 129–138. [Google Scholar] [CrossRef]
- Loizzo, D.; Pandolfo, S.D.; Rogers, D.; Cerrato, C.; Di Meo, N.A.; Autorino, R.; Mirone, V.; Ferro, M.; Porta, C.; Stella, A.; et al. Novel Insights into Autophagy and Prostate Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2022, 23, 3826. [Google Scholar] [CrossRef]
- Grossi, V.; Lucarelli, G.; Forte, G.; Peserico, A.; Matrone, A.; Germani, A.; Rutigliano, M.; Stella, A.; Bagnulo, R.; Loconte, D.; et al. Loss of STK11 Expression Is an Early Event in Prostate Carcinogenesis and Predicts Therapeutic Response to Targeted Therapy against MAPK/P38. Autophagy 2015, 11, 2102–2113. [Google Scholar] [CrossRef] [Green Version]
- Mazzone, M.; Menga, A.; Castegna, A. Metabolism and TAM Functions-It Takes Two to Tango. FEBS J. 2018, 285, 700–716. [Google Scholar] [CrossRef] [Green Version]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-Tumor Activity. Cell. 2016, 167, 829–842.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Reyes, I.; Chandel, N.S. Cancer Metabolism: Looking Forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, G.; Rutigliano, M.; Ferro, M.; Giglio, A.; Intini, A.; Triggiano, F.; Palazzo, S.; Gigante, M.; Castellano, G.; Ranieri, E.; et al. Activation of the Kynurenine Pathway Predicts Poor Outcome in Patients with Clear Cell Renal Cell Carcinoma. Urol. Oncol. Semin. Orig. Investig. 2017, 35, 461.e15–461.e27. [Google Scholar] [CrossRef]
- Riesenberg, R.; Weiler, C.; Spring, O.; Eder, M.; Buchner, A.; Popp, T.; Castro, M.; Kammerer, R.; Takikawa, O.; Hatz, R.A.; et al. Expression of Indoleamine 2,3-Dioxygenase in Tumor Endothelial Cells Correlates with Long-Term Survival of Patients with Renal Cell Carcinoma. Clin. Cancer Res. 2007, 13, 6993–7002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.-B.; Zhu, S.-P.; Liu, T.-P.; Zhao, H.; Chen, P.-F.; Duan, Y.-J.; Hu, R. Cancer Associated Fibroblasts Promote Renal Cancer Progression Through a TDO/Kyn/AhR Dependent Signaling Pathway. Front. Oncol. 2021, 11, 628821. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Quintana, F.J. The Aryl Hydrocarbon Receptor: An Environmental Sensor Integrating Immune Responses in Health and Disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liang, X.; Dong, W.; Fang, Y.; Lv, J.; Zhang, T.; Fiskesund, R.; Xie, J.; Liu, J.; Yin, X.; et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8+ T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell. 2018, 33, 480–494.e7. [Google Scholar] [CrossRef] [Green Version]
- Divella, C.; Stasi, A.; Franzin, R.; Rossini, M.; Pontrelli, P.; Sallustio, F.; Netti, G.S.; Ranieri, E.; Lacitignola, L.; Staffieri, F.; et al. Pentraxin-3-Mediated Complement Activation in a Swine Model of Renal Ischemia/Reperfusion Injury. Aging 2021, 13, 10920–10933. [Google Scholar] [CrossRef]
- Netti, G.S.; Lucarelli, G.; Spadaccino, F.; Castellano, G.; Gigante, M.; Divella, C.; Rocchetti, M.T.; Rascio, F.; Mancini, V.; Stallone, G.; et al. PTX3 Modulates the Immunoflogosis in Tumor Microenvironment and Is a Prognostic Factor for Patients with Clear Cell Renal Cell Carcinoma. Aging 2020, 12, 7585–7602. [Google Scholar] [CrossRef]
- Lucarelli, G.; Netti, G.S.; Rutigliano, M.; Lasorsa, F.; Loizzo, D.; Milella, M.; Schirinzi, A.; Fontana, A.; Di Serio, F.; Tamma, R.; et al. MUC1 Expression Affects the Immunoflogosis in Renal Cell Carcinoma Microenvironment through Complement System Activation and Immune Infiltrate Modulation. Int. J. Mol. Sci. 2023, 24, 4814. [Google Scholar] [CrossRef]
- Hanahan, D.; Folkman, J. Patterns and Emerging Mechanisms of the Angiogenic Switch during Tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasherman, L.; Siu, D.H.W.; Woodford, R.; Harris, C.A. Angiogenesis Inhibitors and Immunomodulation in Renal Cell Cancers: The Past, Present, and Future. Cancers 2022, 14, 1406. [Google Scholar] [CrossRef] [PubMed]
- Presta, L.G.; Chen, H.; O’Connor, S.J.; Chisholm, V.; Meng, Y.G.; Krummen, L.; Winkler, M.; Ferrara, N. Humanization of an Anti-Vascular Endothelial Growth Factor Monoclonal Antibody for the Therapy of Solid Tumors and Other Disorders. Cancer Res. 1997, 57, 4593–4599. [Google Scholar] [PubMed]
- Di Lorenzo, G.; De Placido, S.; Pagliuca, M.; Ferro, M.; Lucarelli, G.; Rossetti, S.; Bosso, D.; Puglia, L.; Pignataro, P.; Ascione, I.; et al. The Evolving Role of Monoclonal Antibodies in the Treatment of Patients with Advanced Renal Cell Carcinoma: A Systematic Review. Expert. Opin. Biol. Ther. 2016, 16, 1387–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.C.; Haworth, L.; Sherry, R.M.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Steinberg, S.M.; Chen, H.X.; Rosenberg, S.A. A Randomized Trial of Bevacizumab, an Anti-Vascular Endothelial Growth Factor Antibody, for Metastatic Renal Cancer. N. Engl. J. Med. 2003, 349, 427–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, M.; Porta, C. Sunitinib in the Treatment of Renal Cell Carcinoma: An Update on Recent Evidence. Ther. Adv. Urol. 2017, 9, 195–207. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Oudard, S.; Negrier, S.; Szczylik, C.; Pili, R.; Bjarnason, G.A.; et al. Overall Survival and Updated Results for Sunitinib Compared with Interferon Alfa in Patients with Metastatic Renal Cell Carcinoma. J. Clin. Oncol. 2009, 27, 3584–3590. [Google Scholar] [CrossRef]
- Verheijen, R.B.; Beijnen, J.H.; Schellens, J.H.M.; Huitema, A.D.R.; Steeghs, N. Clinical Pharmacokinetics and Pharmacodynamics of Pazopanib: Towards Optimized Dosing. Clin. Pharmacokinet. 2017, 56, 987–997. [Google Scholar] [CrossRef] [Green Version]
- Sternberg, C.N.; Davis, I.D.; Mardiak, J.; Szczylik, C.; Lee, E.; Wagstaff, J.; Barrios, C.H.; Salman, P.; Gladkov, O.A.; Kavina, A.; et al. Pazopanib in Locally Advanced or Metastatic Renal Cell Carcinoma: Results of a Randomized Phase III Trial. J. Clin. Oncol. 2023, 41, 1957–1964. [Google Scholar] [CrossRef]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Staehler, M.; Negrier, S.; Chevreau, C.; Desai, A.A.; Rolland, F.; et al. Sorafenib for Treatment of Renal Cell Carcinoma: Final Efficacy and Safety Results of the Phase III Treatment Approaches in Renal Cancer Global Evaluation Trial. J. Clin. Oncol. 2009, 27, 3312–3318. [Google Scholar] [CrossRef]
- Chen, Y.; Tortorici, M.A.; Garrett, M.; Hee, B.; Klamerus, K.J.; Pithavala, Y.K. Clinical Pharmacology of Axitinib. Clin. Pharmacokinet. 2013, 52, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Bukowski, R.M. Axitinib Treatment in Patients with Cytokine-Refractory Metastatic Renal Cell Cancer. Curr. Oncol. Rep. 2009, 11, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Lacy, S.A.; Miles, D.R.; Nguyen, L.T. Clinical Pharmacokinetics and Pharmacodynamics of Cabozantinib. Clin. Pharmacokinet. 2017, 56, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Halabi, S.; Sanford, B.L.; Hahn, O.; Michaelson, M.D.; Walsh, M.K.; Feldman, D.R.; Olencki, T.; Picus, J.; Small, E.J.; et al. Cabozantinib versus Sunitinib As Initial Targeted Therapy for Patients with Metastatic Renal Cell Carcinoma of Poor or Intermediate Risk: The Alliance A031203 CABOSUN Trial. JCO 2017, 35, 591–597. [Google Scholar] [CrossRef]
- Voss, M.H.; Molina, A.M.; Motzer, R.J. MTOR Inhibitors in Advanced Renal Cell Carcinoma. Hematol. Oncol. Clin. N. Am. 2011, 25, 835–852. [Google Scholar] [CrossRef] [Green Version]
- Tannir, N.M.; Msaouel, P.; Ross, J.A.; Devine, C.E.; Chandramohan, A.; Gonzalez, G.M.N.; Wang, X.; Wang, J.; Corn, P.G.; Lim, Z.D.; et al. Temsirolimus versus Pazopanib (TemPa) in Patients with Advanced Clear-Cell Renal Cell Carcinoma and Poor-Risk Features: A Randomized Phase II Trial. Eur. Urol. Oncol. 2020, 3, 687–694. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grünwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N.; et al. Phase 3 Trial of Everolimus for Metastatic Renal Cell Carcinoma: Final Results and Analysis of Prognostic Factors. Cancer 2010, 116, 4256–4265. [Google Scholar] [CrossRef]
- Yang, J.C.; Hughes, M.; Kammula, U.; Royal, R.; Sherry, R.M.; Topalian, S.L.; Suri, K.B.; Levy, C.; Allen, T.; Mavroukakis, S.; et al. Ipilimumab (Anti-CTLA4 Antibody) Causes Regression of Metastatic Renal Cell Cancer Associated with Enteritis and Hypophysitis. J. Immunother. 2007, 30, 825–830. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Escudier, B.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Plimack, E.R.; Procopio, G.; McDermott, D.F.; et al. Nivolumab versus Everolimus in Patients with Advanced Renal Cell Carcinoma: Updated Results with Long-Term Follow-up of the Randomized, Open-Label, Phase 3 CheckMate 025 Trial. Cancer 2020, 126, 4156–4167. [Google Scholar] [CrossRef]
- Jang, A.; Sweeney, P.L.; Barata, P.C.; Koshkin, V.S. PD-L1 Expression and Treatment Implications in Metastatic Clear Cell Renal Cell Carcinoma: A Systematic Review. KCA 2021, 5, 31–46. [Google Scholar] [CrossRef]
- Yasuda, S.; Sho, M.; Yamato, I.; Yoshiji, H.; Wakatsuki, K.; Nishiwada, S.; Yagita, H.; Nakajima, Y. Simultaneous Blockade of Programmed Death 1 and Vascular Endothelial Growth Factor Receptor 2 (VEGFR2) Induces Synergistic Anti-Tumour Effect In Vivo. Clin. Exp. Immunol. 2013, 172, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Powles, T.; Atkins, M.B.; Escudier, B.; McDermott, D.F.; Suarez, C.; Bracarda, S.; Stadler, W.M.; Donskov, F.; Lee, J.L.; et al. Atezolizumab plus Bevacizumab versus Sunitinib in Patients with Previously Untreated Metastatic Renal Cell Carcinoma (IMmotion151): A Multicentre, Open-Label, Phase 3, Randomised Controlled Trial. Lancet 2019, 393, 2404–2415. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef]
- Motzer, R.; Alekseev, B.; Rha, S.-Y.; Porta, C.; Eto, M.; Powles, T.; Grünwald, V.; Hutson, T.E.; Kopyltsov, E.; Méndez-Vidal, M.J.; et al. Lenvatinib plus Pembrolizumab or Everolimus for Advanced Renal Cell Carcinoma. N. Engl. J. Med. 2021, 384, 1289–1300. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Powles, T.; Burotto, M.; Escudier, B.; Bourlon, M.T.; Zurawski, B.; Oyervides Juárez, V.M.; Hsieh, J.J.; Basso, U.; Shah, A.Y.; et al. Nivolumab plus Cabozantinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2021, 384, 829–841. [Google Scholar] [CrossRef]
- Niewada, M.; Macioch, T.; Konarska, M.; Mela, A.; Goszczyński, A.; Przekopińska, B.; Rajkiewicz, K.; Wysocki, P.; Krzakowski, M. Immune Checkpoint Inhibitors Combined with Tyrosine Kinase Inhibitors or Immunotherapy for Treatment-Naïve Metastatic Clear-Cell Renal Cell Carcinoma—A Network Meta-Analysis. Focus on Cabozantinib Combined with Nivolumab. Front. Pharmacol. 2023, 13, 1063178. [Google Scholar] [CrossRef]
- Jonasch, E.; Donskov, F.; Iliopoulos, O.; Rathmell, W.K.; Narayan, V.K.; Maughan, B.L.; Oudard, S.; Else, T.; Maranchie, J.K.; Welsh, S.J.; et al. Belzutifan for Renal Cell Carcinoma in von Hippel-Lindau Disease. N. Engl. J. Med. 2021, 385, 2036–2046. [Google Scholar] [CrossRef]


{kind=link}
| Trial | Drugs | Primary Endpoint | |
|---|---|---|---|
| NCT02420821 | Atezolizumab + Bevacizumab | PFS | [132] |
| NCT02853331 | Pembrolizumab + Axitinib | PFS, OS | [133] |
| NCT02684006 | Avelumab + Axitinib | PFS, OS | [134] |
| NCT02811861 | Pembrolizumab + Lenvatinib | PFS | [135] |
| NCT03141177 | Nivolumab + Cabozantinib | PFS | [136] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lasorsa, F.; Rutigliano, M.; Milella, M.; Ferro, M.; Pandolfo, S.D.; Crocetto, F.; Tataru, O.S.; Autorino, R.; Battaglia, M.; Ditonno, P.; et al. Cellular and Molecular Players in the Tumor Microenvironment of Renal Cell Carcinoma. J. Clin. Med. 2023, 12, 3888. https://doi.org/10.3390/jcm12123888
Lasorsa F, Rutigliano M, Milella M, Ferro M, Pandolfo SD, Crocetto F, Tataru OS, Autorino R, Battaglia M, Ditonno P, et al. Cellular and Molecular Players in the Tumor Microenvironment of Renal Cell Carcinoma. Journal of Clinical Medicine. 2023; 12(12):3888. https://doi.org/10.3390/jcm12123888
Chicago/Turabian StyleLasorsa, Francesco, Monica Rutigliano, Martina Milella, Matteo Ferro, Savio Domenico Pandolfo, Felice Crocetto, Octavian Sabin Tataru, Riccardo Autorino, Michele Battaglia, Pasquale Ditonno, and et al. 2023. "Cellular and Molecular Players in the Tumor Microenvironment of Renal Cell Carcinoma" Journal of Clinical Medicine 12, no. 12: 3888. https://doi.org/10.3390/jcm12123888