
Levodopa-Induced Dyskinesias in Parkinson’s Disease: An Overview on Pathophysiology, Clinical Manifestations, Therapy Management Strategies and Future Directions

 ,
,  ,
,
Abstract
:1. Introduction
2. Levodopa-Induced Dyskinesias
2.1. Clinical Risk Factors
2.2. Genetic Risk Factors
2.3. Epidemiology
2.4. Clinical Manifestations
2.5. Objective LID Monitoring
2.6. Pathophysiology
Levodopa Pharmacokinetics and Pharmacodynamics
2.7. Neurophysiology
2.8. Neurotransmitter Systems
2.8.1. Serotonergic System
2.8.2. Glutamatergic System
2.8.3. Noradrenergic System
2.8.4. Cholinergic System
2.8.5. Opioid System
2.8.6. Endocannabinoid System
2.8.7. Adenosinergic System
2.9. Imaging Studies
3. Therapeutic Options
3.1. Levodopa Therapy Optimization
- -
- Reducing the dose of levodopa and distributing the inter-dose timing.
- -
- Adding an add-on medication, such as Amantadine, can help to reduce the severity of dyskinesias.
- -
- -
- Adjusting the timing of medication doses: spreading out the doses throughout the day can help to maintain more stable medication levels.
- -
- Adding an add-on medication, such as Amantadine, can help reduce the severity of dyskinesias.
- -
- Adjusting the timing and dosage of medication or increasing the dose of levodopa can help maintain more stable medication levels.
- -
- Adding an add-on medication, such as an extended-release dopamine agonist, MAO-B inhibitor or COMT inhibitor, can help to reduce the severity of OFF dystonia by potentiating the dopaminergic stimulation.
- -
- Apomorphine injections or sublingual administration [281] can provide rapid relief from OFF dystonia.
3.2. Non-Dopaminergic Drugs
3.3. Deep Brain Stimulation
3.4. Closed-Loop Therapy
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ehringer, H.; Hornykiewicz, O. Verteilung von Noradrenalin und Dopamin (3-Hydroxytyramin) im Gehirn des Menschen und ihr Verhalten bei Erkrankungen des extrapyramidalen Systems. Klin. Wochenschr. 1960, 38, 1236–1239. [Google Scholar] [CrossRef]
- Cotzias, G.C.; Van Woert, M.H.; Schiffer, L.M. Aromatic amino acids and modification of parkinsonism. N. Engl. J. Med. 1967, 276, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Connolly, B.S.; Lang, A.E. Pharmacological treatment of Parkinson disease: A review. JAMA 2014, 311, 1670–1683. [Google Scholar] [CrossRef] [PubMed]
- Hechtner, M.C.; Vogt, T.; Zollner, Y.; Schroder, S.; Sauer, J.B.; Binder, H.; Singer, S.; Mikolajczyk, R. Quality of life in Parkinson’s disease patients with motor fluctuations and dyskinesias in five European countries. Park. Relat. Disord. 2014, 20, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Cotzias, G.C.; Papavasiliou, P.S.; Gellene, R. Modification of Parkinsonism—Chronic treatment with L-dopa. N. Engl. J. Med. 1969, 280, 337–345. [Google Scholar] [CrossRef]
- Warren Olanow, C.; Kieburtz, K.; Rascol, O.; Poewe, W.; Schapira, A.H.; Emre, M.; Nissinen, H.; Leinonen, M.; Stocchi, F. Factors predictive of the development of Levodopa-induced dyskinesia and wearing-off in Parkinson’s disease. Mov. Disord. 2013, 28, 1064–1071. [Google Scholar] [CrossRef]
- Schrag, A.; Quinn, N. Dyskinesias and motor fluctuations in Parkinson’s disease. A community-based study. Brain A J. Neurol. 2000, 123 Pt 11, 2297–2305. [Google Scholar] [CrossRef] [Green Version]
- Colosimo, C.; De Michele, M. Motor fluctuations in Parkinson’s disease: Pathophysiology and treatment. Eur. J. Neurol. 1999, 6, 1–21. [Google Scholar] [CrossRef]
- Fabbrini, G.; Mouradian, M.M.; Juncos, J.L.; Schlegel, J.; Mohr, E.; Chase, T.N. Motor fluctuations in Parkinson’s disease: Central pathophysiological mechanisms, Part I. Ann. Neurol. 1988, 24, 366–371. [Google Scholar] [CrossRef]
- Mouradian, M.M.; Juncos, J.L.; Fabbrini, G.; Schlegel, J.; Bartko, J.J.; Chase, T.N. Motor fluctuations in Parkinson’s disease: Central pathophysiological mechanisms, Part II. Ann. Neurol. 1988, 24, 372–378. [Google Scholar] [CrossRef]
- Assenza, G.; Capone, F.; di Biase, L.; Ferreri, F.; Florio, L.; Guerra, A.; Marano, M.; Paolucci, M.; Ranieri, F.; Salomone, G. Oscillatory activities in neurological disorders of elderly: Biomarkers to target for neuromodulation. Front. Aging Neurosci. 2017, 9, 189, Corrigendum in Front. Aging Neurosci. 2017, 9, 252. [Google Scholar] [CrossRef] [PubMed]
- Swann, N.C.; de Hemptinne, C.; Miocinovic, S.; Qasim, S.; Wang, S.S.; Ziman, N.; Ostrem, J.L.; San Luciano, M.; Galifianakis, N.B.; Starr, P.A. Gamma oscillations in the hyperkinetic state detected with chronic human brain recordings in Parkinson’s disease. J. Neurosci. 2016, 36, 6445–6458. [Google Scholar] [CrossRef] [PubMed]
- Halje, P.; Tamte, M.; Richter, U.; Mohammed, M.; Cenci, M.A.; Petersson, P. Levodopa-induced dyskinesia is strongly associated with resonant cortical oscillations. J. Neurosci. 2012, 32, 16541–16551. [Google Scholar] [CrossRef] [Green Version]
- Güttler, C.; Altschüler, J.; Tanev, K.; Böckmann, S.; Haumesser, J.K.; Nikulin, V.V.; Kühn, A.A.; van Riesen, C. Levodopa-induced dyskinesia are mediated by cortical gamma oscillations in experimental Parkinsonism. Mov. Disord. 2021, 36, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, M.; Mazzone, P.; Oliviero, A.; Insola, A.; Tonali, P.; Lazzaro, V.D.; Brown, P. Movement-related changes in synchronization in the human basal ganglia. Brain 2002, 125, 1235–1246. [Google Scholar] [CrossRef]
- Alonso-Frech, F.; Zamarbide, I.; Alegre, M.; Rodriguez-Oroz, M.C.; Guridi, J.; Manrique, M.; Valencia, M.; Artieda, J.; Obeso, J.A. Slow oscillatory activity and levodopa-induced dyskinesias in Parkinson’s disease. Brain 2006, 129, 1748–1757. [Google Scholar] [CrossRef] [Green Version]
- Alegre, M.; López-Azcárate, J.; Alonso-Frech, F.; Rodríguez-Oroz, M.C.; Valencia, M.; Guridi, J.; Artieda, J.; Obeso, J.A. Subthalamic activity during diphasic dyskinesias in Parkinson’s disease. Mov. Disord. 2012, 27, 1178–1181. [Google Scholar] [CrossRef]
- Cagnan, H.; Kuhn, A.A.; Brown, P. Co-modulation of finely tuned high-gamma band activity across hemispheres in Parkinson’s disease. Clin. Neurophysiol. 2014, 125, 777–785. [Google Scholar] [CrossRef] [Green Version]
- Wiest, C.; Torrecillos, F.; Tinkhauser, G.; Pogosyan, A.; Morgante, F.; Pereira, E.; Tan, H. Finely-tuned gamma oscillations: Spectral characteristics and links to dyskinesia. Exp. Neurol. 2022, 351, 113999. [Google Scholar] [CrossRef]
- Antonini, A.; Stoessl, A.J.; Kleinman, L.S.; Skalicky, A.M.; Marshall, T.S.; Sail, K.R.; Onuk, K.; Odin, P.L.A. Developing consensus among movement disorder specialists on clinical indicators for identification and management of advanced Parkinson’s disease: A multi-country Delphi-panel approach. Curr. Med. Res. Opin. 2018, 34, 2063–2073. [Google Scholar] [CrossRef]
- Group, P.S. Levodopa and the progression of Parkinson’s disease. New. Engl. J. Med. 2004, 351, 2498–2508. [Google Scholar]
- Oertel, W.H.; Wolters, E.; Sampaio, C.; Gimenez-Roldan, S.; Bergamasco, B.; Dujardin, M.; Grosset, D.G.; Arnold, G.; Leenders, K.L.; Hundemer, H.P.; et al. Pergolide versus levodopa monotherapy in early Parkinson’s disease patients: The PELMOPET study. Mov. Disord. 2006, 21, 343–353. [Google Scholar] [CrossRef]
- Rascol, O.; Brooks, D.J.; Korczyn, A.D.; De Deyn, P.P.; Clarke, C.E.; Lang, A.E. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. N. Engl. J. Med. 2000, 342, 1484–1491. [Google Scholar] [CrossRef] [PubMed]
- Hauser, R.A.; Rascol, O.; Korczyn, A.D.; Jon Stoessl, A.; Watts, R.L.; Poewe, W.; De Deyn, P.P.; Lang, A.E. Ten-year follow-up of Parkinson’s disease patients randomized to initial therapy with ropinirole or levodopa. Mov. Disord. 2007, 22, 2409–2417. [Google Scholar] [CrossRef] [PubMed]
- Holloway, R.G.; Shoulson, I.; Fahn, S.; Kieburtz, K.; Lang, A.; Marek, K.; McDermott, M.; Seibyl, J.; Weiner, W.; Musch, B.; et al. Pramipexole vs levodopa as initial treatment for Parkinson disease: A 4-year randomized controlled trial. Arch. Neurol. 2004, 61, 1044–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, J.; Krafzcyk, S.; Valkovic, P.; Eggert, T.; Claassen, J.; Botzel, K. Objective measurement of muscle rigidity in Parkinsonian patients treated with subthalamic stimulation. Mov. Disord. 2009, 24, 57–63. [Google Scholar] [CrossRef]
- Lees, A.J.; Katzenschlager, R.; Head, J.; Ben-Shlomo, Y. Ten-year follow-up of three different initial treatments in de-novo PD: A randomized trial. Neurology 2001, 57, 1687–1694. [Google Scholar] [CrossRef]
- Stocchi, F.; Rascol, O.; Kieburtz, K.; Poewe, W.; Jankovic, J.; Tolosa, E.; Barone, P.; Lang, A.E.; Olanow, C.W. Initiating levodopa/carbidopa therapy with and without entacapone in early Parkinson disease: The STRIDE-PD study. Ann. Neurol. 2010, 68, 18–27. [Google Scholar] [CrossRef]
- Shoulson, I.; Oakes, D.; Fahn, S.; Lang, A.; Langston, J.W.; LeWitt, P.; Olanow, C.W.; Penney, J.B.; Tanner, C.; Kieburtz, K.; et al. Impact of sustained deprenyl (selegiline) in levodopa-treated Parkinson’s disease: A randomized placebo-controlled extension of the deprenyl and tocopherol antioxidative therapy of parkinsonism trial. Ann. Neurol. 2002, 51, 604–612. [Google Scholar] [CrossRef]
- de Lau, L.M.; Verbaan, D.; Marinus, J.; Heutink, P.; van Hilten, J.J. Catechol-O-methyltransferase Val158Met and the risk of dyskinesias in Parkinson’s disease. Mov. Disord. 2012, 27, 132–135. [Google Scholar] [CrossRef]
- Kaiser, R.; Hofer, A.; Grapengiesser, A.; Gasser, T.; Kupsch, A.; Roots, I.; Brockmöller, J. L -dopa-induced adverse effects in PD and dopamine transporter gene polymorphism. Neurology 2003, 60, 1750–1755. [Google Scholar] [CrossRef] [PubMed]
- Rieck, M.; Schumacher-Schuh, A.F.; Altmann, V.; Francisconi, C.L.; Fagundes, P.T.; Monte, T.L.; Callegari-Jacques, S.M.; Rieder, C.R.; Hutz, M.H. DRD2 haplotype is associated with dyskinesia induced by levodopa therapy in Parkinson’s disease patients. Pharmacogenomics 2012, 13, 1701–1710. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, S.A.; Loonen, A.J.; Pechlivanoglou, P.; Freidin, M.B.; Al Hadithy, A.F.; Rudikov, E.V.; Zhukova, I.A.; Govorin, N.V.; Sorokina, V.A.; Fedorenko, O.Y.; et al. NMDA receptor genotypes associated with the vulnerability to develop dyskinesia. Transl. Psychiatry 2012, 2, e67. [Google Scholar] [CrossRef] [Green Version]
- Foltynie, T.; Cheeran, B.; Williams-Gray, C.H.; Edwards, M.J.; Schneider, S.A.; Weinberger, D.; Rothwell, J.C.; Barker, R.A.; Bhatia, K.P. BDNF val66met influences time to onset of levodopa induced dyskinesia in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2009, 80, 141–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zappia, M.; Annesi, G.; Nicoletti, G.; Arabia, G.; Annesi, F.; Messina, D.; Pugliese, P.; Spadafora, P.; Tarantino, P.; Carrideo, S.; et al. Sex differences in clinical and genetic determinants of levodopa peak-dose dyskinesias in Parkinson disease: An exploratory study. Arch. Neurol. 2005, 62, 601–605. [Google Scholar] [CrossRef] [Green Version]
- Strong, J.A.; Dalvi, A.; Revilla, F.J.; Sahay, A.; Samaha, F.J.; Welge, J.A.; Gong, J.; Gartner, M.; Yue, X.; Yu, L. Genotype and smoking history affect risk of levodopa-induced dyskinesias in Parkinson’s disease. Mov. Disord. 2006, 21, 654–659. [Google Scholar] [CrossRef]
- Falla, M.; Di Fonzo, A.; Hicks, A.A.; Pramstaller, P.P.; Fabbrini, G. Genetic variants in levodopa-induced dyskinesia (LID): A systematic review and meta-analysis. Park. Relat. Disord. 2021, 84, 52–60. [Google Scholar] [CrossRef]
- Duvoisin, R.C. Variations in the ”on-off“ phenomenon. Adv. Neurol. 1974, 5, 339–340. [Google Scholar]
- Rajput, A.H.; Fenton, M.E.; Birdi, S.; Macaulay, R.; George, D.; Rozdilsky, B.; Ang, L.C.; Senthilselvan, A.; Hornykiewicz, O. Clinical–pathological study of levodopa complications. Mov. Disord. 2002, 17, 289–296. [Google Scholar] [CrossRef]
- Van Gerpen, J.A.; Kumar, N.; Bower, J.H.; Weigand, S.; Ahlskog, J.E. Levodopa-associated dyskinesia risk among Parkinson disease patients in Olmsted County, Minnesota, 1976-1990. Arch. Neurol. 2006, 63, 205–209. [Google Scholar] [CrossRef] [Green Version]
- Ahlskog, J.E.; Muenter, M.D. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov. Disord. Off J. Mov. Disord. Soc. 2001, 16, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Group, P.S. Impact of deprenyl and tocopherol treatment on Parkinson’s disease in DATATOP patients requiring levodopa. Ann. Neurol. 1996, 39, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Block, G.; Liss, C.; Reines, S.; Irr, J.; Nibbelink, D. Comparison of immediate-release and controlled release carbidopa/levodopa in Parkinson’s disease. Eur. Neurol. 1997, 37, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Group, P.S.; Group, P.S. Pramipexole vs levodopa as initial treatment for Parkinson disease: A randomized controlled trial. JAMA 2000, 284, 1931–1938. [Google Scholar] [CrossRef] [Green Version]
- Hely, M.A.; Morris, J.G.; Reid, W.G.; Trafficante, R. Sydney multicenter study of Parkinson’s disease: Non-L-dopa–responsive problems dominate at 15 years. Mov. Disord. Off J. Mov. Disord. Soc. 2005, 20, 190–199. [Google Scholar] [CrossRef]
- Zesiewicz, T.A.; Sullivan, K.L.; Hauser, R.A. Levodopa-induced dyskinesia in Parkinson’s disease: Epidemiology, etiology, and treatment. Curr. Neurol. Neurosci. Rep. 2007, 7, 302–310. [Google Scholar] [CrossRef]
- Fox, S.H.; Brotchie, J.M. Levodopa-Induced Dyskinesia in Parkinson’s Disease; Springer: London, UK, 2014. [Google Scholar] [CrossRef]
- Hametner, E.; Seppi, K.; Poewe, W. The clinical spectrum of levodopa-induced motor complications. J. Neurol. 2010, 257 (Suppl. 2), S268–S275. [Google Scholar] [CrossRef]
- Obeso, J.A.; Grandas, F.; Vaamonde, J.; Luquin, M.R.; Artieda, J.; Lera, G.; Rodriguez, M.E.; Martinez-Lage, J.M. Motor complications associated with chronic levodopa therapy in Parkinson’s disease. Neurology 1989, 39 (Suppl. 2), 11–19. [Google Scholar]
- Meloni, M.; Solla, P.; Mascia, M.M.; Marrosu, F.; Cannas, A. Diphasic dyskinesias during levodopa-carbidopa intestinal gel (LCIG) infusion in Parkinson’s disease. Park. Relat. Disord. 2017, 37, 92–96. [Google Scholar] [CrossRef]
- Voon, V.; Fernagut, P.-O.; Wickens, J.; Baunez, C.; Rodriguez, M.; Pavon, N.; Juncos, J.L.; Obeso, J.A.; Bezard, E. Chronic dopaminergic stimulation in Parkinson’s disease: From dyskinesias to impulse control disorders. Lancet Neurol. 2009, 8, 1140–1149. [Google Scholar] [CrossRef]
- Olanow, C.W.; Calabresi, P.; Obeso, J.A. Continuous Dopaminergic Stimulation as a Treatment for Parkinson’s Disease: Current Status and Future Opportunities. Mov. Disord. 2020, 35, 1731–1744. [Google Scholar] [CrossRef] [PubMed]
- di Biase, L.; Di Santo, A.; Caminiti, M.L.; Pecoraro, P.M.; Di Lazzaro, V. Classification of Dystonia. Life 2022, 12, 206. [Google Scholar] [CrossRef]
- di Biase, L.; Di Santo, A.; Caminiti, M.L.; Pecoraro, P.M.; Carbone, S.P.; Di Lazzaro, V. Dystonia Diagnosis: Clinical Neurophysiology and Genetics. J. Clin. Med. 2022, 11, 4184. [Google Scholar] [CrossRef] [PubMed]
- d’Angelis, O.; Di Biase, L.; Vollero, L.; Merone, M. IoT architecture for continuous long term monitoring: Parkinson’s Disease case study. Internet Things 2022, 20, 100614. [Google Scholar] [CrossRef]
- di Biase, L.; Pecoraro, P.M.; Pecoraro, G.; Caminiti, M.L.; Di Lazzaro, V. Markerless radio frequency indoor monitoring for telemedicine: Gait analysis, indoor positioning, fall detection, tremor analysis, vital signs and sleep monitoring. Sensors 2022, 22, 8486. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Ferro, A.; Elshehabi, M.; Godinho, C.; Salkovic, D.; Hobert, M.A.; Domingos, J.; van Uem, J.M.; Ferreira, J.J.; Maetzler, W. New methods for the assessment of Parkinson’s disease (2005 to 2015): A systematic review. Mov. Disord. 2016, 31, 1283–1292. [Google Scholar] [CrossRef] [Green Version]
- Hssayeni, M.D.; Jimenez-Shahed, J.; Burack, M.A.; Ghoraani, B. Dyskinesia estimation during activities of daily living using wearable motion sensors and deep recurrent networks. Sci. Rep. 2021, 11, 7865. [Google Scholar] [CrossRef]
- Lee, S.I.; Daneault, J.-F.; Golabchi, F.N.; Patel, S.; Paganoni, S.; Shih, L.; Bonato, P. A novel method for assessing the severity of levodopa-induced dyskinesia using wearable sensors. In Proceedings of the 2015 37th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), Milano, Italy, 25–29 August 2015; pp. 8087–8090. [Google Scholar]
- Deuschl, G.; Krack, P.; Lauk, M.; Timmer, J. Clinical neurophysiology of tremor. J. Clin. Neurophysiol. 1996, 13, 110–121. [Google Scholar] [CrossRef]
- Di Pino, G.; Formica, D.; Melgari, J.-M.; Taffoni, F.; Salomone, G.; di Biase, L.; Caimo, E.; Vernieri, F.; Guglielmelli, E. Neurophysiological bases of tremors and accelerometric parameters analysis. In Proceedings of the 2012 4th IEEE RAS & EMBS International Conference on Biomedical Robotics and Biomechatronics (BioRob), Rome, Italy, 24–27 June 2012; pp. 1820–1825. [Google Scholar] [CrossRef]
- di Biase, L.; Brittain, J.S.; Shah, S.A.; Pedrosa, D.J.; Cagnan, H.; Mathy, A.; Chen, C.C.; Martin-Rodriguez, J.F.; Mir, P.; Timmerman, L.; et al. Tremor stability index: A new tool for differential diagnosis in tremor syndromes. Brain A J. Neurol. 2017, 140, 1977–1986. [Google Scholar] [CrossRef]
- Di Biase, L.; Brittain, J.-S.; Peter, B.; Di Lazzaro, V.; Shah, S.A. Methods and System for Characterising Tremors. U.S. Patent 11, 576, 592 B2, 14 February 2023. [Google Scholar]
- Stamatakis, J.; Ambroise, J.; Cremers, J.; Sharei, H.; Delvaux, V.; Macq, B.; Garraux, G. Finger tapping clinimetric score prediction in Parkinson’s disease using low-cost accelerometers. Comput. Intell. Neurosci. 2013, 2013, 717853. [Google Scholar] [CrossRef]
- Summa, S.; Tosi, J.; Taffoni, F.; Di Biase, L.; Marano, M.; Rizzo, A.C.; Tombini, M.; Di Pino, G.; Formica, D. Assessing bradykinesia in Parkinson’s disease using gyroscope signals. In Proceedings of the Rehabilitation Robotics (ICORR), London, UK, 17–20 July 2017; pp. 1556–1561. [Google Scholar]
- di Biase, L.; Summa, S.; Tosi, J.; Taffoni, F.; Marano, M.; Cascio Rizzo, A.; Vecchio, F.; Formica, D.; Di Lazzaro, V.; Di Pino, G.; et al. Quantitative Analysis of Bradykinesia and Rigidity in Parkinson’s Disease. Front. Neurol. 2018, 9, 121. [Google Scholar] [CrossRef] [Green Version]
- Endo, T.; Okuno, R.; Yokoe, M.; Akazawa, K.; Sakoda, S. A novel method for systematic analysis of rigidity in Parkinson’s disease. Mov. Disord. 2009, 24, 2218–2224. [Google Scholar] [CrossRef]
- Kwon, Y.; Park, S.H.; Kim, J.W.; Ho, Y.; Jeon, H.M.; Bang, M.J.; Koh, S.B.; Kim, J.H.; Eom, G.M. Quantitative evaluation of parkinsonian rigidity during intra-operative deep brain stimulation. Bio-Med. Mater. Eng. 2014, 24, 2273–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raiano, L.; Di Pino, G.; Di Biase, L.; Tombini, M.; Tagliamonte, N.L.; Formica, D. PDMeter: A Wrist Wearable Device for an at-home Assessment of the Parkinson’s Disease Rigidity. IEEE Trans. Neural Syst. Rehabil. Eng. 2020, 28, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.T.; MacDougall, H.G.; Gracies, J.M.; Cohen, H.S.; Ondo, W.G. Long-term monitoring of gait in Parkinson’s disease. Gait Posture 2007, 26, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Schlachetzki, J.C.M.; Barth, J.; Marxreiter, F.; Gossler, J.; Kohl, Z.; Reinfelder, S.; Gassner, H.; Aminian, K.; Eskofier, B.M.; Winkler, J.; et al. Wearable sensors objectively measure gait parameters in Parkinson’s disease. PLoS ONE 2017, 12, e0183989. [Google Scholar] [CrossRef] [PubMed]
- Tosi, J.; Summa, S.; Taffoni, F.; Biase, L.d.; Marano, M.; Rizzo, A.C.; Tombini, M.; Schena, E.; Formica, D.; Pino, G.D. Feature Extraction in Sit-to-Stand Task Using M-IMU Sensors and Evaluatiton in Parkinson’s Disease. In Proceedings of the 2018 IEEE International Symposium on Medical Measurements and Applications (MeMeA), Rome, Italy, 11–13 June 2018; pp. 1–6. [Google Scholar] [CrossRef]
- Suppa, A.; Kita, A.; Leodori, G.; Zampogna, A.; Nicolini, E.; Lorenzi, P.; Rao, R.; Irrera, F. L-DOPA and freezing of gait in Parkinson’s disease: Objective assessment through a wearable wireless system. Front. Neurol. 2017, 8, 406. [Google Scholar] [CrossRef]
- di Biase, L.; Di Santo, A.; Caminiti, M.L.; De Liso, A.; Shah, S.A.; Ricci, L.; Di Lazzaro, V. Gait analysis in Parkinson’s disease: An overview of the most accurate markers for diagnosis and symptoms monitoring. Sensors 2020, 20, 3529. [Google Scholar] [CrossRef]
- di Biase, L.; Raiano, L.; Caminiti, M.L.; Pecoraro, P.M.; Di Lazzaro, V. Parkinson’s Disease Wearable Gait Analysis: Kinematic and Dynamic Markers for Diagnosis. Sensors 2022, 22, 8773. [Google Scholar] [CrossRef] [PubMed]
- Luis-Martínez, R.; Monje, M.H.G.; Antonini, A.; Sánchez-Ferro, Á.; Mestre, T.A. Technology-Enabled Care: Integrating Multidisciplinary Care in Parkinson’s Disease Through Digital Technology. Front. Neurol. 2020, 11, 575975. [Google Scholar] [CrossRef]
- Odin, P.; Chaudhuri, K.R.; Volkmann, J.; Antonini, A.; Storch, A.; Dietrichs, E.; Pirtošek, Z.; Henriksen, T.; Horne, M.; Devos, D.; et al. Viewpoint and practical recommendations from a movement disorder specialist panel on objective measurement in the clinical management of Parkinson’s disease. NPJ Park. Dis. 2018, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Pfister, F.M.; Um, T.T.; Pichler, D.C.; Goschenhofer, J.; Abedinpour, K.; Lang, M.; Endo, S.; Ceballos-Baumann, A.O.; Hirche, S.; Bischl, B. High-resolution motor state detection in Parkinson’s disease using convolutional neural networks. Sci. Rep. 2020, 10, 5860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.H.; Mestre, T.A.; Fox, S.H.; Taati, B. Automated assessment of levodopa-induced dyskinesia: Evaluating the responsiveness of video-based features. Park. Relat. Disord. 2018, 53, 42–45. [Google Scholar] [CrossRef]
- Alam, M.N.; Garg, A.; Munia, T.T.K.; Fazel-Rezai, R.; Tavakolian, K. Vertical ground reaction force marker for Parkinson’s disease. PLoS ONE 2017, 12, e0175951. [Google Scholar] [CrossRef]
- Cavallo, F.; Moschetti, A.; Esposito, D.; Maremmani, C.; Rovini, E. Upper limb motor pre-clinical assessment in Parkinson’s disease using machine learning. Park. Relat. Disord. 2019, 63, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Pan, Z. A novel ensemble of random forest for assisting diagnosis of Parkinson’s disease on small handwritten dynamics dataset. Int. J. Med. Inform. 2020, 144, 104283. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.M.; Hammerla, N.Y.; Ploetz, T.; Andras, P.; Rochester, L.; Walker, R.W. Unsupervised home monitoring of Parkinson’s disease motor symptoms using body-worn accelerometers. Park. Relat. Disord. 2016, 33, 44–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilias, T.; Filip, B.; Radu, C.; Dag, N.; Marina, S.; Mevludin, M. Using measurements from wearable sensors for automatic scoring of Parkinson’s disease motor states: Results from 7 patients. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 2017, 2017, 131–134. [Google Scholar] [CrossRef]
- di Biase, L.; Raiano, L.; Caminiti, M.L.; Pecoraro, P.M.; Di Lazzaroo, V. Artificial intelligence in Parkinson’s disease—Symptoms identification and monitoring. In Augmenting Neurological Disorder Prediction and Rehabilitation Using Artificial Intelligence; Elsevier: Amsterdam, The Netherlands, 2022; pp. 35–52. [Google Scholar]
- di Biase, L.; Tinkhauser, G.; Martin Moraud, E.; Caminiti, M.L.; Pecoraro, P.M.; Di Lazzaro, V. Adaptive, personalized closed-loop therapy for Parkinson’s disease: Biochemical, neurophysiological, and wearable sensing systems. Expert Rev. Neurother. 2021, 21, 1371–1388. [Google Scholar] [CrossRef]
- Contin, M.; Martinelli, P. Pharmacokinetics of levodopa. J. Neurol. 2010, 257 (Suppl. 2), S253–S261. [Google Scholar] [CrossRef]
- Nutt, J.G.; Fellman, J.H. Pharmacokinetics of levodopa. Clin. Neuropharmacol. 1984, 7, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Juncos, J.L.; Fabbrini, G.; Mouradian, M.M.; Serrati, C.; Chase, T.N. Dietary influences on the antiparkinsonian response to levodopa. Arch. Neurol. 1987, 44, 1003–1005. [Google Scholar] [CrossRef]
- Guebila, M.B.; Thiele, I. Model-based dietary optimization for late-stage, levodopa-treated, Parkinson’s disease patients. NPJ Syst. Biol. Appl. 2016, 2, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Contin, M.; Riva, R.; Martinelli, P.; Albani, F.; Baruzzi, A. Relationship between levodopa concentration, dyskinesias, and motor effect in parkinsonian patients: A 3-year follow-up study. Clin. Neuropharmacol. 1997, 20, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Harder, S.; Baas, H. Concentration-response relationship of levodopa in patients at different stages of Parkinson’s disease. Clin. Pharmacol. Ther. 1998, 64, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Contin, M.; Riva, R.; Martinelli, P.; Cortelli, P.; Albani, F.; Baruzzi, A. Pharmacodynamic modeling of oral levodopa: Clinical application in Parkinson’s disease. Neurology 1993, 43, 367. [Google Scholar] [CrossRef]
- Adamiak, U.; Kaldonska, M.; Klodowska-Duda, G.; Wyska, E.; Safranow, K.; Bialecka, M.; Gawronska-Szklarz, B. Pharmacokinetic-pharmacodynamic modeling of levodopa in patients with advanced Parkinson disease. Clin. Neuropharmacol. 2010, 33, 135–141. [Google Scholar] [CrossRef]
- Fabbrini, G.; Juncos, J.; Mouradian, M.; Serrati, C.; Chase, T. Levodopa pharmacokinetic mechanisms and motor fluctuations in Parkinson’s disease. Ann. Neurol. Off J. Am. Neurol. Assoc. Child Neurol. Soc. 1987, 21, 370–376. [Google Scholar] [CrossRef]
- Gancher, S.T.; Nutt, J.G.; Woodward, W.R. Peripheral pharmacokinetics of levodopa in untreated, stable, and fluctuating parkinsonian patients. Neurology 1987, 37, 940. [Google Scholar] [CrossRef]
- Contin, M.; Riva, R.; Martinelli, P.; Triggs, E.; Albani, F.; Baruzzi, A. Rate of motor response to oral levodopa and the clinical progression of Parkinson’s disease. Neurology 1996, 46, 1055–1058. [Google Scholar] [CrossRef]
- Contin, M.; Riva, R.; Martinelli, P.; Cortelli, P.; Albani, F.; Baruzzi, A. Longitudinal monitoring of the levodopa concentration-effect relationship in Parkinson’s disease. Neurology 1994, 44, 1287. [Google Scholar] [CrossRef] [PubMed]
- Contin, M.; Riva, R.; Martinelli, P.; Albani, F.; Avoni, P.; Baruzzi, A. Levodopa therapy monitoring in patients with Parkinson disease: A kinetic-dynamic approach. Ther. Drug Monit. 2001, 23, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Triggs, E.; Charles, B.; Contin, M.; Martinelli, P.; Cortelli, P.; Riva, R.; Albani, F.; Baruzzi, A. Population pharmacokinetics and pharmacodynamics of oral levodopa in parkinsonian patients. Eur. J. Clin. Pharmacol. 1996, 51, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Nyholm, D. Pharmacokinetic optimisation in the treatment of Parkinson’s disease. Clin. Pharmacokinet. 2006, 45, 109–136. [Google Scholar] [CrossRef]
- Nutt, J.G.; Carter, J.H.; Lea, E.S.; Sexton, G.J. Evolution of the response to levodopa during the first 4 years of therapy. Ann. Neurol. Off J. Am. Neurol. Assoc. Child Neurol. Soc. 2002, 51, 686–693. [Google Scholar] [CrossRef]
- Hughes, A.J.; Frankel, J.P.; Kempster, P.A.; Stern, G.M.; Lees, A.J. Motor response to levodopa in patients with parkinsonian motor fluctuations: A follow-up study over three years. J. Neurol. Neurosurg. Psychiatry 1994, 57, 430–434. [Google Scholar] [CrossRef] [Green Version]
- Contin, M.; Riva, R.; Martinelli, P.; Cortelli, P.; Albani, F.; Baruzzi, A. A levodopa kinetic-dynamic study of the rate of progression in Parkinson’s disease. Neurology 1998, 51, 1075–1080. [Google Scholar] [CrossRef]
- Baas, H.; Zehrden, F.; Selzer, R.; Kohnen, R.; Loetsch, J.; Harder, S. Pharmacokinetic-pharmacodynamic relationship of levodopa with and without tolcapone in patients with Parkinson’s disease. Clin. Pharmacokinet. 2001, 40, 383–393. [Google Scholar] [CrossRef]
- Richter, U.; Halje, P.; Petersson, P. Mechanisms underlying cortical resonant states: Implications for levodopa-induced dyskinesia. Rev. Neurosci. 2013, 24, 415–429. [Google Scholar] [CrossRef]
- Melgari, J.-M.; Curcio, G.; Mastrolilli, F.; Salomone, G.; Trotta, L.; Tombini, M.; Di Biase, L.; Scrascia, F.; Fini, R.; Fabrizio, E. Alpha and beta EEG power reflects L-dopa acute administration in parkinsonian patients. Front. Aging Neurosci. 2014, 6, 302. [Google Scholar] [CrossRef]
- di Biase, L.; Ricci, L.; Caminiti, M.L.; Pecoraro, P.M.; Carbone, S.P.; Di Lazzaro, V. Quantitative High Density EEG Brain Connectivity Evaluation in Parkinson’s Disease: The Phase Locking Value (PLV). J. Clin. Med. 2023, 12, 1450. [Google Scholar] [CrossRef] [PubMed]
- Bastide, M.F.; Meissner, W.G.; Picconi, B.; Fasano, S.; Fernagut, P.O.; Feyder, M.; Francardo, V.; Alcacer, C.; Ding, Y.; Brambilla, R.; et al. Pathophysiology of L-dopa-induced motor and non-motor complications in Parkinson’s disease. Prog. Neurobiol. 2015, 132, 96–168. [Google Scholar] [CrossRef] [PubMed]
- Albin, R.L.; Young, A.B.; Penney, J.B. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989, 12, 366–375. [Google Scholar] [CrossRef] [PubMed]
- DeLong, M.R. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990, 13, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.I.; Verhagen Metman, L.; Ohara, S.; Dougherty, P.M.; Kim, J.H.; Lenz, F.A. Internal pallidal neuronal activity during mild drug-related dyskinesias in Parkinson’s disease: Decreased firing rates and altered firing patterns. J. Neurophysiol. 2007, 97, 2627–2641. [Google Scholar] [CrossRef] [Green Version]
- Levy, R.; Dostrovsky, J.O.; Lang, A.E.; Sime, E.; Hutchison, W.D.; Lozano, A.M. Effects of apomorphine on subthalamic nucleus and globus pallidus internus neurons in patients with Parkinson’s disease. J. Neurophysiol. 2001, 86, 249–260. [Google Scholar] [CrossRef]
- Merello, M.; Balej, J.; Delfino, M.; Cammarota, A.; Betti, O.; Leiguarda, R. Apomorphine induces changes in GPi spontaneous outflow in patients with Parkinson’s disease. Mov. Disord. 1999, 14, 45–49. [Google Scholar] [CrossRef]
- Lozano, A.M.; Lang, A.E.; Levy, R.; Hutchison, W.; Dostrovsky, J. Neuronal recordings in Parkinson’s disease patients with dyskinesias induced by apomorphine. Ann. Neurol. 2000, 47 (Suppl. 1), S141–S146. [Google Scholar]
- Foffani, G.; Ardolino, G.; Meda, B.; Egidi, M.; Rampini, P.; Caputo, E.; Baselli, G.; Priori, A. Altered subthalamo-pallidal synchronisation in parkinsonian dyskinesias. J. Neurol. Neurosurg. Psychiatry 2005, 76, 426–428. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Oroz, M.C.; Lopez-Azcarate, J.; Garcia-Garcia, D.; Alegre, M.; Toledo, J.; Valencia, M.; Guridi, J.; Artieda, J.; Obeso, J.A. Involvement of the subthalamic nucleus in impulse control disorders associated with Parkinson’s disease. Brain A J. Neurol. 2011, 134 Pt 1, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Silberstein, P.; Oliviero, A.; Di Lazzaro, V.; Insola, A.; Mazzone, P.; Brown, P. Oscillatory pallidal local field potential activity inversely correlates with limb dyskinesias in Parkinson’s disease. Exp. Neurol. 2005, 194, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Little, S.; Pogosyan, A.; Kuhn, A.A.; Brown, P. beta band stability over time correlates with Parkinsonian rigidity and bradykinesia. Exp. Neurol. 2012, 236, 383–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Azcarate, J.; Tainta, M.; Rodriguez-Oroz, M.C.; Valencia, M.; Gonzalez, R.; Guridi, J.; Iriarte, J.; Obeso, J.A.; Artieda, J.; Alegre, M. Coupling between beta and high-frequency activity in the human subthalamic nucleus may be a pathophysiological mechanism in Parkinson’s disease. J. Neurosci. 2010, 30, 6667–6677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prescott, I.A.; Liu, L.D.; Dostrovsky, J.O.; Hodaie, M.; Lozano, A.M.; Hutchison, W.D. Lack of depotentiation at basal ganglia output neurons in PD patients with levodopa-induced dyskinesia. Neurobiol. Dis. 2014, 71, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Nadjar, A.; Gerfen, C.R.; Bezard, E. Priming for l-dopa-induced dyskinesia in Parkinson’s disease: A feature inherent to the treatment or the disease? Prog. Neurobiol. 2009, 87, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miocinovic, S.; Swann, N.C.; de Hemptinne, C.; Miller, A.; Ostrem, J.L.; Starr, P.A. Cortical gamma oscillations in isolated dystonia. Park. Relat. Disord. 2018, 49, 104–105. [Google Scholar] [CrossRef]
- Kempf, F.; Brücke, C.; Salih, F.; Trottenberg, T.; Kupsch, A.; Schneider, G.H.; Doyle Gaynor, L.M.; Hoffmann, K.T.; Vesper, J.; Wöhrle, J. Gamma activity and reactivity in human thalamic local field potentials. Eur. J. Neurosci. 2009, 29, 943–953. [Google Scholar] [CrossRef]
- Benarroch, E.E. Serotonergic modulation of basal ganglia circuits: Complexity and therapeutic opportunities. Neurology 2009, 73, 880–886. [Google Scholar] [CrossRef]
- Berger, M.; Gray, J.A.; Roth, B.L. The expanded biology of serotonin. Annu. Rev. Med. 2009, 60, 355–366. [Google Scholar] [CrossRef] [Green Version]
- Lavoie, B.; Parent, A. Immunohistochemical study of the serotoninergic innervation of the basal ganglia in the squirrel monkey. J. Comp. Neurol. 1990, 299, 1–16. [Google Scholar] [CrossRef]
- Fox, S.H.; Chuang, R.; Brotchie, J.M. Serotonin and Parkinson’s disease: On movement, mood, and madness. Mov. Disord. Off J. Mov. Disord. Soc. 2009, 24, 1255–1266. [Google Scholar] [CrossRef] [PubMed]
- Arai, R.; Karasawa, N.; Geffard, M.; Nagatsu, I. L-DOPA is converted to dopamine in serotonergic fibers of the striatum of the rat: A double-labeling immunofluorescence study. Neurosci. Lett. 1995, 195, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Arai, R.; Karasawa, N.; Geffard, M.; Nagatsu, T.; Nagatsu, I. Immunohistochemical evidence that central serotonin neurons produce dopamine from exogenousl-DOPA in the rat, with reference to the involvement of aromaticl-amino acid decarboxylase. Brain Res. 1994, 667, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Kannari, K.; Maeda, T.; Tomiyama, M.; Suda, T.; Matsunaga, M. Role of serotonergic neurons in L-DOPA-derived extracellular dopamine in the striatum of 6-OHDA-lesioned rats. Neuroreport 1999, 10, 631–634. [Google Scholar] [CrossRef]
- Carta, M.; Carlsson, T.; Kirik, D.; Björklund, A. Dopamine released from 5-HT terminals is the cause of L-DOPA-induced dyskinesia in parkinsonian rats. Brain 2007, 130, 1819–1833. [Google Scholar] [CrossRef] [Green Version]
- Eskow, K.L.; Dupre, K.B.; Barnum, C.J.; Dickinson, S.O.; Park, J.Y.; Bishop, C. The role of the dorsal raphe nucleus in the development, expression, and treatment of L-dopa-induced dyskinesia in hemiparkinsonian rats. Synapse 2009, 63, 610–620. [Google Scholar] [CrossRef] [Green Version]
- Rylander, D.; Parent, M.; O’Sullivan, S.S.; Dovero, S.; Lees, A.J.; Bezard, E.; Descarries, L.; Cenci, M.A. Maladaptive plasticity of serotonin axon terminals in levodopa-induced dyskinesia. Ann. Neurol. 2010, 68, 619–628. [Google Scholar] [CrossRef]
- Tronci, E.; Lisci, C.; Stancampiano, R.; Fidalgo, C.; Collu, M.; Devoto, P.; Carta, M. 5-Hydroxy-tryptophan for the treatment of L-DOPA-induced dyskinesia in the rat Parkinson’s disease model. Neurobiol. Dis. 2013, 60, 108–114. [Google Scholar] [CrossRef]
- Conti, M.M.; Ostock, C.Y.; Lindenbach, D.; Goldenberg, A.A.; Kampton, E.; Dell’isola, R.; Katzman, A.C.; Bishop, C. Effects of prolonged selective serotonin reuptake inhibition on the development and expression of L-DOPA-induced dyskinesia in hemi-parkinsonian rats. Neuropharmacology 2014, 77, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Bishop, C.; George, J.A.; Buchta, W.; Goldenberg, A.A.; Mohamed, M.; Dickinson, S.O.; Eissa, S.; Eskow Jaunarajs, K.L. Serotonin transporter inhibition attenuates l-DOPA-induced dyskinesia without compromising l-DOPA efficacy in hemi-parkinsonian rats. Eur. J. Neurosci. 2012, 36, 2839–2848. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Zhu, W.-M.; Stanic, D.; Finkelstein, D.I.; Horne, M.H.; Henderson, J.; Lawrence, A.J.; O’Connor, L.; Tomas, D.; Drago, J. Sprouting of dopamine terminals and altered dopamine release and uptake in Parkinsonian dyskinaesia. Brain 2008, 131, 1574–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, I.; Bose, S.K.; Pavese, N.; Ramlackhansingh, A.; Turkheimer, F.; Hotton, G.; Hammers, A.; Brooks, D.J. Glutamate NMDA receptor dysregulation in Parkinson’s disease with dyskinesias. Brain 2011, 134, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.; Mercuri, N.; Sancesario, G.; Bernardi, G. Electrophysiology of dopamine-denervated striatal neurons. Implications for Parkinson’s disease. Brain A J. Neurol. 1993, 116, 433–452. [Google Scholar]
- Hallett, P.J.; Dunah, A.; Ravenscroft, P.; Zhou, S.; Bezard, E.; Crossman, A.R.; Brotchie, J.M.; Standaert, D.G. Alterations of striatal NMDA receptor subunits associated with the development of dyskinesia in the MPTP-lesioned primate model of Parkinson’s disease. Neuropharmacology 2005, 48, 503–516. [Google Scholar] [CrossRef]
- Picconi, B.; Gardoni, F.; Centonze, D.; Mauceri, D.; Cenci, M.A.; Bernardi, G.; Calabresi, P.; Di Luca, M. Abnormal Ca2+-calmodulin-dependent protein kinase II function mediates synaptic and motor deficits in experimental parkinsonism. J. Neurosci. 2004, 24, 5283–5291. [Google Scholar] [CrossRef]
- Gubellini, P.; Picconi, B.; Bari, M.; Battista, N.; Calabresi, P.; Centonze, D.; Bernardi, G.; Finazzi-Agro, A.; Maccarrone, M. Experimental parkinsonism alters endocannabinoid degradation: Implications for striatal glutamatergic transmission. J. Neurosci. 2002, 22, 6900–6907. [Google Scholar] [CrossRef] [Green Version]
- Calabresi, P.; Pisani, A.; Mercuri, N.B.; Bernardi, G. Heterogeneity of metabotropic glutamate receptors in the striatum: Electrophysiological evidence. Eur. J. Neurosci. 1993, 5, 1370–1377. [Google Scholar] [CrossRef]
- Greenamyre, J.T. Glutamatergic influences on the basal ganglia. Clin. Neuropharmacol. 2001, 24, 65–70. [Google Scholar] [CrossRef]
- Duty, S. Targeting glutamate receptors to tackle the pathogenesis, clinical symptoms and levodopa-induced dyskinesia associated with Parkinson’s disease. CNS Drugs 2012, 26, 1017–1032. [Google Scholar] [CrossRef]
- Gubellini, P.; Saulle, E.; Centonze, D.; Bonsi, P.; Pisani, A.; Bernardi, G.; Conquet, F.; Calabresi, P. Selective involvement of mGlu1 receptors in corticostriatal LTD. Neuropharmacology 2001, 40, 839–846. [Google Scholar] [CrossRef]
- Gubellini, P.; Saulle, E.; Centonze, D.; Costa, C.; Tropepi, D.; Bernardi, G.; Conquet, F.; Calabresi, P. Corticostriatal LTP requires combined mGluR1 and mGluR5 activation. Neuropharmacology 2003, 44, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Rüdiger, M.; Maria, F.; Rodney, C.; Adele, G.; Kari, J.; Carmela, M.; Richard, S.; Karl, S.; Pier, S.; Stuck, B.E. (International Commission on Non-Ionizing Radiation Protection). ICNIRP Guidelines for Limiting Exposure to Electric Fields Induced by Movement of the Human Body in a Static Magnetic Field and by Time-Varying Magnetic Fields below 1 Hz. Health Phys. 2014, 106, 418–425. [Google Scholar]
- Sgambato-Faure, V.; Cenci, M.A. Glutamatergic mechanisms in the dyskinesias induced by pharmacological dopamine replacement and deep brain stimulation for the treatment of Parkinson’s disease. Prog. Neurobiol. 2012, 96, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Picconi, B.; Centonze, D.; Håkansson, K.; Bernardi, G.; Greengard, P.; Fisone, G.; Cenci, M.A.; Calabresi, P. Loss of bidirectional striatal synaptic plasticity in L-DOPA–induced dyskinesia. Nat. Neurosci. 2003, 6, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Vastagh, C.; Gardoni, F.; Bagetta, V.; Stanic, J.; Zianni, E.; Giampà, C.; Picconi, B.; Calabresi, P.; Di Luca, M. N-methyl-D-aspartate (NMDA) receptor composition modulates dendritic spine morphology in striatal medium spiny neurons. J. Biol. Chem. 2012, 287, 18103–18114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.-Y.; Lee, C.-H.; Shih, C.-C.; Liou, H.-H. Paraquat inhibits postsynaptic AMPA receptors on dopaminergic neurons in the substantia nigra pars compacta. Biochem. Pharmacol. 2008, 76, 1155–1164. [Google Scholar] [CrossRef]
- Bernard, V.; Gardiol, A.; Faucheux, B.; Bloch, B.; Agid, Y.; Hirsch, E.C. Expression of glutamate receptors in the human and rat basal ganglia: Effect of the dopaminergic denervation on AMPA receptor gene expression in the striatopallidal complex in Parkinson’s disease and rat with 6-OHDA lesion. J. Comp. Neurol. 1996, 368, 553–568. [Google Scholar] [CrossRef]
- Betarbet, R.; Porter, R.H.; Greenamyre, J.T. GluR1 glutamate receptor subunit is regulated differentially in the primate basal ganglia following nigrostriatal dopamine denervation. J. Neurochem. 2000, 74, 1166–1174. [Google Scholar] [CrossRef]
- Santini, E.; Valjent, E.; Usiello, A.; Carta, M.; Borgkvist, A.; Girault, J.-A.; Hervé, D.; Greengard, P.; Fisone, G. Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in L-DOPA-induced dyskinesia. J. Neurosci. 2007, 27, 6995–7005. [Google Scholar] [CrossRef] [Green Version]
- Berridge, C.W.; Waterhouse, B.D. The locus coeruleus–noradrenergic system: Modulation of behavioral state and state-dependent cognitive processes. Brain Res. Rev. 2003, 42, 33–84. [Google Scholar] [CrossRef]
- Mason, S.T.; Fibiger, H.C. Regional topography within noradrenergic locus coeruleus as revealed by retrograde transport of horseradish peroxidase. J. Comp. Neurol. 1979, 187, 703–724. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, P. Anatomy of the Noradrenergic Pathways in the Primate Brain Alteration in Parkinson’s Disease. In Noradrenergic Mechanisms in Parkinson’s Disease; CRC Press: Boca Raton, FL, USA, 1994; pp. 74–88. [Google Scholar]
- Margules, D.; Lewis, M.J.; Dragovich, J.A.; Margules, A.S. Hypothalamic norepinephrine: Circadian rhythms and the control of feeding behavior. Science 1972, 178, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Pfaus, J.G. Pathways of sexual desire. J. Sex. Med. 2009, 6, 1506–1533. [Google Scholar] [CrossRef]
- Alachkar, A.; Brotchie, J.M.; Jones, O.T. Changes in the mRNA levels of α 2A and α 2C adrenergic receptors in rat models of parkinson’s disease and L-DOPA-induced dyskinesia. J. Mol. Neurosci. 2012, 46, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Ribas, C.; Miralles, A.; Busquets, X.; García-Sevilla, J.A. Brain α2-adrenoceptors in monoamine-depleted rats: Increased receptor density, G coupling proteins, receptor turnover and receptor mRNA. Br. J. Pharmacol. 2001, 132, 1467–1476. [Google Scholar] [CrossRef] [Green Version]
- Rommelfanger, K.; Edwards, G.; Freeman, K.; Liles, L.; Miller, G.; Weinshenker, D. Norepinephrine loss produces more profound motor deficits than MPTP treatment in mice. Proc. Natl. Acad. Sci. USA 2007, 104, 13804–13809. [Google Scholar] [CrossRef] [Green Version]
- Perez, V.; Marín, C.; Rubio, A.; Aguilar, E.; Barbanoj, M.; Kulisevsky, J. Effect of the additional noradrenergic neurodegeneration to 6-OHDA-lesioned rats in levodopa-induced dyskinesias and in cognitive disturbances. J. Neural. Transm. 2009, 116, 1257–1266. [Google Scholar] [CrossRef]
- Miguelez, C.; Aristieta, A.; Cenci, M.A.; Ugedo, L. The locus coeruleus is directly implicated in L-DOPA-induced dyskinesia in parkinsonian rats: An electrophysiological and behavioural study. PLoS ONE 2011, 6, e24679. [Google Scholar] [CrossRef]
- Buck, K.; Ferger, B. Comparison of intrastriatal administration of noradrenaline and l-DOPA on dyskinetic movements: A bilateral reverse in vivo microdialysis study in 6-hydroxydopamine-lesioned rats. Neuroscience 2009, 159, 16–20. [Google Scholar] [CrossRef]
- Arai, A.; Tomiyama, M.; Kannari, K.; Kimura, T.; Suzuki, C.; Watanabe, M.; Kawarabayashi, T.; Shen, H.; Shoji, M. Reuptake of L-DOPA-derived extracellular DA in the striatum of a rodent model of Parkinson’s disease via norepinephrine transporter. Synapse 2008, 62, 632–635. [Google Scholar] [CrossRef]
- Sommermeyer, H.; Frielingsdorf, J.; Knorr, A. Effects of prazosin on the dopaminergic neurotransmission in rat brain. Eur. J. Pharmacol. 1995, 276, 267–270. [Google Scholar] [CrossRef]
- Henry, B.; Fox, S.H.; Peggs, D.; Crossman, A.R.; Brotchie, J.M. The α2-adrenergic receptor antagonist idazoxan reduces dyskinesia and enhances anti-parkinsonian actions of L-dopa in the MPTP-lesioned primate model of Parkinson’s disease. Mov. Disord. Off J. Mov. Disord. Soc. 1999, 14, 744–753. [Google Scholar] [CrossRef]
- Savola, J.M.; Hill, M.; Engstrom, M.; Merivuori, H.; Wurster, S.; McGuire, S.G.; Fox, S.H.; Crossman, A.R.; Brotchie, J.M. Fipamezole (JP-1730) is a potent α2 adrenergic receptor antagonist that reduces levodopa-induced dyskinesia in the MPTP-lesioned primate model of Parkinson’s disease. Mov. Disord. Off J. Mov. Disord. Soc. 2003, 18, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.H.; Henry, B.; Hill, M.P.; Peggs, D.; Crossman, A.R.; Brotchie, J.M. Neural mechanisms underlying peak-dose dyskinesia induced by levodopa and apomorphine are distinct: Evidence from the effects of the alpha2 adrenoceptor antagonist idazoxan. Mov. Disord. Off J. Mov. Disord. Soc. 2001, 16, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Johnston, T.H.; Fox, S.H.; Piggott, M.J.; Savola, J.M.; Brotchie, J.M. The α2 adrenergic antagonist fipamezole improves quality of levodopa action in Parkinsonian primates. Mov. Disord. 2010, 25, 2084–2093. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, A.; Bonnet, A.; Vidailhet, M.; Agid, Y. Improvement of levodopa-induced dyskinesia by propranolol in Parkinson’s disease. Neurology 1996, 46, 1548–1551. [Google Scholar] [CrossRef] [PubMed]
- Millar, N.S.; Gotti, C. Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology 2009, 56, 237–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albuquerque, E.X.; Pereira, E.F.; Alkondon, M.; Rogers, S.W. Mammalian nicotinic acetylcholine receptors: From structure to function. Physiol. Rev. 2009, 89, 73–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quik, M.; Wonnacott, S. α6β2* and α4β2* nicotinic acetylcholine receptors as drug targets for Parkinson’s disease. Pharmacol. Rev. 2011, 63, 938–966. [Google Scholar] [CrossRef] [Green Version]
- Quik, M.; Cox, H.; Parameswaran, N.; O’Leary, K.; Langston, J.W.; Di Monte, D. Nicotine reduces levodopa-induced dyskinesias in lesioned monkeys. Ann. Neurol. Off J. Am. Neurol. Assoc. Child Neurol. Soc. 2007, 62, 588–596. [Google Scholar] [CrossRef]
- Quik, M.; Mallela, A.; Ly, J.; Zhang, D. Nicotine reduces established levodopa-induced dyskinesias in a monkey model of Parkinson’s disease. Mov. Disord. 2013, 28, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Quik, M.; Mallela, A.; Chin, M.; McIntosh, J.M.; Perez, X.A.; Bordia, T. Nicotine-mediated improvement in L-dopa-induced dyskinesias in MPTP-lesioned monkeys is dependent on dopamine nerve terminal function. Neurobiol. Dis. 2013, 50, 30–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.Z.; Grady, S.R.; Quik, M. Nicotine reduces L-DOPA-induced dyskinesias by acting at β2* nicotinic receptors. J. Pharmacol. Exp. Ther. 2011, 338, 932–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quik, M.; Park, K.M.; Hrachova, M.; Mallela, A.; Huang, L.Z.; McIntosh, J.M.; Grady, S.R. Role for α6 nicotinic receptors in l-dopa-induced dyskinesias in parkinsonian mice. Neuropharmacology 2012, 63, 450–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quik, M.; Campos, C.; Grady, S.R. Multiple CNS nicotinic receptors mediate L-dopa-induced dyskinesias: Studies with parkinsonian nicotinic receptor knockout mice. Biochem. Pharmacol. 2013, 86, 1153–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caulfield, M.P.; Birdsall, N.J. International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol. Rev. 1998, 50, 279–290. [Google Scholar]
- Langmead, C.J.; Watson, J.; Reavill, C. Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol. Ther. 2008, 117, 232–243. [Google Scholar] [CrossRef]
- Gomez-Mancilla, B.; Bedard, P.J. Effect of nondopaminergic drugs on L-dopa-induced dyskinesias in MPTP-treated monkeys. Clin. Neuropharmacol. 1993, 16, 418–427. [Google Scholar] [CrossRef]
- Huot, P.; Johnston, T.H.; Koprich, J.B.; Fox, S.H.; Brotchie, J.M. The pharmacology of L-DOPA-induced dyskinesia in Parkinson’s disease. Pharmacol. Rev. 2013, 65, 171–222. [Google Scholar] [CrossRef]
- Hanrieder, J.; Ljungdahl, A.; Fälth, M.; Mammo, S.E.; Bergquist, J.; Andersson, M. L-DOPA-induced dyskinesia is associated with regional increase of striatal dynorphin peptides as elucidated by imaging mass spectrometry. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [Green Version]
- Ljungdahl, A.; Hanrieder, J.; Fälth, M.; Bergquist, J.; Andersson, M. Imaging mass spectrometry reveals elevated nigral levels of dynorphin neuropeptides in L-DOPA-induced dyskinesia in rat model of Parkinson’s disease. PLoS ONE 2011, 6, e25653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourdenx, M.; Nilsson, A.; Wadensten, H.; Fälth, M.; Li, Q.; Crossman, A.R.; Andrén, P.E.; Bezard, E. Abnormal structure-specific peptide transmission and processing in a primate model of Parkinson’s disease and L-DOPA-induced dyskinesia. Neurobiol. Dis. 2014, 62, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Samadi, P.; Grégoire, L.; Bédard, P.J. Opioid antagonists increase the dyskinetic response to dopaminergic agents in parkinsonian monkeys: Interaction between dopamine and opioid systems. Neuropharmacology 2003, 45, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Samadi, P.; Grégoire, L.; Tahar, A.H.; Di Paolo, T.; Rouillard, C.; Bédard, P.J. Naltrexone in the short-term decreases antiparkinsonian response to l-Dopa and in the long-term increases dyskinesias in drug-naive parkinsonian monkeys. Neuropharmacology 2005, 49, 165–173. [Google Scholar] [CrossRef]
- Samadi, P.; Grégoire, L.; Rouillard, C.; Bédard, P.J. Dyskinesias occur in response to saline and naltrexone alone after priming with combination of dopaminergic agents and naltrexone in the MPTP parkinsonian monkeys. Neurobiol. Dis. 2005, 19, 266–272. [Google Scholar] [CrossRef]
- Ikeda, K.; Yoshikawa, S.; Kurokawa, T.; Yuzawa, N.; Nakao, K.; Mochizuki, H. TRK-820, a selective kappa opioid receptor agonist, could effectively ameliorate L-DOPA-induced dyskinesia symptoms in a rat model of Parkinson’s disease. Eur. J. Pharmacol. 2009, 620, 42–48. [Google Scholar] [CrossRef]
- Kano, M.; Ohno-Shosaku, T.; Hashimotodani, Y.; Uchigashima, M.; Watanabe, M. Endocannabinoid-mediated control of synaptic transmission. Physiol. Rev. 2009, 89, 309–380. [Google Scholar] [CrossRef]
- O’sullivan, S. Cannabinoids go nuclear: Evidence for activation of peroxisome proliferator-activated receptors. Br. J. Pharmacol. 2007, 152, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Devane, W.A.; Hanuš, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef]
- Di Marzo, V.; Fontana, A.; Cadas, H.; Schinelli, S.; Cimino, G.; Schwartz, J.-C.; Piomelli, D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 1994, 372, 686–691. [Google Scholar] [CrossRef] [Green Version]
- Mechoulam, R.; Ben-Shabat, S.; Hanus, L.; Ligumsky, M.; Kaminski, N.E.; Schatz, A.R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.R. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995, 50, 83–90. [Google Scholar] [CrossRef] [PubMed]
- López-Moreno, J.A.; González-Cuevas, G.; Moreno, G.; Navarro, M. The pharmacology of the endocannabinoid system: Functional and structural interactions with other neurotransmitter systems and their repercussions in behavioral addiction. Addict. Biol. 2008, 13, 160–187. [Google Scholar] [CrossRef] [PubMed]
- Console-Bram, L.; Marcu, J.; Abood, M.E. Cannabinoid receptors: Nomenclature and pharmacological principles. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2012, 38, 4–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallmichrath, I.; Szabo, B. Cannabinoids inhibit striatonigral GABAergic neurotransmission in the mouse. Neuroscience 2002, 113, 671–682. [Google Scholar] [CrossRef]
- Martín, A.B.; Fernandez-Espejo, E.; Ferrer, B.; Gorriti, M.A.; Bilbao, A.; Navarro, M.; Rodriguez de Fonseca, F.; Moratalla, R. Expression and function of CB1 receptor in the rat striatum: Localization and effects on D1 and D2 dopamine receptor-mediated motor behaviors. Neuropsychopharmacology 2008, 33, 1667–1679. [Google Scholar] [CrossRef] [Green Version]
- Morera-Herreras, T.; Ruiz-Ortega, J.; Gomez-Urquijo, S.; Ugedo, L. Involvement of subthalamic nucleus in the stimulatory effect of Δ9-tetrahydrocannabinol on dopaminergic neurons. Neuroscience 2008, 151, 817–823. [Google Scholar] [CrossRef]
- Gerdeman, G.L.; Ronesi, J.; Lovinger, D.M. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat. Neurosci. 2002, 5, 446–451. [Google Scholar] [CrossRef]
- Morgese, M.G.; Cassano, T.; Cuomo, V.; Giuffrida, A. Anti-dyskinetic effects of cannabinoids in a rat model of Parkinson’s disease: Role of CB1 and TRPV1 receptors. Exp. Neurol. 2007, 208, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Giuffrida, A.; Parsons, L.; Kerr, T.; De Fonseca, F.R.; Navarro, M.; Piomelli, D. Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat. Neurosci. 1999, 2, 358–363. [Google Scholar] [CrossRef]
- Masserano, J.; Karoum, F.; Wyatt, R. SR 141716A, a CB1 cannabinoid receptor antagonist, potentiates the locomotor stimulant effects of amphetamine and apomorphine. Behav. Pharmacol. 1999, 10, 429–432. [Google Scholar] [CrossRef]
- Kreitzer, A.C.; Malenka, R.C. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature 2007, 445, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.H.; Henry, B.; Hill, M.; Crossman, A.; Brotchie, J. Stimulation of cannabinoid receptors reduces levodopa-induced dyskinesia in the MPTP-lesioned nonhuman primate model of Parkinson’s disease. Mov. Disord. Off J. Mov. Disord. Soc. 2002, 17, 1180–1187. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.; Macheda, T.; Morgese, M.G.; Trabace, L.; Giuffrida, A. The cannabinoid agonist WIN55212-2 decreases L-DOPA-induced PKA activation and dyskinetic behavior in 6-OHDA-treated rats. Neurosci. Res. 2012, 72, 236–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, S.; Gorman, A.M.; Finn, D.P.; Dowd, E. The effects of cannabinoid drugs on abnormal involuntary movements in dyskinetic and non-dyskinetic 6-hydroxydopamine lesioned rats. Brain Res. 2010, 1363, 40–48. [Google Scholar] [CrossRef]
- Sieradzan, K.; Fox, S.; Hill, M.; Dick, J.; Crossman, A.; Brotchie, J. Cannabinoids reduce levodopa-induced dyskinesia in Parkinson’s disease: A pilot study. Neurology 2001, 57, 2108–2111. [Google Scholar] [CrossRef] [PubMed]
- Hermann, H.; Marsicano, G.; Lutz, B. Coexpression of the cannabinoid receptor type 1 with dopamine and serotonin receptors in distinct neuronal subpopulations of the adult mouse forebrain. Neuroscience 2002, 109, 451–460. [Google Scholar] [CrossRef]
- Mathur, B.N.; Capik, N.A.; Alvarez, V.A.; Lovinger, D.M. Serotonin induces long-term depression at corticostriatal synapses. J. Neurosci. 2011, 31, 7402–7411. [Google Scholar] [CrossRef] [Green Version]
- Nakazi, M.; Bauer, U.; Nickel, T.; Kathmann, M.; Schlicker, E. Inhibition of serotonin release in the mouse brain via presynaptic cannabinoid CB 1 receptors. Naunyn-Schmiedebergs Arch. Pharmacol. 2000, 361, 19–24. [Google Scholar]
- Linden, J. Molecular approach to adenosine receptors: Receptor-mediated mechanisms of tissue protection. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 775–787. [Google Scholar] [CrossRef]
- Cunha, R.A. Neuroprotection by adenosine in the brain: From A 1 receptor activation to A 2A receptor blockade. Purinergic Signal. 2005, 1, 111–134. [Google Scholar] [CrossRef] [Green Version]
- Gomes, C.V.; Kaster, M.P.; Tomé, A.R.; Agostinho, P.M.; Cunha, R.A. Adenosine receptors and brain diseases: Neuroprotection and neurodegeneration. Biochim. Biophys. Acta 2011, 1808, 1380–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- V Lopes, L.; M Sebastiao, A.; A Ribeiro, J. Adenosine and related drugs in brain diseases: Present and future in clinical trials. Curr. Top. Med. Chem. 2011, 11, 1087–1101. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Varani, K. Adenosine as a multi-signalling guardian angel in human diseases: When, where and how does it exert its protective effects? Trends Pharmacol. Sci. 2016, 37, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic signalling: Therapeutic developments. Front. Pharmacol. 2017, 8, 661. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, K.A.; Merighi, S.; Varani, K.; Borea, P.A.; Baraldi, S.; Aghazadeh Tabrizi, M.; Romagnoli, R.; Baraldi, P.G.; Ciancetta, A.; Tosh, D.K. A3 adenosine receptors as modulators of inflammation: From medicinal chemistry to therapy. Med. Res. Rev. 2018, 38, 1031–1072. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, K.; Ferré, S.; Zoli, M.; Agnati, L.F. Integrated events in central dopamine transmission as analyzed at multiple levels. Evidence for intramembrane adenosine A2A/dopamine D2 and adenosine A1/dopamine D1 receptor interactions in the basal ganglia. Brain Res. Rev. 1998, 26, 258–273. [Google Scholar] [CrossRef]
- Fuxe, K.; Ferré, S.; Genedani, S.; Franco, R.; Agnati, L.F. Adenosine receptor–dopamine receptor interactions in the basal ganglia and their relevance for brain function. Physiol. Behav. 2007, 92, 210–217. [Google Scholar] [CrossRef]
- Hauser, R.A.; Cantillon, M.; Pourcher, E.; Micheli, F.; Mok, V.; Onofrj, M.; Huyck, S.; Wolski, K. Preladenant in patients with Parkinson’s disease and motor fluctuations: A phase 2, double-blind, randomised trial. Lancet Neurol. 2011, 10, 221–229. [Google Scholar] [CrossRef]
- Pourcher, E.; Hauser, R.A. Adenosinergic Receptor Antagonists: Clinical Experience in Parkinson’s Disease. In The Adenosinergic System: A Non-Dopaminergic Target in Parkinson’s Disease; Springer: Cham, Switzerland, 2015; pp. 291–307. [Google Scholar]
- LeWitt, P.A.; Guttman, M.; Tetrud, J.W.; Tuite, P.J.; Mori, A.; Chaikin, P.; Sussman, N.M.; Group, U.S. Adenosine A2A receptor antagonist istradefylline (KW-6002) reduces “off” time in Parkinson’s disease: A double-blind, randomized, multicenter clinical trial (6002-US-005). Ann. Neurol. 2008, 63, 295–302. [Google Scholar] [CrossRef]
- Hauser, R.A.; Shulman, L.M.; Trugman, J.M.; Roberts, J.W.; Mori, A.; Ballerini, R.; Sussman, N.M. Study of istradefylline in patients with Parkinson’s disease on levodopa with motor fluctuations. Mov. Disord. Off J. Mov. Disord. Soc. 2008, 23, 2177–2185. [Google Scholar] [CrossRef]
- Stacy, M.; Silver, D.; Mendis, T.; Sutton, J.; Mori, A.; Chaikin, P.; Sussman, N. A 12-week, placebo-controlled study (6002-US-006) of istradefylline in Parkinson disease. Neurology 2008, 70, 2233–2240. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; Kondo, T.; Group, J.I.S. Adenosine A2A receptor antagonist istradefylline reduces daily OFF time in Parkinson’s disease. Mov. Disord. 2013, 28, 1138–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, R.A.; Olanow, C.W.; Kieburtz, K.D.; Pourcher, E.; Docu-Axelerad, A.; Lew, M.; Kozyolkin, O.; Neale, A.; Resburg, C.; Meya, U. Tozadenant (SYN115) in patients with Parkinson’s disease who have motor fluctuations on levodopa: A phase 2b, double-blind, randomised trial. Lancet Neurol. 2014, 13, 767–776. [Google Scholar] [CrossRef]
- Therapies, B. Biotie’s tozadenant (SYN115) meets primary and multiple secondary endpoints in phase 2b study in Parkinson’s disease. Press. Release Dec. 2012, 11. [Google Scholar]
- Politis, M. Neuroimaging in Parkinson disease: From research setting to clinical practice. Nat. Rev. Neurol. 2014, 10, 708–722. [Google Scholar] [CrossRef]
- Au, W.L.; Adams, J.R.; Troiano, A.R.; Stoessl, A.J. Parkinson’s disease: In vivo assessment of disease progression using positron emission tomography. Brain Res. Mol. Brain Res. 2005, 134, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.J.; Frey, K.A.; Marek, K.L.; Oakes, D.; Paty, D.; Prentice, R.; Shults, C.W.; Stoessl, A.J. Assessment of neuroimaging techniques as biomarkers of the progression of Parkinson’s disease. Exp. Neurol. 2003, 184 (Suppl. 1), S68–S79. [Google Scholar] [CrossRef]
- Sossi, V.; Doudet, D.J.; Holden, J.E. A reversible tracer analysis approach to the study of effective dopamine turnover. J. Cereb. Blood Flow. Metab. 2001, 21, 469–476. [Google Scholar] [CrossRef] [Green Version]
- Linazasoro, G.; Antonini, A.; Maguire, R.P.; Leenders, K.L. Pharmacological and PET studies in patient’s with Parkinson’s disease and a short duration-motor response: Implications in the pathophysiology of motor complications. J. Neural Transm. 2004, 111, 497–509. [Google Scholar] [CrossRef]
- Hong, J.Y.; Oh, J.S.; Lee, I.; Sunwoo, M.K.; Ham, J.H.; Lee, J.E.; Sohn, Y.H.; Kim, J.S.; Lee, P.H. Presynaptic dopamine depletion predicts levodopa-induced dyskinesia in de novo Parkinson disease. Neurology 2014, 82, 1597–1604. [Google Scholar] [CrossRef]
- de la Fuente-Fernández, R.; Lu, J.Q.; Sossi, V.; Jivan, S.; Schulzer, M.; Holden, J.E.; Lee, C.S.; Ruth, T.J.; Calne, D.B.; Stoessl, A.J. Biochemical variations in the synaptic level of dopamine precede motor fluctuations in Parkinson’s disease: PET evidence of increased dopamine turnover. Ann. Neurol. 2001, 49, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Stoessl, A.J. Central pharmacokinetics of levodopa: Lessons from imaging studies. Mov. Disord. 2015, 30, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Pavese, N.; Evans, A.H.; Tai, Y.F.; Hotton, G.; Brooks, D.J.; Lees, A.J.; Piccini, P. Clinical correlates of levodopa-induced dopamine release in Parkinson disease: A PET study. Neurology 2006, 67, 1612–1617. [Google Scholar] [CrossRef] [PubMed]
- Doudet, D.J.; Chan, G.L.; Holden, J.E.; McGeer, E.G.; Aigner, T.A.; Wyatt, R.J.; Ruth, T.J. 6-[18F]Fluoro-L-DOPA PET studies of the turnover of dopamine in MPTP-induced parkinsonism in monkeys. Synapse 1998, 29, 225–232. [Google Scholar] [CrossRef]
- Sossi, V.; de La Fuente-Fernandez, R.; Holden, J.E.; Doudet, D.J.; McKenzie, J.; Stoessl, A.J.; Ruth, T.J. Increase in dopamine turnover occurs early in Parkinson’s disease: Evidence from a new modeling approach to PET 18 F-fluorodopa data. J. Cereb. Blood Flow. Metab. 2002, 22, 232–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sossi, V.; de la Fuente-Fernandez, R.; Holden, J.E.; Schulzer, M.; Ruth, T.J.; Stoessl, J. Changes of dopamine turnover in the progression of Parkinson’s disease as measured by positron emission tomography: Their relation to disease-compensatory mechanisms. J. Cereb. Blood Flow. Metab. 2004, 24, 869–876. [Google Scholar] [CrossRef] [Green Version]
- Sossi, V.; de la Fuente-Fernandez, R.; Schulzer, M.; Adams, J.; Stoessl, J. Age-related differences in levodopa dynamics in Parkinson’s: Implications for motor complications. Brain 2006, 129, 1050–1058. [Google Scholar] [CrossRef] [Green Version]
- Golbe, L.I. Young-onset Parkinson’s disease: A clinical review. Neurology 1991, 41 Pt 1, 168–173. [Google Scholar] [CrossRef]
- Grandas, F.; Galiano, M.L.; Tabernero, C. Risk factors for levodopa-induced dyskinesias in Parkinson’s disease. J. Neurol. 1999, 246, 1127–1133. [Google Scholar] [CrossRef]
- Quinn, N.; Critchley, P.; Marsden, C.D. Young onset Parkinson’s disease. Mov. Disord. 1987, 2, 73–91. [Google Scholar] [CrossRef]
- Kumar, N.; Van Gerpen, J.A.; Bower, J.H.; Ahlskog, J.E. Levodopa-dyskinesia incidence by age of Parkinson’s disease onset. Mov. Disord. 2005, 20, 342–344. [Google Scholar] [CrossRef]
- Piccini, P.; Weeks, R.A.; Brooks, D.J. Alterations in opioid receptor binding in Parkinson’s disease patients with levodopa-induced dyskinesias. Ann. Neurol. 1997, 42, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Mishina, M.; Ishiwata, K.; Naganawa, M.; Kimura, Y.; Kitamura, S.; Suzuki, M.; Hashimoto, M.; Ishibashi, K.; Oda, K.; Sakata, M.; et al. Adenosine A(2A) receptors measured with [C]TMSX PET in the striata of Parkinson’s disease patients. PLoS ONE 2011, 6, e17338. [Google Scholar] [CrossRef] [Green Version]
- Hirano, S.; Asanuma, K.; Ma, Y.; Tang, C.; Feigin, A.; Dhawan, V.; Carbon, M.; Eidelberg, D. Dissociation of metabolic and neurovascular responses to levodopa in the treatment of Parkinson’s disease. J. Neurosci. Off J. Soc. Neurosci. 2008, 28, 4201–4209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faucheux, B.A.; Bonnet, A.M.; Agid, Y.; Hirsch, E.C. Blood vessels change in the mesencephalon of patients with Parkinson’s disease. Lancet 1999, 353, 981–982. [Google Scholar] [CrossRef] [PubMed]
- Ohlin, K.E.; Francardo, V.; Lindgren, H.S.; Sillivan, S.E.; O’Sullivan, S.S.; Luksik, A.S.; Vassoler, F.M.; Lees, A.J.; Konradi, C.; Cenci, M.A. Vascular endothelial growth factor is upregulated by L-dopa in the parkinsonian brain: Implications for the development of dyskinesia. Brain 2011, 134, 2339–2357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-Y.; Seo, S.; Lee, J.S.; Kim, H.-J.; Kim, Y.K.; Jeon, B.S. Putaminal serotonergic innervation: Monitoring dyskinesia risk in Parkinson disease. Neurology 2015, 85, 853–860. [Google Scholar] [CrossRef]
- Stocchi, F.; Tagliati, M.; Olanow, C.W. Treatment of levodopa-induced motor complications. Mov. Disord. 2008, 23 (Suppl. 3), S599–S612. [Google Scholar] [CrossRef]
- Obeso, J.A.; Olanow, C.W.; Nutt, J.G. Levodopa motor complications in Parkinson’s disease. Trends Neurosci. 2000, 23, S2–S7. [Google Scholar] [CrossRef]
- Del Sorbo, F.; Albanese, A. Levodopa-induced dyskinesias and their management. J. Neurol. 2008, 255 (Suppl. 4), 32–41. [Google Scholar] [CrossRef]
- Olanow, C.W.; Kieburtz, K.; Odin, P.; Espay, A.J.; Standaert, D.G.; Fernandez, H.H.; Vanagunas, A.; Othman, A.A.; Widnell, K.L.; Robieson, W.Z.; et al. Continuous intrajejunal infusion of levodopa-carbidopa intestinal gel for patients with advanced Parkinson’s disease: A randomised, controlled, double-blind, double-dummy study. Lancet Neurol. 2014, 13, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Salomone, G.; Marano, M.; di Biase, L.; Melgari, J.M.; Di Lazzaro, V. Dopamine dysregulation syndrome and punding in levodopa-carbidopa intestinal gel (LCIG) infusion: A serious but preventable complication. Park. Relat. Disord. 2015, 21, 1124–1125. [Google Scholar] [CrossRef]
- Melgari, J.M.; Salomone, G.; di Biase, L.; Marano, M.; Scrascia, F.; Di Lazzaro, V. Dyskinesias during levodopa-carbidopa intestinal gel (LCIG) infusion: Management inclinical practice. Park. Relat. Disord. 2015, 21, 327–328. [Google Scholar] [CrossRef] [PubMed]
- Marano, M.; Naranian, T.; di Biase, L.; Di Santo, A.; Poon, Y.Y.; Arca, R.; Cossu, G.; Marano, P.; Di Lazzaro, V.; Fasano, A. Complex dyskinesias in Parkinson patients on levodopa/carbidopa intestinal gel. Park. Relat. Disord. 2019, 69, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Marano, M.; Pizzicannella, M.; di Biase, L.; Rea, R.; Di Santo, A.; Martino, M.; Andrisani, G.; Pandolfi, M.; Di Matteo, F.M.; Di Lazzaro, V. Jejunal pulling syndrome: A peculiar LCIG complication. Park. Relat. Disord. 2018, 52, 113–114. [Google Scholar] [CrossRef] [PubMed]
- Manson, A.J.; Turner, K.; Lees, A.J. Apomorphine monotherapy in the treatment of refractory motor complications of Parkinson’s disease: Long-term follow-up study of 64 patients. Mov. Disord. 2002, 17, 1235–1241. [Google Scholar] [CrossRef]
- Di Lazzaro, V.; Rothwell, J.C. Corticospinal activity evoked and modulated by non-invasive stimulation of the intact human motor cortex. J. Physiol. 2014, 592, 4115–4128. [Google Scholar] [CrossRef]
- Lefaucheur, J.P.; Aleman, A.; Baeken, C.; Benninger, D.H.; Brunelin, J.; Di Lazzaro, V.; Filipović, S.R.; Grefkes, C.; Hasan, A.; Hummel, F.C.; et al. Evidence-based guidelines on the therapeutic use of repetitive transcranial magnetic stimulation (rTMS): An update (2014-2018). Clin. Neurophysiol. 2020, 131, 474–528. [Google Scholar] [CrossRef]
- di Biase, L.; Falato, E.; Di Lazzaro, V. Transcranial Focused Ultrasound (tFUS) and Transcranial Unfocused Ultrasound (tUS) Neuromodulation: From Theoretical Principles to Stimulation Practices. Front. Neurol. 2019, 10, 549. [Google Scholar] [CrossRef] [Green Version]
- di Biase, L.; Falato, E.; Caminiti, M.L.; Pecoraro, P.M.; Narducci, F.; Di Lazzaro, V. Focused Ultrasound (FUS) for Chronic Pain Management: Approved and Potential Applications. Neurol. Res. Int. 2021, 2021, 8438498. [Google Scholar] [CrossRef]
- di Biase, L.; Munhoz, R.P. Deep brain stimulation for the treatment of hyperkinetic movement disorders. Expert Rev. Neurother. 2016, 16, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Kern, D.S.; Picillo, M.; Thompson, J.A.; Sammartino, F.; di Biase, L.; Munhoz, R.P.; Fasano, A. Interleaving Stimulation in Parkinson’s Disease, Tremor, and Dystonia. Stereotact. Funct. Neurosurg. 2018, 96, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Sandoe, C.; Krishna, V.; Basha, D.; Sammartino, F.; Tatsch, J.; Picillo, M.; di Biase, L.; Poon, Y.Y.; Hamani, C.; Reddy, D.; et al. Predictors of deep brain stimulation outcome in tremor patients. Brain Stimul. 2018, 11, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Krack, P.; Volkmann, J.; Tinkhauser, G.; Deuschl, G. Deep Brain Stimulation in Movement Disorders: From Experimental Surgery to Evidence-Based Therapy. Mov. Disord. 2019, 34, 1795–1810. [Google Scholar] [CrossRef] [PubMed]
- Rizzone, M.; Fasano, A.; Daniele, A.; Zibetti, M.; Merola, A.; Rizzi, L.; Piano, C.; Piccininni, C.; Romito, L.; Lopiano, L. Long-term outcome of subthalamic nucleus DBS in Parkinson’s disease: From the advanced phase towards the late stage of the disease? Park. Relat. Disord. 2014, 20, 376–381. [Google Scholar] [CrossRef]
- di Biase, L.; Fasano, A. Low-frequency deep brain stimulation for Parkinson’s disease: Great expectation or false hope? Mov. Disord. 2016, 31, 962–967. [Google Scholar] [CrossRef]
- Tinkhauser, G.; Pogosyan, A.; Debove, I.; Nowacki, A.; Shah, S.A.; Seidel, K.; Tan, H.; Brittain, J.S.; Petermann, K.; di Biase, L.; et al. Directional local field potentials: A tool to optimize deep brain stimulation. Mov. Disord. 2018, 33, 159–164. [Google Scholar] [CrossRef]
- di Biase, L.; Piano, C.; Bove, F.; Ricci, L.; Caminiti, M.L.; Stefani, A.; Viselli, F.; Modugno, N.; Cerroni, R.; Calabresi, P. Intraoperative Local Field Potential Beta Power and Three-Dimensional Neuroimaging Mapping Predict Long-Term Clinical Response to Deep Brain Stimulation in Parkinson Disease: A Retrospective Study. Neuromodulation Technol. Neural Interface 2023. [Google Scholar] [CrossRef]
- Espay, A.J.; Morgante, F.; Merola, A.; Fasano, A.; Marsili, L.; Fox, S.H.; Bezard, E.; Picconi, B.; Calabresi, P.; Lang, A.E. Levodopa-induced dyskinesia in Parkinson disease: Current and evolving concepts. Ann. Neurol. 2018, 84, 797–811. [Google Scholar] [CrossRef] [Green Version]
- Kwon, D.K.; Kwatra, M.; Wang, J.; Ko, H.S. Levodopa-Induced Dyskinesia in Parkinson’s Disease: Pathogenesis and Emerging Treatment Strategies. Cells 2022, 11, 3736. [Google Scholar] [CrossRef]
- Vijayakumar, D.; Jankovic, J. Drug-Induced Dyskinesia, Part 1: Treatment of Levodopa-Induced Dyskinesia. Drugs 2016, 76, 759–777. [Google Scholar] [CrossRef]
- Hislop, J.; Margolesky, J.; Shpiner, D.S. Sublingual apomorphine in treatment of Parkinson’s disease: A review. Int. J. Neurosci. 2022, 1–7. [Google Scholar] [CrossRef]
- LeWitt, P.A. Levodopa therapy for Parkinson’s disease: Pharmacokinetics and pharmacodynamics. Mov. Disord. 2015, 30, 64–72. [Google Scholar] [CrossRef]
- Poewe, W.; Antonini, A. Novel formulations and modes of delivery of levodopa. Mov. Disord. Off. J. Mov. Disord. Soc. 2015, 30, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Antonini, A.; Odin, P.; Opiano, L.; Tomantschger, V.; Pacchetti, C.; Pickut, B.; Gasser, U.E.; Calandrella, D.; Mancini, F.; Zibetti, M.; et al. Effect and safety of duodenal levodopa infusion in advanced Parkinson’s disease: A retrospective multicenter outcome assessment in patient routine care. J. Neural Transm. 2013, 120, 1553–1558. [Google Scholar] [CrossRef] [PubMed]
- Antonini, A.; Isaias, I.U.; Canesi, M.; Zibetti, M.; Mancini, F.; Manfredi, L.; Dal Fante, M.; Lopiano, L.; Pezzoli, G. Duodenal levodopa infusion for advanced Parkinson’s disease: 12-month treatment outcome. Mov. Disord. 2007, 22, 1145–1149. [Google Scholar] [CrossRef] [PubMed]
- Antonini, A.; Poewe, W.; Chaudhuri, K.R.; Jech, R.; Pickut, B.; Pirtošek, Z.; Szasz, J.; Valldeoriola, F.; Winkler, C.; Bergmann, L. Levodopa-carbidopa intestinal gel in advanced Parkinson’s: Final results of the GLORIA registry. Park. Relat. Disord. 2017, 45, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Szász, J.A.; Constantin, V.A.; Orbán-Kis, K.; Bancu, L.A.; Ciorba, M.; Mihály, I.; Nagy, E.E.; Szász, R.M.; Kelemen, K.; Simu, M.A. Management challenges of severe, complex dyskinesia. Data from a large cohort of patients treated with Levodopa-carbidopa intestinal gel for advanced Parkinson’s disease. Brain Sci. 2021, 11, 826. [Google Scholar] [CrossRef]
- Hauser, R.A.; Hsu, A.; Kell, S.; Espay, A.J.; Sethi, K.; Stacy, M.; Ondo, W.; O’Connell, M.; Gupta, S.; investigators, I.A.-P. Extended-release carbidopa-levodopa (IPX066) compared with immediate-release carbidopa-levodopa in patients with Parkinson’s disease and motor fluctuations: A phase 3 randomised, double-blind trial. Lancet Neurol. 2013, 12, 346–356. [Google Scholar] [CrossRef]
- Stocchi, F.; Hsu, A.; Khanna, S.; Ellenbogen, A.; Mahler, A.; Liang, G.; Dillmann, U.; Rubens, R.; Kell, S.; Gupta, S. Comparison of IPX066 with carbidopa-levodopa plus entacapone in advanced PD patients. Park. Relat. Disord. 2014, 20, 1335–1340. [Google Scholar] [CrossRef] [Green Version]
- Lewitt, P.A.; Ellenbogen, A.; Chen, D.; Lal, R.; McGuire, K.; Zomorodi, K.; Luo, W.; Huff, F.J. Actively transported levodopa prodrug XP21279: A study in patients with Parkinson disease who experience motor fluctuations. Clin. Neuropharmacol. 2012, 35, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, V.; Fabrizio, E.; Cipriani, R.; Vanacore, N.; Meco, G. Buspirone in levodopa-induced dyskinesias. Clin. Neuropharmacol. 1994, 17, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Politis, M.; Wu, K.; Loane, C.; Brooks, D.J.; Kiferle, L.; Turkheimer, F.E.; Bain, P.; Molloy, S.; Piccini, P. Serotonergic mechanisms responsible for levodopa-induced dyskinesias in Parkinson’s disease patients. J. Clin. Investig. 2014, 124, 1340–1349. [Google Scholar] [CrossRef] [PubMed]
- Goetz, C.G.; Damier, P.; Hicking, C.; Laska, E.; Muller, T.; Olanow, C.W.; Rascol, O.; Russ, H. Sarizotan as a treatment for dyskinesias in Parkinson’s disease: A double-blind placebo-controlled trial. Mov. Disord. 2007, 22, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Bezard, E.; Munoz, A.; Tronci, E.; Pioli, E.Y.; Li, Q.; Porras, G.; Bjorklund, A.; Carta, M. Anti-dyskinetic effect of anpirtoline in animal models of L-DOPA-induced dyskinesia. Neurosci. Res. 2013, 77, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Bezard, E.; Tronci, E.; Pioli, E.Y.; Li, Q.; Porras, G.; Bjorklund, A.; Carta, M. Study of the antidyskinetic effect of eltoprazine in animal models of levodopa-induced dyskinesia. Mov. Disord. 2013, 28, 1088–1096. [Google Scholar] [CrossRef]
- Svenningsson, P.; Rosenblad, C.; af Edholm Arvidsson, K.; Wictorin, K.; Keywood, C.; Shankar, B.; Lowe, D.A.; Björklund, A.; Widner, H. Eltoprazine counteracts l-DOPA-induced dyskinesias in Parkinson’s disease: A dose-finding study. Brain 2015, 138, 963–973. [Google Scholar] [CrossRef] [Green Version]
- Tronci, E.; Fidalgo, C.; Zianni, E.; Collu, M.; Stancampiano, R.; Morelli, M.; Gardoni, F.; Carta, M. Effect of memantine on L-DOPA-induced dyskinesia in the 6-OHDA-lesioned rat model of Parkinson’s disease. Neuroscience 2014, 265, 245–252. [Google Scholar] [CrossRef]
- Clarke, C.E.; Cooper, J.A.; Holdich, T.A.H.; Study, G. A Randomized, Double-blind, Placebo-controlled, Ascending-dose Tolerability and Safety Study of Remacemide as Adjuvant Therapy in Parkinson’s Disease with Response Fluctuations. Clin. Neuropharmacol. 2001, 24, 133–138. [Google Scholar] [CrossRef]
- Giuffra, M.E.; Sethy, V.H.; Davis, T.L.; Mouradian, M.M.; Chase, T.N. Milacemide therapy for Parkinson’s disease. Mov. Disord. 1993, 8, 47–50. [Google Scholar] [CrossRef]
- Nutt, J.G.; Gunzler, S.A.; Kirchhoff, T.; Hogarth, P.; Weaver, J.L.; Krams, M.; Jamerson, B.; Menniti, F.S.; Landen, J.W. Effects of a NR2B selective NMDA glutamate antagonist, CP-101,606, on dyskinesia and parkinsonism. Mov. Disord. 2008, 23, 1860–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Blercom, N.; Lasa, A.; Verger, K.; Masramón, X.; Sastre, V.M.; Linazasoro, G. Effects of Gabapentin on the Motor Response to Levodopa: A Double-Blind, Placebo-Controlled, Crossover Study in Patients With Complicated Parkinson Disease. Clin. Neuropharmacol. 2004, 27, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Wolz, M.; Löhle, M.; Strecker, K.; Schwanebeck, U.; Schneider, C.; Reichmann, H.; Grählert, X.; Schwarz, J.; Storch, A. Levetiracetam for levodopa-induced dyskinesia in Parkinson’s disease: A randomized, double-blind, placebo-controlled trial. J. Neural Transm. 2010, 117, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Iwata, Y.; Irie, S.; Uchida, H.; Suzuki, T.; Watanabe, K.; Iwashita, S.; Mimura, M. Effects of zonisamide on tardive dyskinesia: A preliminary open-label trial. J. Neurol. Sci. 2012, 315, 137–140. [Google Scholar] [CrossRef]
- Durif, F.; Debilly, B.; Galitzky, M.; Morand, D.; Viallet, F.; Borg, M.; Thobois, S.; Broussolle, E.; Rascol, O. Clozapine improves dyskinesias in Parkinson disease: A double-blind, placebo-controlled study. Neurology 2004, 62, 381–388. [Google Scholar] [CrossRef]
- Katzenschlager, R.; Manson, A.; Evans, A.; Watt, H.; Lees, A. Low dose quetiapine for drug induced dyskinesias in Parkinson’s disease: A double blind cross over study. J. Neurol. Neurosurg. Psychiatry 2004, 75, 295–297. [Google Scholar]
- Manson, A.; Schrag, A.; Lees, A. Low-dose olanzapine for levodopa induced dyskinesias. Neurology 2000, 55, 795–799. [Google Scholar] [CrossRef]
- di Biase, L. Method and Device for the Objective Characterization of Symptoms of Parkinson’s Disease. WO Patent WO2022054112A1, 17 March 2022. [Google Scholar]
- di Biase, L. Method for the Management of Oral Therapy in Parkinson’s Disease. WO Patent WO2022054113A1, 17 March 2022. [Google Scholar]
- di Biase, L. Adaptive Method and System for a Personalized Daily Infusion Therapy of Parkinson’s Disease. WO Patent WO2022054111A1, 17 March 2022. [Google Scholar]
- Baston, C.; Contin, M.; Calandra Buonaura, G.; Cortelli, P.; Ursino, M. A mathematical model of levodopa medication effect on basal ganglia in parkinson’s Disease: An application to the alternate finger tapping task. Front. Hum. Neurosci. 2016, 10, 280. [Google Scholar] [CrossRef]
- Ursino, M.; Magosso, E.; Lopane, G.; Calandra-Buonaura, G.; Cortelli, P.; Contin, M. Mathematical modeling and parameter estimation of levodopa motor response in patients with parkinson disease. PLoS ONE 2020, 15, e0229729. [Google Scholar] [CrossRef] [Green Version]
- Chan, P.L.; Nutt, J.G.; Holford, N.H. Modeling the short-and long-duration responses to exogenous levodopa and to endogenous levodopa production in Parkinson’s disease. J. Pharmacokinet. Pharmacodyn. 2004, 31, 243–268. [Google Scholar] [CrossRef]
- Ursino, M.; Baston, C. Aberrant learning in Parkinson’s disease: A neurocomputational study on bradykinesia. Eur. J. Neurosci. 2018, 47, 1563–1582. [Google Scholar] [CrossRef] [PubMed]
- Deleu, D.; Sarre, S.; Ebinger, G.; Michotte, Y. Simultaneous monitoring of levodopa, dopamine and their metabolites in skeletal muscle and subcutaneous tissue in different pharmacological conditions using microdialysis. J. Pharm. Biomed. Anal. 1993, 11, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Goud, K.Y.; Moonla, C.; Mishra, R.K.; Yu, C.; Narayan, R.; Litvan, I.; Wang, J. Wearable electrochemical microneedle sensor for continuous monitoring of levodopa: Toward Parkinson management. ACS Sens. 2019, 4, 2196–2204. [Google Scholar] [CrossRef] [PubMed]
- Dizdar, N.; Kullman, A.; Norlander, B.; Olsson, J.-E.; Kagedal, B. Human pharmacokinetics of L-3, 4-dihydroxyphenylalanine studied with microdialysis. Clin. Chem. 1999, 45, 1813–1820. [Google Scholar] [CrossRef] [Green Version]
- Van Gompel, J.J.; Chang, S.-Y.; Goerss, S.J.; Kim, I.Y.; Kimble, C.; Bennet, K.E.; Lee, K.H. Development of intraoperative electrochemical detection: Wireless instantaneous neurochemical concentration sensor for deep brain stimulation feedback. Neurosurg. Focus 2010, 29, E6. [Google Scholar] [CrossRef] [Green Version]
- Rosin, B.; Slovik, M.; Mitelman, R.; Rivlin-Etzion, M.; Haber, S.N.; Israel, Z.; Vaadia, E.; Bergman, H. Closed-loop deep brain stimulation is superior in ameliorating parkinsonism. Neuron 2011, 72, 370–384. [Google Scholar] [CrossRef] [Green Version]
- Little, S.; Pogosyan, A.; Neal, S.; Zavala, B.; Zrinzo, L.; Hariz, M.; Foltynie, T.; Limousin, P.; Ashkan, K.; FitzGerald, J. Adaptive deep brain stimulation in advanced Parkinson disease. Ann. Neurol. 2013, 74, 449–457. [Google Scholar] [CrossRef] [Green Version]
- Brown, P.; Oliviero, A.; Mazzone, P.; Insola, A.; Tonali, P.; Di Lazzaro, V. Dopamine dependency of oscillations between subthalamic nucleus and pallidum in Parkinson’s disease. J. Neurosci. 2001, 21, 1033–1038. [Google Scholar] [CrossRef] [Green Version]
- Hammond, C.; Bergman, H.; Brown, P. Pathological synchronization in Parkinson’s disease: Networks, models and treatments. Trends Neurosci. 2007, 30, 357–364. [Google Scholar] [CrossRef]
- Neumann, W.J.; Degen, K.; Schneider, G.H.; Brücke, C.; Huebl, J.; Brown, P.; Kühn, A.A. Subthalamic synchronized oscillatory activity correlates with motor impairment in patients with Parkinson’s disease. Mov. Disord. 2016, 31, 1748–1751. [Google Scholar] [CrossRef] [Green Version]
- Godinho, C.; Domingos, J.; Cunha, G.; Santos, A.T.; Fernandes, R.M.; Abreu, D.; Gonçalves, N.; Matthews, H.; Isaacs, T.; Duffen, J. A systematic review of the characteristics and validity of monitoring technologies to assess Parkinson’s disease. J. Neuroeng. Rehabil. 2016, 13, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]

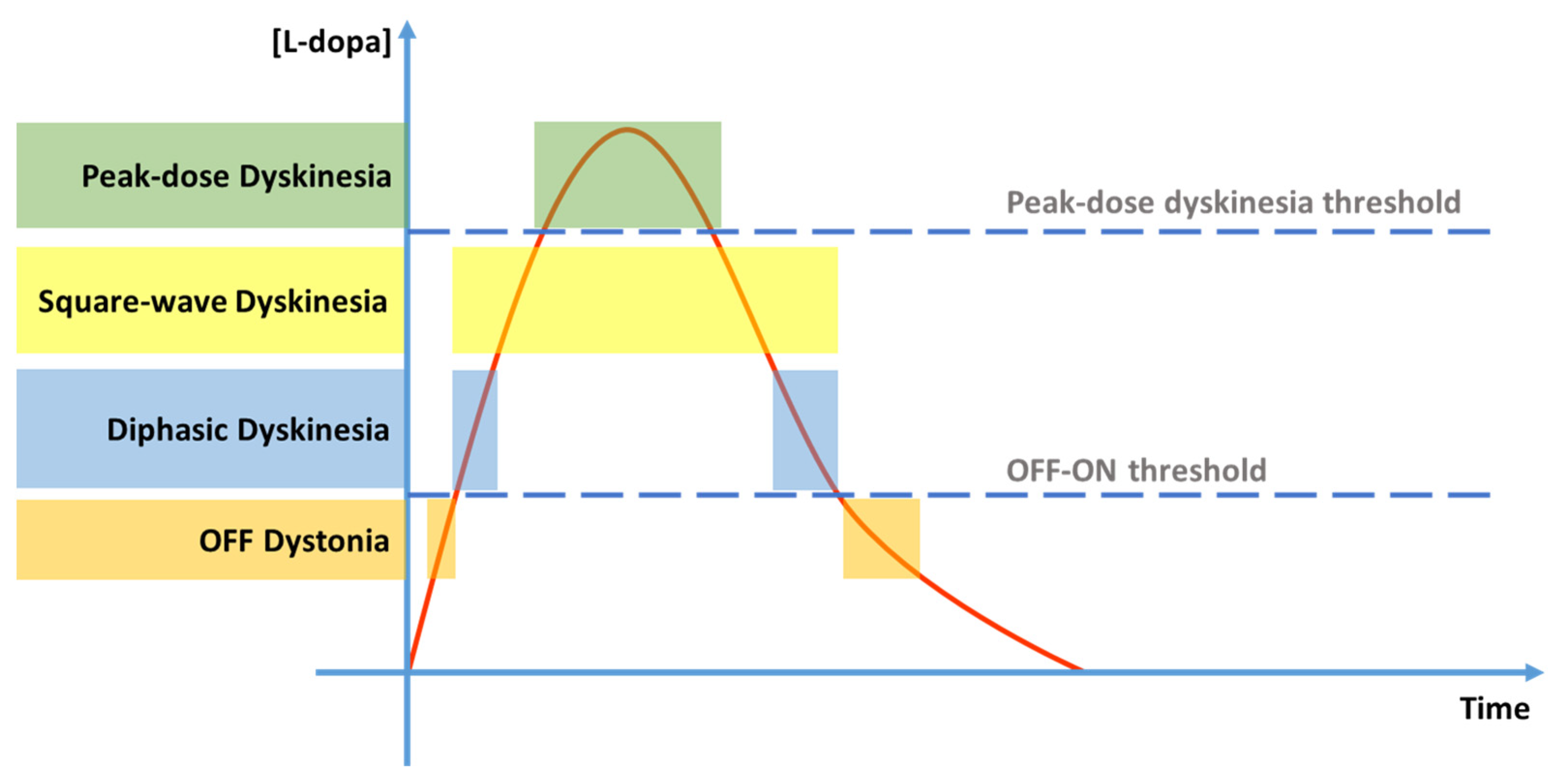
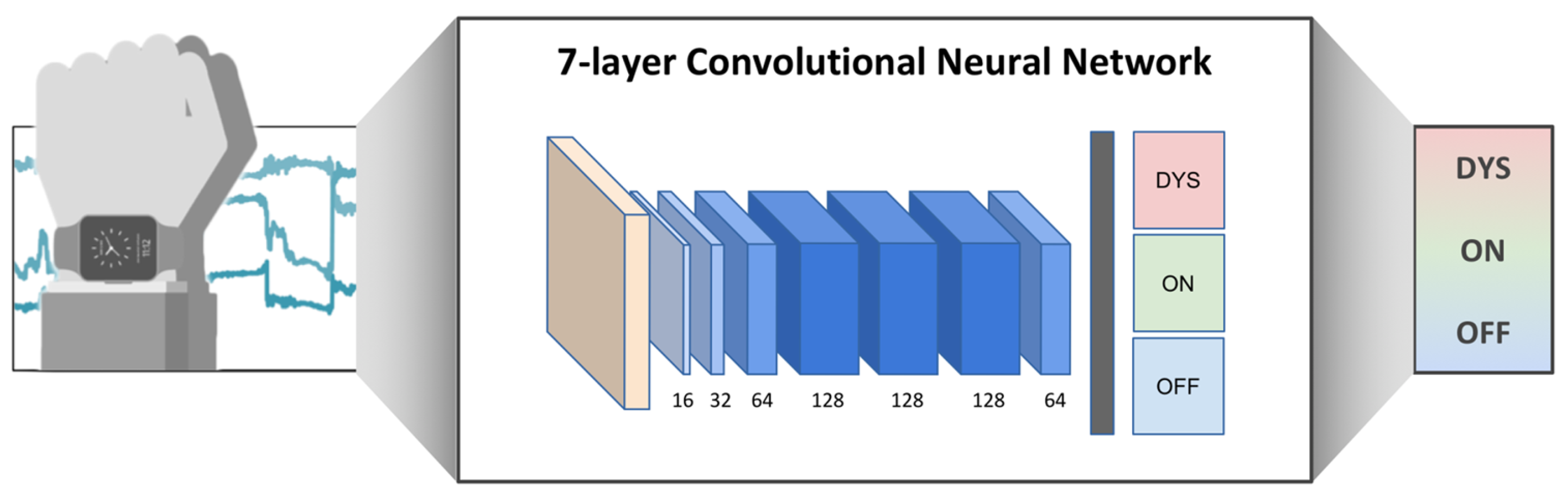





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Function | Gene Variant | Ref. |
|---|---|---|---|
| COMT | Involved in the metabolism of dopamine | Val158Met | [30] |
| DRD2 | Encodes the D2 subtype of the dopamine receptor | ANKK1 TTCTA haplotype | [32] |
| BDNF | Involved in neuronal survival and synaptic plasticity | Val66Met | [34] |
| SLC6A3 | Dopamine transporter gene (DAT) | 40-bp VNTR | [31] |
| GRIN2A | Encodes a subunit of the NMDA receptor, involved in glutamate transmission | rs7192557 and rs8057394 | [33] |
| Type of Study | N° Patients | Levodopa Exposure | % Dyskinetic Patients | Ref. |
|---|---|---|---|---|
| Meta-analysis | 335 (pre-levodopa era) | 3–6 w | 23.3 | [41] |
| 606 (pre-levodopa era) | 2–4 m | 34.5 | ||
| 606 (pre-levodopa era) | 5–6 m | 53.4 | ||
| 2645 (pre-levodopa era) | 7–12 m | 54.8 | ||
| 982 (pre-levodopa era) | 1–2 y | 71.9 | ||
| 297(pre-levodopa era) | 2.5–3.5 y | 56.7 | ||
| 432 | 7–12 m | 7 | ||
| 575 | 1–2 y | 28.7 | ||
| 747 | 2.5–3.5 y | 26.9 | ||
| 1599 | 4–6 y | 36.2 | ||
| 514 | 9–15+ y | 87.8 | ||
| Prospective, double-blind, randomized clinical trial | 88 | 5 y | 45 | [24] |
| 27 | 10 y | 77.8 | ||
| Clinico-pathological | 42 | 6.4 y | 31 | [39] |
| 14.3 y | 61.9 | |||
| Community-based | 87 | <5 y | 11 | [7] |
| 6–9 y | 32 | |||
| >10 y | 89 | |||
| Community-based | 126 | 5 y | 30 | [40] |
| 10 y | 59 | |||
| DATATOP | 352 | 20.5 m | 30 | [42] |
| FIRST | 187 on IR | 5 y | 20.6 | [43] |
| 193 on CR | 5 y | 21.8 | ||
| 056-study | 45 | 5 y | 45 | [23] |
| CALM-PD study | 131 | 3 y | 54 | [44] |
| PELMOPET study | 90 | 3 y | 26 | [22] |
| ELLDOPA study | 92 (150 mg/die) | 8 m | 3 | [21] |
| 88 (300 mg/die) | 8 m | 2 | ||
| 91 (600 mg/die) | 8 m | 16 | ||
| STRIDE-PD study | 372 (Ldopa) | 2 y | 32 | [28] |
| 373 (Ldopa + entecapone) | 38 | |||
| SIDNEY study | 52 | 15 y | 94% (12% severe dyskinesias) | [45] |
| Neurotransmitter | Receptor | Therapeutic Target | Effect on LIDs | |
|---|---|---|---|---|
| Serotonergic system | Serotonin | 5-HT1A 5-HT1B | 5-HT1A and 5-HT1B Receptor Agonists | - Density of serotonergic terminals in the striatum directly correlates with the severity of LIDs - Serotonergic neurons convert exogenous levodopa into dopamine and release it without autoregulatory feedback |
| 5-HT2A 5-HT2C | 5-HT2A Receptors Antagonists | |||
| 5-HT3 |
| |||
| Glutamatergic system | Glutamate | mGluR | MGluR antagonist | - Altered trafficking - Hyperactive |
| NMDA | NMDA receptor antagonist (GluN2A/B subunit) | - Altered trafficking - Alteration of subunit composition - Supersensitivity in the putamen following long-term levodopa | ||
| AMPA | AMPA receptor antagonist | - Altered trafficking - Increased index of rectification (IR) of AMPA current in striatal medium spiny neurons - Increased activity of Ca2+ -permeable AMPAR due to hyperphosphorylation of GluR1 subunit | ||
| Noradrenergic system | Noradrenaline | α-1/2 | α receptor antagonist | - NA loss causes parkinsonism and spontaneous dyskinesias in DBH knock-out mice - NA infusion in the striatum promotes LID in hemiparkinsonian rats - NAT activity should re-uptake DA and reduce LIDs - Controversial evidence |
| β-1/2 | β receptor antagonist | |||
| Cholinergic system | Acetylcholine | nAChR (α4β2* and α6β2* subtypes) | β2* nAChR agonist | β2 subtype reduces LIDs, but nAChR vary over the course of PD |
| mAChR (m1 to m5) | Variable results with muscarinic antagonists | Not established | ||
| Opioid system | Enkephalin | δ | δ—receptor selective antagonist | - Elevated levels of dynorphin B, α-neoendorphin and Dynorphin A in the dorsolateral striatum and SN - μ and δ receptors promote LIDs - κ receptor reduces LIDs |
| β-endorphin Endomorphin | μ | μ—receptor selective antagonist | ||
| Dynorphin A Dynorphin B α-neoendorphin β-neoendorphin | κ | κ—receptor selective agonist | ||
| Endocannabinoid system | Anandamide 2-AG | CB1/2 | CB-receptor agonist | - The stimulation of the CB1 receptors reduces LIDs by: ● Desensitization of DA receptors ● Normalizing aberrant glutamate release - Net anti-dyskinetic effect - CB1 receptors can also promote LIDs by dopamine synthesis in serotonergic raphe-striatal fibers |
| TRP | Not established | Not established | ||
| PPAR | Not established | Not established | ||
| Adenosinergic system | Adenosine | A2A | - A2A receptor antagonist | - Not clearly established, but the activation of this receptor in the striatum regulates amplification of dopamine and glutamate release - A direct anti-dyskinetic effect seems unlikely |
| A2B | Not established | Not established (poorly expressed in CNS) | ||
| A3 | Not established | Not established (poorly expressed in CNS) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
di Biase, L.; Pecoraro, P.M.; Carbone, S.P.; Caminiti, M.L.; Di Lazzaro, V. Levodopa-Induced Dyskinesias in Parkinson’s Disease: An Overview on Pathophysiology, Clinical Manifestations, Therapy Management Strategies and Future Directions. J. Clin. Med. 2023, 12, 4427. https://doi.org/10.3390/jcm12134427
di Biase L, Pecoraro PM, Carbone SP, Caminiti ML, Di Lazzaro V. Levodopa-Induced Dyskinesias in Parkinson’s Disease: An Overview on Pathophysiology, Clinical Manifestations, Therapy Management Strategies and Future Directions. Journal of Clinical Medicine. 2023; 12(13):4427. https://doi.org/10.3390/jcm12134427
Chicago/Turabian Styledi Biase, Lazzaro, Pasquale Maria Pecoraro, Simona Paola Carbone, Maria Letizia Caminiti, and Vincenzo Di Lazzaro. 2023. "Levodopa-Induced Dyskinesias in Parkinson’s Disease: An Overview on Pathophysiology, Clinical Manifestations, Therapy Management Strategies and Future Directions" Journal of Clinical Medicine 12, no. 13: 4427. https://doi.org/10.3390/jcm12134427