
Normal-Tension Glaucoma and Potential Clinical Links to Alzheimer’s Disease


1
Eugene and Marilyn Glick Eye Institute, Department of Ophthalmology, Indiana University School of Medicine, Indianapolis, IN 46202, USA
2
Pharmacology and Toxicology, Indiana University School of Medicine, Indianapolis, IN 46202, USA
3
Stark Neurosciences Research Institute, Indianapolis, IN 46202, USA
*
Author to whom correspondence should be addressed.
J. Clin. Med. 2024, 13(7), 1948; https://doi.org/10.3390/jcm13071948
Submission received: 19 February 2024
/
Revised: 21 March 2024
/
Accepted: 25 March 2024
/
Published: 27 March 2024
(This article belongs to the Special Issue Latest Advances in Glaucoma, Cataract and Refractive Surgery: Expert Views)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Glaucoma is a group of optic neuropathies and the world’s leading cause of irreversible blindness. Normal-tension glaucoma (NTG) is a subtype of glaucoma that is characterized by a typical pattern of peripheral retinal loss, in which the patient’s intraocular pressure (IOP) is considered within the normal range (<21 mmHg). Currently, the only targetable risk factor for glaucoma is lowering IOP, and patients with NTG continue to experience visual field loss after IOP-lowering treatments. This demonstrates the need for a better understanding of the pathogenesis of NTG and underlying mechanisms leading to neurodegeneration. Recent studies have found significant connections between NTG and cerebral manifestations, suggesting NTG as a neurodegenerative disease beyond the eye. Gaining a better understanding of NTG can potentially provide new Alzheimer’s Disease diagnostics capabilities. This review identifies the epidemiology, current biomarkers, altered fluid dynamics, and cerebral and ocular manifestations to examine connections and discrepancies between the mechanisms of NTG and Alzheimer’s Disease.
1. Background
Glaucoma is a group of optic neuropathies and the leading cause of irreversible blindness worldwide [1,2,3,4,5]. It is characterized by retinal ganglion cell (RGC) death and a distinct pattern of progressive peripheral visual field loss [3,6,7]. Primary open-angle glaucoma (POAG) is the most prevalent form of glaucoma and comprises almost 74% of all glaucoma cases [5,8]. The major risk factor for POAG is elevated intraocular pressure (IOP) [3], although a proportion of POAG patients present with the characteristic visual loss and IOP lower than 21 mmHg [9]. These patients are classified as having normal tension glaucoma (NTG), which represents 20–30% of glaucoma cases [9,10,11]. Since IOP levels lower than 21 mmHg are considered normal, these NTG patients are consequently diagnosed at later disease stages once notable visual loss has occurred [12]. Although some NTG patients benefit from therapeutics that lower IOP by 30%; in the collaborative normal-tension glaucoma study group, one-third of treated patients still experienced continued visual field loss [13]. Further, lowering IOP is currently the only targetable therapeutic for all glaucoma types [14]. This demonstrates the need for understanding IOP-independent pathological mechanisms of NTG to effectively develop neuroprotective treatments. Investigations into the nuances of NTG presents a wealth of data that NTG may have associations with another neurodegenerative disease, Alzheimer’s Disease (AD).
Recent neuroimaging studies have found that NTG neuronal degeneration expands beyond the eye and visual pathways within the brain [15,16]. These non-ocular manifestations support the hypothesis of IOP-independent mechanisms of NTG, and differing disease pathophysiology compared to POAG. The exploration of this disease within the domains of neuro-ophthalmology and neurodegenerative research has sparked significant debate and is emerging as a highly intriguing topic of global interest. Several studies have found NTG to be significantly associated with impaired cognitive functioning [17,18] and a higher incidence of AD [19,20,21,22], while others have disputed these associations [23,24]. In addition to a higher prevalence of NTG [25,26], predisposing genetic factors [27] and evidence of an association with cognitive changes [17,28] are stronger in studies with east Asians.
From the standpoint of AD, there is a growing interest in the study of ocular manifestations of AD. With a projected 150 million people living with dementia by 2050 [29], the need for early and noninvasive methods of diagnosis is integral. As an integral part of the central nervous system, the eye offers potential insights to examine cerebral neurodegeneration through retinal manifestations [30,31]. This review will describe the current knowledge on common pathophysiological mechanisms between NTG and AD, including biomarkers, cerebrospinal fluid dynamics, vascular dysfunction, cerebral and retinal changes, and the missing links between the two diseases (Figure 1).
2. Progressive Trends in NTG and AD Epidemiology
Both NTG and AD are progressive neurodegenerative disorders with risk factors of older age and the female sex [32]. NTG is a form of glaucoma more commonly found in the elderly, primarily because NTG often goes unnoticed until significant vision loss has occurred. Without identifiable IOP changes, it is highly underdiagnosed in younger populations and is most frequently diagnosed at approximately the age of 60 [12]. It is essential to note that this delay in diagnosis is not necessarily due to a later age of onset but rather a later age of detection [33]. On the other hand, AD typically onsets in older populations, with its prevalence increasing from 10% in those aged 65, to 40% in those over the age of 85 [34]. Visual disturbances are among the earliest reported symptoms of AD [35,36], and could serve as a potential early spectrum indicator of AD.
Furthermore, women are disproportionately affected by both NTG [33] and AD [37]. Women are twice as likely to develop AD, and this increased risk is thought to result from a combination of factors including sex chromosomes, hormones, brain structure, and a longer average lifespan [38]. Recent research has been focusing on sex differences in microglial response to neuroinflammation and subsequent neurodegeneration [39]. Similar mechanisms may explain the higher prevalence of NTG in females as well as their increased incidence of vascular dysregulation conditions such as Raynaud’s phenomenon [40,41,42], migraines, vasospasms [43], and low blood pressure [44,45,46].
Another risk factor for NTG is race. In populations diagnosed with glaucoma, NTG is seen at the highest proportions in east Asian populations, ranging from 77% to 92% [47,48,49,50]. This is a much higher frequency compared to approximately the 30% NTG proportion seen in Caucasian POAG populations [10,51]. African populations also have a higher NTG prevalence than Caucasians, with 30–57.1% of African POAG patients diagnosed with NTG [52,53]. However, African patients still have significantly lower frequencies of NTG cases compared to Asian NTG prevalence. This suggests that race may contribute to the pathogenesis of NTG, but it remains unclear whether this association is influenced by genetic factors or environmental factors such as socioeconomic factors leading to lack of eye care.
Beyond age, gender, and race, there are several diseases identified as potential risk factors for both NTG and AD. NTG is highly associated with systemic vascular diseases [54] including migraines [55], low systemic blood pressure [56], low diastolic ocular perfusion pressure [57], and AD [19,20,21,22,28] as mentioned previously. AD risk is also closely associated with vascular diseases [58] and vascular risk factors such as diabetes, hypertension, and dyslipidemia [59,60]. Those with migraine history have a stronger risk of developing AD than those without migraine history, with higher significance, if comorbidly obese [61]. Retinal vessel abnormalities have been found in both early stages of NTG and AD patients [21], suggesting low perfusion pressure in retinal and cerebral microvasculature [58,62] could potentially be indicative of disease pathology.
The risk of AD in conjunction with vascular diseases [45] and vascular risk factors such as diabetes, hypertension, and dyslipidemia [46,47] is closely associated in Asian populations. A large-scale Taiwanese, retrospective cohort study included over 15,300 NTG subjects and 61,000 demographically matched subjects without glaucoma to examine the cumulative risk of AD between the two groups. The NTG group had a higher prevalence of diabetes, HTN, hyperlipidemia, coronary artery disease, and stroke, similar to AD. However, adjusting for these diseases using Cox regression still showed over 50% greater risk of developing AD in those with NTG than in comparable groups (p < 0.0001) [28]. A 10-year nationwide cohort study completed in Korea with 1469 NTG and 7345 controls also found a significantly higher risk of developing AD (p < 0.001), but not Parkinson’s disease (p = 0.983) [63]. Conversely, a predominantly Caucasian-based study in Denmark consisting of 69 NTG patients showed no increase in developing AD during a 12.7-year follow-up [64]. More research may be needed to examine the discrepancies seen between Asian and Caucasian NTG in conjunction with AD associations.
Data are more limited in associations between NTG and cognitive impairment. A 2021 Australasian study compared T-MoCA scores in 144 NTG and 144 high-tension glaucoma (HTG) participants over the age of 65, matched for demographics and ocular parameters. They found cognitive impairment was significantly more prevalent in the NTG cohort [17]. Another study completed in the United States found no significant difference in executive function, learning, and memory between 50 NTG and 50 control subjects aged over 50 years [23]. These conflicting findings may be due to differences in size, age group, and race/genetic makeup of the samples. To determine a true connection between cognitive impairment and NTG, future studies are needed to follow-up a large cohort with repeat testing.
3. Comparing Genetic Biomarkers in NTG and AD: A Comprehensive Analysis
Certain conserved biomarkers in AD and NTG could potentially indicate genetic predispositions and disease stage. These biomarkers include Amyloid b (Aβ) peptide, tau protein, Apolipoprotein (APOE), Optineurin (OPTN), and TANK Binding Kinase 1 (TBK1). Biomarkers classically associated with AD are Aβ peptide, tau protein, and APOE [65,66,67,68,69,70,71,72], while biomarkers classically associated with glaucoma include TBK1, OPTN and myocilin [73,74,75,76,77,78,79,80,81,82,83]. Detection of these biomarkers at early stages of AD or NTG is useful for both diagnostics and insight into pharmaceutical targets. Identifying correlative biomarkers could provide potential tools for early detection, treatment, and prevention of a variety of neurodegenerative diseases including AD and NTG.
3.1. Insights into Aβ and Tau as Biomarkers in NTG and AD
The relationship between Aβ and Tau with AD cases has been extensively studied; known AD pathology indicates the concurrence of extracellular deposits of Aβ peptide fibrils and intracellular neurofibrillary tangle-induced degeneration from hyperphosphorylated tau [65,66,69,70]. The amyloid hypothesis suggests that the accumulation of Aβ peptide plaque deposits cause neuronal changes and eventual cell death that is observed in AD [65,68,84]. The Aβ peptide is made from the amyloid precursor protein (APP) which is primarily found in neurons, astrocytes, microglia, endothelial cells, and meninges [85,86,87]. The APP is cleaved to produce Aβ peptide [88]. Aβ peptide residues 1-42 (Aβ42) have been found to have differing conformational states compared to Aβ peptides residues 1-40 (Aβ40), that increase their inclination towards plaque formation [68,89,90]. It is suggested that Aβ42 deposits start to occur years before the clinical signs or symptoms of AD appear [68,91]. In glaucoma, APP and the Aβ peptide can be indicative of RGC death. Under normal conditions, APP can be transported anterogradely and retrogradely between the brain and optic nerve, where it can ultimately accumulate in retinal neurons [92,93]. However, in glaucoma there is an inhibition of anterograde and retrograde transport [94,95,96]. This has been exemplified in an ocular hypertensive mouse model which showed increased levels of Aβ and APP in the pial/dural complex, the optic nerve, and the RGC layer [93]. The accumulation of APP can drive metabolic changes and alter APP homeostasis, ultimately impacting neuronal viability [93]. However, to the extent of our knowledge, there is little understanding for the APP transport in NTG models.
3.2. Role of APOE4 Allele in AD and NTG: A Genetic Investigation
A factor associated with accumulation of Aβ peptides is the APOE gene e4 allele (APOE4) [97,98,99,100,101]. APOE4 does not bind as tightly to Aβ42 as the APOE e2 and 3 allelic forms, which blocks APOE4 clearance and results in its accumulation along with the Aβ peptide [68,72]. The APOE4 frequency is approximately 40% in central Africa, 24% in Malaysian aboriginals, 26% in Australia where it is especially prevalent among Australian aboriginals, and up to 28% in certain Native American tribes [100,101]. Further, the correlation between the incidence of APOE4 and AD is higher in Japanese populations compared to Caucasians [100]. In addition, a heterozygote with the APOE4 has a 3-fold greater chance of developing AD, and a homozygote has up to a 15-fold greater chance [102,103]. Alternatively, the APOE e2 allele (APOE2) has been shown to have protective effects against AD [100]. In the eye, APOE is expressed in muller cells, retinal ganglion cells, and retinal pigment epithelium of the retina [104,105]. The relationship between APOE4 and glaucoma continues to have conflicting evidence with multiple studies showing either positive or negative associations [27,106,107,108,109,110]. Evidence in support of the positive association between APOE4 and glaucoma was found in studies of Tasmanian patients with NTG and HTG. NTG patients had slightly higher frequencies of APOE4 inheritance at 38%, compared to the frequency of 34% of HTG patients with APOE4 [108]. Evidence in support of the negative association has been shown in a 2011 study of glaucoma subjects of Chinese descent. The genotyping analysis showed a decreased frequency of APOE4 in NTG patients, which lead the authors to conclude that APOE4 could have protective effects against NTG [27]. In widespread genome-wide association studies collected from the Mass Eye and Ear Infirmary and the National Eye Institute Glaucoma Human Genetics Collaboration in 2020, the findings reported that there was a statistically significant association for APOE4 with both HTG and NTG. Due to the highly significant association between APOE4 and NTG patients, the report concluded that the mechanism of APOE’s role was directly associated with retinal cells, rather than dependence on IOP changes [106]. With so many variances between publications on whether APOE4 is protective or detrimental to glaucoma development, a possible explanation of the differences in these studies can be attributed to diet. Many of the populations described to have higher frequencies of APOE4 are less predisposed to Western diets. The effects of a Western diet can result in higher cholesterol levels which both stimulate APOE activity and result in further cholesterol production in the central nervous system (CNS) [101,111]. Additionally, APOE4 is the allelic form that is least effective at exporting cholesterol, which increases the quantity of cholesterol present at the plasma membrane that can interact with the Aβ peptide, which can result in toxic Aβ accumulation [112,113]. Discrepancies between APOE regulation in AD and NTG can potentially be attributed to interaction with monocyte chemoattractant protein-1 (MCP-1/CCL2) [106]. MCP-1/CCL2 is a type of chemokine responsible for monocyte and macrophage migration and infiltration [114]. A reduction in inflammatory response within the retina of patients with the APOE4 can be protective because of decreased immune-mediated damage. However, reduced inflammation might be harmful in AD because a robust immunological reaction is required to address Aβ42-induced cytotoxicity and plaque accumulation [106,115,116]. Collectively, the identification of APOE4 indicates the likelihood of Aβ plaques that can cause AD. However, elucidating the link between APOE4 and NTG, especially in different racial groups, remains a necessity.
Oxidative stress conditions can further modulate the activity of APOE4, which can alter its role in AD and glaucoma. APOE4 is vulnerable to the molecule 4-hydroxynonenal (4-HNE); 4-HNE is a product of lipid peroxidation under stress conditions [117,118]. Ultimately, APOE4 cannot remove 4-HNE [117,118,119]. Oxidative stress causes an upregulation of APOE4, which has inherently low antioxidant properties and is more susceptible to oxidative damage compared to the other APOE alleles [119,120,121]. This effect has been observed in human AD patients with APOE4 having higher incidences of plasma oxidation compared to other APOE alleles [104,122,123,124,125]. In AD, 4-HNE has been shown to expedite Aβ depositions and cytotoxicity. Overall assessments of AD patients’ brains show considerable levels of oxidative stress, lipid peroxidation, and low glucose metabolism [119,126,127,128,129,130,131,132]. These cellular changes are indicative of AD, which outline possible biomarkers for detection of AD [131,133]. In a study of Caucasian NTG patients there was higher total cholesterol, triglyceride, hyperlipidemia indicators, total oxidant status, and oxidative stress index levels compared to pseudoexfoliation glaucoma patients [134,135]. Hyperlipidemia can cause oxidative stress through elevated levels of reactive oxygen species (ROS), which further explains the oxidative load observed [134,136,137]. Cholesterol elevations can be explained by APOE’s role in astrocytes; APOE and cholesterol are co-secreted by astrocytes, resulting in additional neuronal secretion of cholesterol, which can cause ocular increases in cholesterol [111]. Patients with AD and NTG are both predisposed to elevated levels of oxidative stress through APOE4, which can result in metabolic- and stress-related changes that can further promote disease progression. However, especially seen in AD, this can also provide a tool to potentially predict disease in earlier stages. Early predictions of AD using oxidative stress and glucose metabolism analysis can potentially lead to identification of NTG at early stages as well.
3.3. Optineurin in Focus: Bridging the Gap between NTG and AD
The combined impacts of cholesterol and oxidative stress can have inhibitory effects on autophagy that can drive AD development, specifically, with accumulation of the autophagy receptor, Optineurin (OPTN) [138]. The dysregulation of autophagy in AD and NTG indicates that OPTN can be a conserved biomarker that can be used to indicate neurodegeneration stemming from oxidative load, cholesterol levels, or neuroinflammation [77,138,139,140]. There is observed to be correlation between Aβ accumulation and increased cholesterol in the gray matter of AD brains [112,141,142,143]. In mouse models, it has been established that excess cholesterol will result in mitochondrial oxidative stress caused by excess Aβ accumulation. Experimentally, excess cholesterol loading in animal models can be accomplished by depleting the antioxidant glutathione. Depletion of glutathione has been found in AD, NTG, and POAG, indicating an increase in oxidative damage, glutamate neurotoxicity, and cholesterol [123,143,144,145,146,147,148,149,150]. The presence of the autophagy receptor, OPTN, has been found to be accumulated in aged AD models along with corresponding levels of elevated mitochondrial cholesterol levels, thus indicating that OPTN can be used to monitor the successful clearance of ROS and other cargo in AD [138].
In addition to the known role of OPTN in autophagy, the OPTN gene is known for its historical association with glaucoma [73,74,77]. It is believed that OPTN mutations may account for nearly 17% of hereditary forms of NTG [77]. The missense mutation of E50K to the OPTN gene is associated with autosomal dominant inheritance of early-onset familial NTG [73,151,152,153]. An additional OPTN mutation is the variant M98K, which is especially prevalent in NTG patients in Japan [153,154,155]. In vitro and in vivo models illustrate that the E50K mutation and M98K variant can result in RGC death by impaired autophagy pathways [156,157,158,159].
A specific relationship that works to promote autophagy is OPTN and TBK1. OPTN and TBK1 contribute to approximately 2–3% of NTG cases. Patients with these mutations usually develop severe NTG disease by age 40. The E50K mutation enhances TBK1 and OPTN binding and consequently leads to cell death [79,160]. Initiation of autophagy is caused by TBK1 phosphorylation of OPTN, promoting recruitment of MAP1LC3B (LC3B), which is required for autophagy flux [73,79,161]. A hypothesis for TBK1’s involvement in glaucoma pathogenesis is that it disrupts either autophagy or NF-kB signaling, which can lead to RGC apoptosis, further driving the glaucoma phenotype [162]. Overall, in AD and NTG, OPTN can provide evidence of dysregulated autophagy pathways that can cause accumulation of inflammatory or oxidative materials.
4. Vascular Dysregulation and Impaired Pressure Dynamics in NTG and AD: A Unifying Perspective
In addition to higher sensitivity to normal IOP, three other theories aid in explaining NTG pathophysiology. These include vascular dysregulation, an abnormal translaminar pressure gradient, and impaired cerebrospinal fluid (CSF) circulation. No singular theory comprehensively explains NTG pathogenesis; therefore, it is currently believed that a combination of these pathological changes may cause glaucomatous optic disc neuropathy and RGC apoptosis. In this section, we will discuss these NTG mechanisms and their possible connections to AD pathogenesis [163].
4.1. Vascular Flow and the Link to NTG in AD
A leading mechanism for NTG pathogenesis is vascular dysregulation, resulting in decreasing perfusion that can contribute to retinal cell death [13,62,164,165]. Reduced blood flow to the retina, choroid, iris, and optic nerve has been examined in NTG patients [62]. Morphologically, this can be explained by significantly smaller retinal vessel diameter [166,167] and reduced density [168] in NTG patients compared to HTG patients. Similarly, early-stage AD patients are associated with retinal vessel abnormalities such as narrowed veins and decreased retinal blood flow in these veins [169]. Retinal arteriovenous nicking, consistent with narrowed veins, has been identified as predictive of long-term risk of cerebral atrophy, a risk factor for AD [170]. Suboptimal retinal vascular geometry is associated with impaired cognition [171]. Since brain microvascular abnormalities precede cognitive changes in AD, retinal vessel changes could potentially be used as indicators for cerebral microvasculature and thus early diagnosis of AD [58].
Another compounding issue in vascular dysregulation in AD and NTG is angiogenesis. Angiogenesis is the process in which new blood vessels are formed. Angiogenesis is physiologically vital for normal development and wound repair [172,173]. Several molecules are responsible for inducing angiogenesis, including vascular endothelial growth factor (VEGF) [173,174]. Hypoxic environments have been demonstrated to increase VEGF and activate endothelial cells [175]. This has been exemplified in the retina where mRNA expression in retinal pigment epithelial cells and retinal pericytes caused a significant increase in retinal endothelial cell growth [176]. Upregulated activation of endothelial cells contributes to a noxious environment by secretion of proteases and inflammatory factors that may promote neuronal cell death [175]. In terms of NTG, this can indicate how already demonstrated reduced blood flow to the retina and optic nerve [62], can result in ischemia that can drive VEGF activity in the retina [177]. Hypoxia-driven damage has also been observed in AD and other ocular neurodegenerative diseases, such as neovascular Age-Related Macular Degeneration (AMD). In these diseases, hypoxia can drive oxidative stress that can result in cell damage and death [178]. Additionally, VEGF has also been implicated in AD, with evidence that VEGF co-aggregates with AB plaques [179] and increased serum VEGF correlates with increased AD severity in human Apoe4 carriers [180]. In AD mouse models, VEGF paradoxically contributes to reduced cerebral blood flow, likely accelerating cognitive decline [181]. Recent studies targeting VEGF receptor-2, a more specific receptor for vascular VEGF signaling than its counterpart VEGF receptor-1 [182], show promising results in cortical capillary clearance in AD mice models [183,184]. Due to the significant impacts of VEGF, it has become a pharmacologically relevant molecule. Pharmaceutically available VEGF-inhibitors can be delivered as intravitreal injection treatments for neovascular AMD and diabetic macular edema (DME) [178,185]. A specific example of a VEGF-inhibitor that has been used in both treatment of AD and NTG is bevacizumab (Avastin) [186,187]. In an AD mouse model, Avastin was able to restore long-term memory, decrease glial activation, and reverse genetic changes associated with blood–brain barrier dysfunction, as seen in AD models [187]. Alternatively, in clinical trials using Avastin in patients with branch retinal vein occlusion with or without NTG, those with NTG had worsened visual acuity even with Avastin treatment. Further, repeated intravitreal injections of Avastin in those with AMD and DME have been shown to cause sustained increased IOP, which can increase the risk for glaucoma development or accelerate existing glaucoma [188]. Additional studies support the finding that repeated Anti-VEGF injections can lead to elevated IOP in cohorts of patients diagnosed with DME, neovascular AMD [189,190], and non-diabetic patients [191], thus indicating the complicated interplay between NTG and efficacy of anti-VEGF treatments. Furthermore, the risk for intravitreal anti-VEGF treatments extends outside of the eye and has been implicated in possible Parkinson’s-like events, hypertension-induced brain hemorrhage, and dementia [192,193,194]. Further studies examining the implication of VEGF signaling in NTG and AD are necessary to determine targeted anti-VEGF therapeutic efficacy in these diseases.
In addition to vascular morphology changes seen in AD and NTG, decreased retinal perfusion may be due to vessel dropout. The choroid is the layer of blood vessels between the retina and sclera, providing nourishment to RGCs and regulated by the autonomic nervous system. A large-scale study found that compared to controls, the AD group had significantly thinner choroid at all analyzed points except for the temporal macula [30], which is consistent with previous studies [195,196,197,198]. Since choroid thinning is associated with aging, demographically matched control groups were a strength of the study. Similarly, NTG is associated with reduced choroid vascular index (CVI) and thickness, predominantly in Haller’s layer, the larger-sized vessel layer of the choroid [199]. There was no statistical difference between NTG and controls for CVI of choriocapillaris and Sattler’s Layer, the small-to-medium choroidal vessel layer. The breakdown of choroidal layers has not been studied for AD, to the extent of our knowledge. Of note, retinal vessel density is positively correlated with overall cognition, memory, executive function, and visual–spatial perception functions [200]. More studies are needed to compare the coincidental findings of thinning choroid vessels in both diseases and pathological implications for the eye.
Furthermore, poor autoregulation [201,202] is more common in NTG patients than in HTG patients, causing transient episodes of ischemia and reperfusion injury [203]. This dysregulation is more evident at night when blood pressure (BP) naturally dips, further worsening low baseline diastolic BP and ocular perfusion pressure in NTG patients. Low nocturnal diastolic ocular perfusion pressure has been identified as an independent predictor of glaucomatous visual field progression in NTG patients [57]. These nocturnal pressure drops are especially evident in patients being treated for hypertension, particularly with systemic beta blockers [204]. The duration and amplitude of diastolic BP lowering during sleep are implicated in worsening of disease progression [56,205,206,207]. Poor vascular regulation is thought to influence glaucoma progression by the deprivation of RGC nutrients, increasing optic nerve sensitivity to IOP [62]. A study examined nocturnal BP variation in those with mild cognitive impairment (MCI) and normal controls, finding abnormal nocturnal BP patterns significantly higher in the MCI group [208]. MCI is defined as a transitional group between healthy aging and dementia, so early findings may be indicative of AD pathogenesis. While normal nocturnal BP dips slightly, the MCI group included significant abnormal patterns such as extreme dippers, non-dippers, and risers [208]. This suggests that abnormal autoregulation in nocturnal BP profiles is a strong indicator of MCI [208], and the extreme dipping in nocturnal pressures seen in NTG may partially explain the association of cognitive impairment with NTG [57]. Although there are differing abnormal patterns seen between MCI and NTG, the mechanism of vascular dysregulation may relate to a shared underlying pathogenesis, demonstrating the need for more research to better understand the two diseases.
Commonalities in vascular abnormalities are not limited to the eye. As mentioned, in the Section 2, both NTG and AD are associated with abnormalities in systemic vasculature and highly comorbid with systemic vascular diseases such as migraine, blood pressure dysregulation, or vascular risk factors [54,58,61]. Additionally, a prior study examined cerebral blood flow in patients with NTG and control subjects using single photon emission computed tomography scans to compare NTG perfusion patterns with AD-like vascular patterns [22]. This pattern is characteristic of lower regional cerebral blood flow from the middle cerebral artery, causing lower perfusion pressures. Although none of the patients were diagnosed with AD, 7 of the 31 NTG patients exhibited AD-like perfusion pattern, which is much higher than the 1% incidence of AD in the normal population aged 75 and over. Additionally, a 2-year follow-up period found that NTG patients with these AD-like patterns had more rapidly progressing visual field loss than those without the patterns.
These common vascular abnormalities should be examined at the cellular level. One potential example: NTG patients have a higher prevalence of altered endothelial cell function [209,210] and increased plasma levels of endothelin-1 (ET-1) compared to controls [211,212,213]. ET-1 is a potent vasoconstrictor, mainly secreted by endothelial cells. High levels of ET-1 can induce fibrosis of vascular cells and stimulate the production of ROS [214]. However, a Japanese study showed no difference in ET-1 levels in those under the age of 60 between NTG, POAG, and healthy controls [215]. It is therefore possible that the ET-1 elevation is related to aging or later progression of disease. Interestingly, AD is also associated with an elevation in ET-1 plasma levels, and Aβ is found to significantly upregulate secretion [216]. ET-1 should be further characterized in the vascular dysregulation process by comparing NTG and AD patients at different stages of disease.
This evidence is consistent with an interaction in the pathogenesis of the two diseases; however, it is still unclear if reduced blood flow is primary or secondary to glaucomatous optic nerve damage.
4.2. Exploring the Relationship of the Translaminar Pressure Gradient within NTG and AD
A translaminar pressure gradient is another pathogenic element implicated in NTG. This hypothesis states that the pathogenesis of NTG is due to an abnormally high-pressure gradient across the lamina cribrosa, leading to pressure-induced neurodegeneration [217].
The lamina cribrosa is a sieve-like structure through which RGC axons exit the eye from the intraocular into the retrolaminar region of the optic nerve head to make direct connections to the brain. These neuronal axons are impacted by the intraocular pressure (IOP) and intracranial pressure (ICP) zones in this region which is called the translaminar pressure gradient (difference between IOP and intracranial pressure (ICP)) [218]. Thus, an increase in IOP or a decrease in ICP could potentially cause an increased pressure differential, which could result in neuronal cell damage and death. A higher translaminar gradient has been found in NTG compared to HTG and control groups [217]. This may be due to 33% lower cerebrospinal fluid pressure (CSFP) in glaucomatous individuals compared to healthy controls [219], and even lower CSFP in NTG than HTG patients [220,221]. However, meta-analysis has shown marked overlapping in lumbar CSFP measurements between NTG and healthy subjects [222].
A substantial proportion of AD patients have lower CSFP than controls [223]. These results potentially explain the greater risk of AD patients developing glaucoma in lieu of the translaminar pressure theory for glaucoma pathogenesis [224]. There could potentially be numerous causes of the CSFP or translaminar pressure differentials. Measuring CSFP through lumbar puncture to assess intracranial pressure is difficult as it is invasive and may not be representative of the local pressure around the lamina cribrosa. This can be evidenced by differential molecular markers, such as IgG and albumin, seen in CSF from optic nerve subarachnoid spaces compared to lumbar puncture [225]. Recent development of B-scan ultrasounds can examine optic nerve sheave diameter to successfully measure ICP noninvasively and in real time [226]. Using this technology, future studies can observe the connection of translaminar pressure gradients in NTG and AD.
4.3. Altered CSF Fluid Dynamics and Bio-Active Molecules in NTG and AD Pathology
Another theory for NTG pathogenesis is the impairment of CSF circulation in the optic nerve sheath compartment leading to neurodegeneration [227,228]. The optic nerve is surrounded by CSF within the optic nerve sheath, making it sensitive to not only CSF pressure fluctuations, but also its contents. The CSF dynamics theory for NTG postulates reduced CSF clearance at the optic nerve leading to accumulation of neurotoxins and glaucomatous neurodegeneration [229,230]. The computed tomography cisternography imaging of the optic nerve in NTG patients showed high intracranial density of CSF in the intracranial spaces and reduced CSF turnover at the subarachnoid space of the optic nerve [231]. These findings are consistent with impaired CSF circulation, mimicking the compartment syndrome for the optic nerve. Stagnation of CSF may also play a role in the pathophysiology of neurodegenerative diseases such as AD [60,232,233]. A proposed mechanism and risk factor is reduced CSF turnover, initiating dysregulated levels of Aβ peptide and tau protein levels, and their consequential toxic molecular changes [67,234]. In NTG, a similar process is thought to occur as Aβ and tau have been identified in the CSF and deposited in the retina (Figure 2) [235,236,237,238,239]. According to this theory, glaucoma and AD neurodegeneration may occur due to an imbalance of production and clearance of these neurotoxins.
A fluid pathway closely associated with impaired CSF fluid dynamics is the glymphatic system. The glymphatic system distributes CSF throughout the brain and clears metabolites, including Aβ peptides [229,240,241,242]. Changes to the glymphatic system flow can be modulated by aquaporin (AQP) concentrations [243]. There is indication of a relationship between aquaporin-4 (AQP4) and Aβ clearance in the CSF that has been shown in rodents with depleted AQP4. The loss of AQP4 resulted in a drastic decrease in Aβ peptide clearance from the CSF and interstitial fluid [240,241,244,245,246,247]. Therefore, these results indicate that glymphatic system and concentration of AQP4 in CSF clearance can be another contributing factor to the accumulation of Aβ that is pathologically seen in AD patients. The glymphatic system has also been shown to interact with the optic nerve; AQP4s present in the optic nerve can transport CSF into the nerve. Impairment of Aβ clearance from the CSF in AD might correlate with Aβ accumulation also seen in glaucoma [222,229,247]. This indicates that overall CSF fluid dynamics connecting both the brain and eye might be integrated by AQP4 concentration in the glymphatic system.
Another cause of reduced CSF circulation may be explained by reduced optic canal diameter. In evaluations of optic canal size and severity of papilledema in intracranial idiopathic hypertension patients, the severity of papilledema modified the size of the optic canal cross-sectional area. From this study, they inferred that the optic canal diameter can influence the inflow of CSF to the subarachnoid space and impact CSF turnover [248]. This concept is applicable in NTG, which is associated with a smaller optic canal diameter than normal controls [249]. In addition to reduced CSF circulation, smaller optic nerve sheath diameter has been associated with lowered ICP [250,251]. These findings can also be integrated into the translaminar theory as history of lower ICP in NTG patients [221] due to decreased CSF density in the subarachnoid leads to an imbalance of translaminar pressure differential and eventual axonal death [231].
A closer look into the molecular changes between CSF and the optic nerve details the presence of biologically active molecules that have the potential to cause biochemical injury. A specific active molecule at high concentrations in the CSF are the beta trace proteins, which are lipocalin-type prostaglandin D synthases (L-PGDS) [225,252,253,254,255,256]. In non-glaucomatous rodent models of the eye, there is evidence of high localization of L-PGDS in the retinal pigment epithelium of the retina [257]. In the retina, it is suspected that in these cells, L-PGDS can have a role as a retinoid transporter or recycler and a lipophilic transporter [257]. In studies with NTG patients, elevated concentrations in the perioptic CSF have been found compared to patients without NTG [258]. Additionally, higher concentrations of L-PGDS have been found in the compartmentalized optic nerve CSF compared to the lumbar CSF in NTG patients with optic nerve sheath compartmentation [258]. The differential L-PGDS concentrations discovered to be secondary to CSF dynamics corroborate the findings from the computed tomography cisternography imaging of NTG patients [231]. L-PGDS’s role as a lipophilic transporter can allow it to bind and clear neurotoxic molecules, for instance Aβ and tau [259,260]. This has been exemplified in patients with idiopathic normal pressure hydrocephalus who were shown to have significant clearance relationship between total tau proteins and L-PGDS [260]. However, accumulation of L-PGDS in the blocked compartments of the CSF can cause damage to tissues in the affected area through deleterious effects on mitochondria and apoptosis-inducing properties at the optic nerve [225,261,262]. In relation to AD, the plasma of patients diagnosed with AD had greater concentrations of L-PGDS compared to controls. L-PGDS was found to have apoptotic properties due to the upregulation of inflammatory and oxidative stress factors, such as COX-2, ROS, and reduced antioxidants [263,264].
Another biologically active molecule in the CSF is the brain-derived neurotrophic factor (BDNF), which can connect CSF dynamics to other neurological capabilities [265]. BDNF is needed for the organization of neuronal networks and synaptic plasticity [265,266]. In non-AD human subjects, BDNF has been reported to be decreased in the CSF due to aging. Those with lower levels of BDNF in the CSF had poorer memory and diminished executive function [265]. The phenomenon of decreased CSF BDNF has been confirmed in patients with AD [266,267,268]. There are inconsistencies on whether BDNF levels in the CSF are higher or lower in females compared to males, this is interesting considering the evidence that women are more likely to develop AD [38,265,269]. However, the evidence does show a correlation between decreased body mass index and decreased plasma BDNF in females [269]. It was recently discovered that those with NTG have significantly lower systemic BDNF levels compared to HTG and controls, identifying a potential IOP-independent mechanism of pathogenesis [270] A limitation to this connection is that the BDNF levels were measured from serum and not the CSF; however, a study on rodent models suggested low BDNF plasma correlated to low brain and ocular BDNF [271]. More research is needed to elucidate whether this finding translates to human levels of BDNF, but this would explain current NTG therapeutics that are believed to improve BDNF production [272,273].The facilitative role of CSF with BDNF is still speculated [274,275]; it is plausible that loss of CSF-diffused BDNF at the optic nerve results in retinal neuron changes [275].
Similarly, CSF protein aggregates activate Toll-like receptors on microglial cells, promoting neuroinflammation [276]. As the first line of immune defense in the CNS, microglia will release inflammatory factors such as nitric oxide, TNFa, IL-6 and others, further activating downstream inflammatory processes [277]. Increased protein aggregates and myelin debris concentrations in the CSF and deposition in retinal tissue cause microglial cells to release cytotoxic factors which may potentiate neurodegeneration in AD and NTG [278]. Overall, neurons within the optic sheath are extremely sensitive to concentration changes in biologically active molecules in the CSF, and these effects must be further studied to fully comprehend their role in the pathogenesis of NTG.
4.4. Ocular Manifestations in the Context of NTG and AD: An Investigative Analysis
Despite the varying theories primarily influencing NTG pathogenesis, several retinal manifestations are similar in NTG and AD patients (Figure 3). Optical coherence tomography (OCT) is a noninvasive imaging method that observes retinal cell degeneration through changes in macular thickness and peripapillary retinal nerve fiber layer (RNFL) [30,279,280,281,282,283]. The macular ganglion cell complex (GCC) thickness is a parameter of great interest, including the three innermost retinal layers preferentially involved in glaucoma. The GCC includes axons, cell bodies, and dendrites of RGCs from the macular RNFL, the ganglion cell layer, and the inner plexiform layer [283]. A study examined peripapillary RNFL, macular GCC, and global loss volume in NTG, AD, and healthy control subjects [284]. Global loss volume refers to the sum of negative fractional deviation in retinal thickness compared to healthy controls [285]. They found significant reductions in RNFL thickness and macular GCC, as well as significant increases in global loss volume rate in NTG and AD patients compared to controls [284]. There was no significant difference in RNFL or GCC parameters between the NTG and AD groups despite the difference in the mean age of groups as 53.6 and 73.6, respectively (p > 0.05) [284]. The most significant reductions in retinal thickness in AD and NTG are found in the superior-nasal quadrant, consistent with AD postmortem retinal analysis [286]. RNFL thickness is dependent on age, with 10 years of aging associated with an approximate 4µm decrease [287]. Thus, it is noteworthy that the AD groups were significantly lower compared to the control group matched for age (p < 0.05) [284]. This shows that the decrease in RNFL thickness in the NTG and AD groups is due to disease pathogenesis and not aging alone. Electroretinogram study has shown direct correlation between thinning of RNFL and retinal dysfunction in those with glaucoma and AD compared to controls [288]. In this study, RNFL thickness was not correlated with IOP or aging, consistent with retinal dysfunction from another mechanism causing retinal cell loss [288]. Reduced overall RNFL and GCC thickness in AD compared to controls is consistent with pathological studies [169,195,280,282,289,290,291,292,293,294,295,296,297,298,299,300]. The study also determined the duration of disease was moderately correlated with RNFL thinning, and strongly correlated with decreased GCC thickness and increased global loss volume for AD patients [284]. In this study, no significant relationship was found between mini-mental state examination (MMSE) score (a measurement of AD disease severity) and OCT parameters, suggesting that ocular changes are found early in the progression of AD [284]. This supports the use of OCT in early diagnosis of AD to assess retinal changes and determine the duration of disease. On the other hand, several studies have found a significant association between thinning in RNFL, GCC, and macula and cognitive decline [30,200,279,295,301,302,303,304,305,306,307,308,309,310,311]. Metanalysis of 25 studies comparing AD and MCI patients to healthy controls found lower peripapillary RNFL and decreased total macular thickness in AD patients [312]. These studies show the utility of OCT and OCT-Angiography in detecting early retina and capillary changes that precede cognitive changes, acting as a potential noninvasive biomarker for early AD.
The retinal cell loss and optic nerve degeneration seen in AD [286,300,313,314,315,316] has been partly implicated in visual dysfunction [317,318,319] and electroretinogram abnormalities [282,292,320] described in the clinical presentation of AD. Interestingly, retinal cell loss from Aβ deposition inside and around melanopsin RGCs (mRGCs) is thought to be implicated in sleep disturbances observed in AD as well [300]. The mRGCs intrinsically express the photopigment melanopsin and contribute to the photoentrainment of circadian rhythms through projections to suprachiasmatic nucleus [321]. Findings show a significant loss and abnormal mRGC morphology postmortem in AD compared to controls, causing circadian rhythm misalignment [300]. The variable degrees of rest and activity in circadian rhythm dysfunction in AD patients are thought to have a bidirectional relationship with pathogenesis along with many other factors [322]. The same pathophysiology in part influences NTG progression and sleep dysregulation as well [323,324,325]; however, mRGC loss is associated with the severe stages of NTG [326,327]. This is consistent with sleep disturbances in late-stage NTG and may also suggest NTG as an early spectrum of AD progression.
There are many optic nerve head morphology patterns seen in AD that may manifest it as an ocular disease. A study of 30 AD patients found thinner disc rim, increased cup volume, and increased cup-to-disc ratio compared to age-matched controls [328]. These results correlated significantly with Alzheimer’s Disease Assessment Scale scores and longer durations of disease. Although no significant difference could be made between AD patients and controls comparing the pallor area-to-disc area ratio, AD patients with higher pallor area-to-disc ratio had higher Alzheimer’s Disease Assessment Scale scores and longer duration of disease [328]. This is suggestive that optic nerve head involvement may subgroup AD patients and be useful for monitoring progression and assessment of treatment efficacy. Additionally, a recent report demonstrated significantly thinner rim volume in NTG than HTG [329]. They also suggest many similarities between AD and NTG optic nerve patterns, including thinner disc rim, increased cup volume, and increased cup to disc ratio. More findings are needed to directly discern whether AD optic nerve manifestations are similar to NTG.
4.5. A Silent Connection: Exploring Cerebral Manifestations in NTG and AD
Early-stage POAG has been associated with widespread brain abnormalities [330] including decreased anatomical connectivity through white matter tracts [331], altered resting state functional connectivity signaling [332], and grey matter atrophy [333] compared to normal controls. Although initially hypothesized to be due to pathologically high IOP and consequent retinal degeneration, recent studies have also compared HTG and NTG MRI analysis to determine if neurodegeneration beyond the eye is independent of elevated IOP [15]. A multimodal brain MRI study found functional connectivity altered in NTG at short-range visual network levels, and in HTG at long-range levels between the limbic network and secondary visual network [15]. There is a lower overall functional connectivity in NTG compared to HTG of both visual and executive networks, and increased atrophy of the visual cortex with higher axial diffusivity in HTG [15]. Lower axial diffusivity refers to decreased parallel diffusion in fiber tracts, suggestive of more axonal injury causing less coherent orientation of axons in NTG [334]. Compared to controls, NTG is also associated with decreased white matter within the corpus callosum and parietal lobe, areas beyond explanation of propagated retinal or pre-geniculate damage [16]. These findings suggest changes are, at least in part, IOP-independent and are more consistent with glaucoma as a neurodegenerative disorder.
In AD, the brain findings are identified earliest as Aβ accumulation in the neocortex and cortical atrophy in the medial temporal region [335,336,337]. Additionally, Aβ accumulation in the hippocampus leads to hippocampal volume loss, found early in the asymptomatic and MCI stages [338]. As AD progresses, white matter [339,340,341] and grey matter [338,342] degeneration, alterations in functional connectivity [343,344,345,346,347], and visual and auditory functional MRI signal changes [348,349,350,351] have been identified. Specific connectivity pattern changes in some white and grey matter areas have been detected in AD progression [352]. These functional connectivity changes may be utilized to predict AD, but more research is needed to elucidate specific white matter connections. Functional connectivity should be compared between early AD and NTG to address any overlapping patterns. Furthermore, there are no current postmortem studies that examine the extent of AD pathology in brains of those with NTG. Given the extent of cerebral manifestations seen in NTG, this study would definitively demonstrate whether there is a correlation between the two diseases (Figure 3).
5. Conclusions
NTG and AD are neurodegenerative diseases that may share a common mechanistic pathogenesis due to similar biomarkers, retinal manifestations, and diffuse brain atrophy (Figure 4). Vascular dysregulation, a high translaminar pressure gradient, and impaired CSF dynamics are currently research areas in proposed NTG pathogenesis. The exact mechanisms are still unclear, as none of these theories can comprehensively explain the development of the disease. New findings of Aβ and tau depositions within the vasculature, retina, and CSF may connect NTG to AD through a neuroinflammatory process. By considering the processes of neurodegeneration in AD, the pathophysiology of NTG may be better understood.
Despite strong evidence suggesting discrete connections between NTG and AD, there are still many missing links. Future research is needed to study very low ICP groups in AD in relation to CSF dynamics and optic nerve sheath diameter. There is a need for more research on racial discrepancies seen in high proportions of the Asian population with NTG and differential retinal and cerebral manifestations. Future research should also address the age-related spectrum of disease in NTG and comorbidity with AD. Despite the efforts to slow progression of NTG with IOP-lowering treatments, no ocular medications have shown neuroprotective impact in lowering AD risk [28]. Discrepancies with nocturnal blood pressure patterns and conflicting cognitive decline association studies may suggest NTG is not an early manifestation of AD. However, the cerebral involvement of NTG cannot be ignored as it classifies NTG as a disorder beyond the eye. This review brings to light the correlative studies that identify links between NTG and AD to better understand pathogenesis and ultimately develop comprehensive therapeutics for both diseases.
Author Contributions
K.H. and N.E.B. have compiled, generated content and are major contributors in writing of the review. T.P.S. is a major contributor for the ideation and compilation of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This project was funded by NIH 1R01EY034174-01A1 (T.P.S.). This study was supported in part by an unrestricted grant from Research to Prevent Blindness, Inc. to the Indiana University School of Medicine, Department of Ophthalmology. This review was supported by startup funds from Indiana University School of Medicine, Department of Ophthalmology.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Conflicts of Interest
Author T.P.S. is the inventor of patent “Devices, Systems, and Methods for Modeling Ocular Translaminar Pressure Gradients” (US Patent # US20190327958A1). Authors NEB and KO declare that they have no competing interests. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
| NTG | Normal-tension glaucoma |
| HTG | High-tension glaucoma |
| POAG | Primary open-angle glaucoma |
| ALS | Amyotrophic lateral sclerosis |
| AD | Alzheimer’s Disease |
| IOP | Intraocular pressure |
| ICP | Intracranial pressure |
| RGC | Retinal ganglion cell |
| mRGCs | Melanopsin RGCs |
| RNFL | Retinal nerve fiber layer |
| GCC | Ganglion cell complex |
| MMSE | Mini-mental state examination |
| CVI | Choroid vascular index |
| MCI | Mild cognitive impairment |
| APOE | Apolipoprotein e4 allele |
| OPTN | Optineurin |
| TBK1 | TANK Binding Kinase 1 |
| Aβ | Amyloid β |
| Aβ42 | Aβ peptide residues 1-42 |
| Aβ40 | Aβ peptides residues 1-40 |
| APP | Amyloid precursor protein |
| IL | Interleukins |
| TNFa | Tumor necrosis factor a |
| NF-kB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| MCP-1/CCL2 | Monocyte chemoattractant protein-1 |
| 4-HNE | 4-hydroxynonenal |
| LC3B/MAP1LC3B | Microtubule associated protein 1 light chain 3 beta |
| ET-1 | Endothelin-1 |
| TLPG | Translaminar pressure gradient |
| CSFP | Cerebrospinal fluid pressure |
| AQP | Aquaporin |
| AQP-4 | Aquaporin-4 |
| L-PGDS | Lipocalin-type prostaglandin D synthases |
| BDNF | Brain-derived neurotrophic factor |
| T-MoCA | Telephone Montreal Cognitive Assessment |
References
- Quigley, H.A.; Broman, A.T. The number of people with glaucoma worldwide in 2010 and 2020. Br. J. Ophthalmol. 2006, 90, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Chen, A.; Zou, M.; Zhang, Y.; Jin, L.; Li, Y.; Zheng, D.; Jin, G.; Congdon, N. Time trends, associations and prevalence of blindness and vision loss due to glaucoma: An analysis of observational data from the Global Burden of Disease Study 2017. BMJ Open 2022, 12, e053805. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, R.N.; Aung, T.; Medeiros, F.A. The pathophysiology and treatment of glaucoma: A review. JAMA 2014, 311, 1901–1911. [Google Scholar] [CrossRef] [PubMed]
- Coleman, A.L.; Brigatti, L. The glaucomas. Minerva Med. 2001, 92, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, R.N.; Khaw, P.T. Primary open-angle glaucoma. Lancet 2004, 363, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Kerrigan, L.A.; Zack, D.J.; Quigley, H.A.; Smith, S.D.; Pease, M.E. TUNEL-positive ganglion cells in human primary open-angle glaucoma. Arch. Ophthalmol. 1997, 115, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- Parekh, A.S.; Tafreshi, A.; Dorairaj, S.K.; Weinreb, R.N. Clinical applicability of the International Classification of Disease and Related Health Problems (ICD-9) glaucoma staging codes to predict disease severity in patients with open-angle glaucoma. Eur. J. Gastroenterol. Hepatol. 2014, 23, e18–e22. [Google Scholar] [CrossRef] [PubMed]
- Kapetanakis, V.V.; Chan, M.P.Y.; Foster, P.J.; Cook, D.G.; Owen, C.G.; Rudnicka, A.R. Global variations and time trends in the prevalence of primary open angle glaucoma (POAG): A systematic review and meta-analysis. Br. J. Ophthalmol. 2015, 100, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Esporcatte, B.L.B.; Tavares, I.M. Normal-tension glaucoma: An update. Arq. Bras. Oftalmol. 2016, 79, 270–276. [Google Scholar] [CrossRef]
- Dielemans, I.; Vingerling, J.R.; Wolfs, R.C.; Hofman, A.; Grobbee, D.E.; de Jong, P.T. The prevalence of primary open-angle glaucoma in a population-based study in The Netherlands. Ophthalmology 1994, 101, 1851–1855. [Google Scholar] [CrossRef]
- Kim, K.E.; Park, K.-H. Update on the Prevalence, Etiology, Diagnosis, and Monitoring of Normal-Tension Glaucoma. Asia-Pac. J. Ophthalmol. 2016, 5, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.K.; Kee, C. Population-based glaucoma prevalence studies in Asians. Surv. Ophthalmol. 2014, 59, 434–447. [Google Scholar] [CrossRef] [PubMed]
- The effectiveness of intraocular pressure reduction in the treatment of normal-tension glaucoma. Collaborative Normal-Tension Glaucoma Study Group. Am. J. Ophthalmol. 1998, 126, 498–505. [Google Scholar]
- Caprioli, J.; Song, B. New directions in the treatment of normal tension glaucoma. Indian J. Ophthalmol. 2014, 62, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, A.; Zhang, J.; Costantino, F.; De Stefano, N.; Frezzotti, P. Diffuse brain damage in normal tension glaucoma. Hum. Brain Mapp. 2018, 39, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Boucard, C.C.; Hanekamp, S.; Ćurčić-Blake, B.; Ida, M.; Yoshida, M.; Cornelissen, F.W. Neurodegeneration beyond the primary visual pathways in a population with a high incidence of normal-pressure glaucoma. Ophthalmic Physiol. Opt. 2016, 36, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Mullany, S.; Xiao, L.; Qassim, A.; Marshall, H.; Gharahkhani, P.; MacGregor, S.; Hassall, M.M.; Siggs, O.M.; Souzeau, E.; E Craig, J. Normal-tension glaucoma is associated with cognitive impairment. Br. J. Ophthalmol. 2022, 106, 952–956. [Google Scholar] [CrossRef] [PubMed]
- Yochim, B.P.; Mueller, A.E.; Kane, K.D.; Kahook, M.Y. Prevalence of cognitive impairment, depression, and anxiety symptoms among older adults with glaucoma. Eur. J. Gastroenterol. Hepatol. 2012, 21, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Tamura, H.; Kawakami, H.; Kanamoto, T.; Kato, T.; Yokoyama, T.; Sasaki, K.; Izumi, Y.; Matsumoto, M.; Mishima, H.K. High frequency of open-angle glaucoma in Japanese patients with Alzheimer’s disease. J. Neurol. Sci. 2006, 246, 79–83. [Google Scholar] [CrossRef]
- Bayer, A.; Ferrari, F.; Erb, C. High occurrence rate of glaucoma among patients with Alzheimer’s disease. Eur. Neurol. 2002, 47, 165–168. [Google Scholar] [CrossRef]
- Tian, T.; Liu, Y.-H. Normal-tension glaucoma and Alzheimer’s disease: Retinal vessel signs as a possible common underlying risk factor. Med. Hypotheses 2011, 77, 466. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Utsunomiya, K.; Ota, H.; Ogura, Y.; Narabayashi, I.; Ikeda, T. Comparative study of cerebral blood flow in patients with normal-tension glaucoma and control subjects. Arch. Ophthalmol. 2006, 141, 394–396. [Google Scholar] [CrossRef]
- Cui, Q.N.; Jethi, M.; Driver, T.; Porco, T.C.; Kuo, J.; Lin, S.C.; Stamper, R.L.; Han, Y.; Chiu, C.S.; Ramanathan, S.; et al. Individuals with and without normal tension glaucoma exhibit comparable performance on tests of cognitive function. Int. J. Ophthalmol. 2021, 14, 1721–1728. [Google Scholar] [CrossRef]
- Kessing, L.V.M.; Lopez, A.G.M.; Andersen, P.K.M.; Kessing, S.V.M. No increased risk of developing Alzheimer disease in patients with glaucoma. Eur. J. Gastroenterol. Hepatol. 2007, 16, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Pekmezci, M.; Vo, B.; Lim, A.K.; Hirabayashi, D.R.; Tanaka, G.H.; Weinreb, R.N.; Lin, S.C. The characteristics of glaucoma in Japanese Americans. JAMA Ophthalmol. 2009, 127, 167. [Google Scholar] [CrossRef]
- Brahma, M.M.; Takahashi, K.; Namekata, K.; Harada, T.; Goshima, Y.; Ohshima, T. Genetic inhibition of collapsin response mediator protein-2 phosphorylation ameliorates retinal ganglion cell death in normal-tension glaucoma models. Genes. Cells 2022, 27, 526–536. [Google Scholar] [CrossRef]
- Lam, C.Y.; Fan, B.J.; Wang, D.Y.; Tam, P.O.S.; Tham, C.C.Y.; Leung, D.Y.L.; Fan, D.S.P.; Lam, D.S.C.; Pang, C.P. Association of apolipoprotein E polymorphisms with normal tension glaucoma in a Chinese population. Eur. J. Gastroenterol. Hepatol. 2006, 15, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-Y.; Lai, Y.-J.; Yen, Y.-F.; Shen, Y.-C.; Wang, C.-Y.; Liang, C.-Y.; Lin, K.-H.; Fan, L.-W. Association between normal tension glaucoma and the risk of Alzheimer’s disease: A nationwide population-based cohort study in Taiwan. BMJ Open 2018, 8, e022987. [Google Scholar] [CrossRef]
- Global Burden of Disease Dementia Forecast Collaborators. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [Google Scholar] [CrossRef]
- Salobrar-Garcia, E.; Méndez-Hernández, C.; Hoz, R.D.; Ramírez, A.I.; López-Cuenca, I.; Fernández-Albarral, J.A.; Rojas, P.; Wang, S.; García-Feijoo, J.; Gil, P.; et al. Ocular Vascular Changes in Mild Alzheimer’s Disease Patients: Foveal Avascular Zone, Choroidal Thickness, and ONH Hemoglobin Analysis. J. Pers. Med. 2020, 10, 231. [Google Scholar] [CrossRef]
- Mirzaei, N.; Shi, H.; Oviatt, M.; Doustar, J.; Rentsendorj, A.; Fuchs, D.-T.; Sheyn, J.; Black, K.L.; Koronyo, Y.; Koronyo-Hamaoui, M. Alzheimer’s Retinopathy: Seeing Disease in the Eyes. Front. Neurosci. 2020, 14, 921. [Google Scholar] [CrossRef] [PubMed]
- Stojcic, M.; Hentova-Sencanic, P.; Stojcic, B.; Sencanic, I. Comparison of normal tension and high tension glaucoma patients (corrected) according to age and sex. Srp. Arh. Celok. Lek. 2012, 140, 699–703. [Google Scholar] [CrossRef]
- Mallick, J.; Devi, L.; Malik, P.; Mallick, J. Update on Normal Tension Glaucoma. J. Ophthalmic Vis. Res. 2016, 11, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sidhu, J.; Goyal, A.; Tsao, J. Alzheimer Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Katz, B.; Rimmer, S. Ophthalmologic manifestations of Alzheimer’s disease. Surv. Ophthalmol. 1989, 34, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Sadun, A.A.; Borchert, M.; DeVita, E.; Hinton, D.R.; Bassi, J. Assessment of visual impairment in patients with Alzheimer’s disease. Am. J. Ophthalmol. 1987, 104, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, S.; Wolf, P.A.; Beiser, A.; Au, R.; McNulty, K.; White, R.; D’Agostino, R.B. Lifetime risk of dementia and Alzheimer’s disease. The impact of mortality on risk estimates in the Framingham Study. Neurology 1997, 49, 1498–1504. [Google Scholar] [CrossRef]
- Mielke, M.M. Sex and Gender Differences in Alzheimer’s Disease Dementia. Psychiatr. Times 2018, 35, 14–17. [Google Scholar] [PubMed]
- Coales, I.; Tsartsalis, S.; Fancy, N.; Weinert, M.; Clode, D.; Owen, D.; Matthews, P.M. Alzheimer’s disease-related transcriptional sex differences in myeloid cells. J. Neuroinflamm. 2022, 19, 1–13. [Google Scholar] [CrossRef]
- Garner, R.; Kumari, R.; Lanyon, P.; Doherty, M.; Zhang, W. Prevalence, risk factors and associations of primary Raynaud’s phenomenon: Systematic review and meta-analysis of observational studies. BMJ Open 2015, 5, e006389. [Google Scholar] [CrossRef]
- Henry, E.; E Newby, D.; Webb, D.J.; O’Brien, C. Peripheral endothelial dysfunction in normal pressure glaucoma. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1710–1714. [Google Scholar]
- Drance, S.; Douglas, G.; Wijsman, K.; Schulzer, M.; Britton, R. Response of blood flow to warm and cold in normal and low-Tension glaucoma patients. Arch. Ophthalmol. 1988, 105, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Gramer, G.; Weber, B.H.; Gramer, E. Migraine and Vasospasm in Glaucoma: Age-Related Evaluation of 2027 Patients With Glaucoma or Ocular Hypertension. Investig. Ophthalmol. Vis. Sci. 2015, 56, 7999–8007. [Google Scholar] [CrossRef]
- Owens, P.; Lyons, S.; O’brien, E. Arterial hypotension: Prevalence of low blood pressure in the general population using ambulatory blood pressure monitoring. J. Hum. Hypertens. 2000, 14, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Binggeli, T.; Schoetzau, A.; Konieczka, K. In glaucoma patients, low blood pressure is accompanied by vascular dysregulation. EPMA J. 2018, 9, 387–391. [Google Scholar] [CrossRef]
- Kaiser, H.J.; Flammer, J.; Graf, T.; Stümpfig, D. Systemic blood pressure in glaucoma patients. Graefes Arch. Clin. Exp. Ophthalmol. 1993, 231, 677–680. [Google Scholar] [CrossRef]
- Iwase, A.; Suzuki, Y.; Araie, M.; Yamamoto, T.; Abe, H.; Shirato, S.; Kuwayama, Y.; Mishima, H.; Shimuzu, H.; Tomita, G.; et al. The prevalence of primary open-angle glaucoma in Japanese: The Tajimi Study. Ophthalmology 2004, 111, 1641–1648. [Google Scholar] [CrossRef] [PubMed]
- Vijaya, L.; George, R.; Baskaran, M.; Arvind, H.; Raju, P.; Ramesh, S.V.; Kumaramanickavel, G.; McCarty, C. Prevalence of primary open-angle glaucoma in an urban south Indian population and comparison with a rural population: The chennai glaucoma study. Ophthalmology 2008, 115, 648–654.e1. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Xu, L.; Yang, H.; Jonas, J.B. Prevalence of glaucoma in north China: The Beijing Eye study. Arch. Ophthalmol. 2010, 150, 917–924. [Google Scholar] [CrossRef]
- Kim, C.-S.; Seong, G.J.; Lee, N.-H.; Song, K.-C. Prevalence of primary open-angle glaucoma in central South Korea: The Namil study. Ophthalmology 2011, 118, 1024–1030. [Google Scholar] [CrossRef]
- Bonomi, L.; Marchini, G.; Marraffa, M.; Bernardi, P.; De Franco, I.; Perfetti, S.; Varotto, A.; Tenna, V. Prevalence of glaucoma and intraocular pressure distribution in a defined population: The Egna-Neumarkt study. Ophthalmology 1998, 105, 209–215. [Google Scholar] [CrossRef]
- Leske, M.C.; Connell, A.M.; Wu, S.Y.; Hyman, L.G.; Schachat, A.P. Risk factors for open-angle glaucoma: The Barbados Eye Study. Arch. Ophthalmol. 1995, 113, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Rotchford, A.P.; Johnson, G.J. Glaucoma in Zulus: A population-based cross-sectional survey in a rural district in South Africa. Arch. Ophthalmol. 2002, 120, 471–478. [Google Scholar] [CrossRef]
- Fan, N.; Wang, P.; Tang, L.; Liu, X. Ocular Blood Flow and Normal Tension Glaucoma. BioMed Res. Int. 2015, 2015, 308505. [Google Scholar] [CrossRef] [PubMed]
- Furlanetto, R.; De Moraes, C.G.; Teng, C.C.; Liebmann, J.M.; Greenfield, D.S.; Gardiner, S.; Ritch, R.; Krupin, T. Low-pressure glaucoma treatment study group. Risk factors for optic disc hemorrhage in the low-pressure glaucoma treatment study. Am. J. Ophthalmol. 2014, 157, 945–952.e1. [Google Scholar] [CrossRef] [PubMed]
- Charlson, M.E.; de Moraes, C.G.; Link, A.; Wells, M.T.; Harmon, G.; Peterson, J.C.; Ritch, R.; Liebmann, J.M. Nocturnal systemic hypotension increases the risk of glaucoma progression. Ophthalmology 2014, 121, 2004–2012. [Google Scholar] [CrossRef] [PubMed]
- Raman, P.; Suliman, N.B.; Zahari, M.; Kook, M.; Ramli, N. Low nocturnal diastolic ocular perfusion pressure as a risk factor for NTG progression: A 5-year prospective study. Eye 2018, 32, 1183–1189. [Google Scholar] [CrossRef]
- de la Torre, J.C. Alzheimer disease as a vascular disorder: Nosological evidence. Stroke 2002, 33, 1152–1162. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kim, K.; Lee, Y.C.; Kim, S.; Won, H.-H.; Yu, T.Y.; Lee, E.-M.; Kang, J.M.; Lewis, M.; Kim, D.K.; et al. Associations between vascular risk factors and subsequent Alzheimer’s disease in older adults. Alzheimers Res. Ther. 2020, 12, 117. [Google Scholar] [CrossRef] [PubMed]
- Serot, J.M.; Zmudka, J.; Jouanny, P. A possible role for CSF turnover and choroid plexus in the pathogenesis of late onset Alzheimer’s disease. J. Alzheimers Dis. 2012, 30, 17–26. [Google Scholar] [CrossRef]
- Kim, J.; Ha, W.S.; Park, S.H.; Han, K.; Baek, M.S. Association between migraine and Alzheimer’s disease: A nationwide cohort study. Front. Aging Neurosci. 2023, 15, 1196185. [Google Scholar] [CrossRef]
- Flammer, J.; Orgül, S.; Costa, V.P.; Orzalesi, N.; Krieglstein, G.K.; Serra, L.M.; Renard, J.-P.; Stefánsson, E. The impact of ocular blood flow in glaucoma. Prog. Retin. Eye Res. 2002, 21, 359–393. [Google Scholar] [CrossRef]
- Moon, J.Y.; Kim, H.J.; Park, Y.H.; Park, T.K.; Park, E.-C.; Kim, C.Y.; Lee, S.H. Association between Open-Angle Glaucoma and the Risks of Alzheimer’s and Parkinson’s Diseases in South Korea: A 10-year Nationwide Cohort Study. Sci. Rep. 2018, 8, 11161. [Google Scholar] [CrossRef]
- Bach-Holm, D.; Kessing, S.V.; Mogensen, U.; Forman, J.L.; Andersen, P.K.; Kessing, L.V. Normal tension glaucoma and Alzheimer disease: Comorbidity? Acta Ophthalmol. 2012, 90, 683–685. [Google Scholar] [CrossRef] [PubMed]
- Duyckaerts, C.; Delatour, B.; Potier, M.-C. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009, 118, 5–36. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease and Down’s syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 1984, 122, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Drummond, E.; Pires, G.; MacMurray, C.; Askenazi, M.; Nayak, S.; Bourdon, M.; Safar, J.; Ueberheide, B.; Wisniewski, T. Phosphorylated tau interactome in the human Alzheimer’s disease brain. Brain 2020, 143, 2803–2817. [Google Scholar] [CrossRef]
- Pîrşcoveanu, D.F.V.; Pirici, I.; Tudorică, V.; Balseanu, T.-A.; Albu, V.C.; Bondari, S.; Bumbea, A.M.; Pîrşcoveanu, M. Tau protein in neurodegenerative diseases—A review. Rom. J. Morphol. Embryol. 2017, 58, 1141–1150. [Google Scholar]
- Orr, M.E.; Sullivan, A.C.; Frost, B. A Brief Overview of Tauopathy: Causes, Consequences, and Therapeutic Strategies. Trends Pharmacol. Sci. 2017, 38, 637–648. [Google Scholar] [CrossRef]
- Schmechel, D.E.; Saunders, A.M.; Strittmatter, W.J.; Crain, B.J.; Hulette, C.M.; Joo, S.H.; Pericak-Vance, M.A.; Goldgaber, D.; Roses, A.D. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 9649–9653. [Google Scholar] [CrossRef]
- Wiggs, J.L.; Pasquale, L.R. Genetics of glaucoma. Hum. Mol. Genet. 2017, 26, R21–R27. [Google Scholar] [CrossRef]
- Liu, Y.; Allingham, R.R. Major review: Molecular genetics of primary open-angle glaucoma. Exp. Eye Res. 2017, 160, 62–84. [Google Scholar] [CrossRef]
- Alward, W.L.M.; van der Heide, C.; Khanna, C.L.; Roos, B.R.; Sivaprasad, S.; Kam, J.; Ritch, R.; Lotery, A.; Igo, R.P.; Bailey, J.N.C.; et al. Myocilin Mutations in Patients with Normal-Tension Glaucoma. JAMA Ophthalmol 2019, 137, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Weisschuh, N.; Neumann, D.; Wolf, C.; Wissinger, B.; Gramer, E. Prevalence of myocilin and optineurin sequence variants in German normal tension glaucoma patients. Mol. Vis. 2005, 11, 284–287. [Google Scholar] [PubMed]
- Rezaie, T.; Child, A.; Hitchings, R.; Brice, G.; Miller, L.; Coca-Prados, M.; Héon, E.; Krupin, T.; Ritch, R.; Kreutzer, D.; et al. Adult-Onset primary open-angle glaucoma caused by mutations in optineurin. Science 2002, 295, 1077–1079. [Google Scholar] [CrossRef] [PubMed]
- Liuska, P.J.; Harju, M.; Kivelä, T.T.; Turunen, J.A. Prevalence of MYOC risk variants for glaucoma in different populations. Acta Ophthalmol. 2021, 99, E1090–E1097. [Google Scholar] [CrossRef]
- Swarup, G.; Sayyad, Z. Altered Functions and Interactions of Glaucoma-Associated Mutants of Optineurin. Front. Immunol. 2018, 9, 1287. [Google Scholar] [CrossRef]
- Sakurada, Y.; Mabuchi, F. Advances in glaucoma genetics. Prog. Brain Res. 2015, 220, 107–126. [Google Scholar]
- Challa, P. Glaucoma genetics. Int. Ophthalmol. Clin. 2008, 48, 73–94. [Google Scholar] [CrossRef]
- Fingert, J.H. Primary open-angle glaucoma genes. Eye 2011, 25, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Sirohi, K.; Swarup, G. Defects in autophagy caused by glaucoma-associated mutations in optineurin. Exp. Eye Res. 2016, 144, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Forloni, G.; Demicheli, F.; Giorgi, S.; Bendotti, C.; Angeretti, N. Expression of amyloid precursor protein mRNAs in endothelial, neuronal and glial cells: Modulation by interleukin-1. Mol. Brain Res. 1992, 16, 128–134. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, A.; Chen, H.; Autilio-Gambetti, L.; Gambetti, P. Differential APP gene expression in rat cerebral cortex, meninges, and primary astroglial, microglial and neuronal cultures. FEBS Lett. 1991, 292, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Hung, A.; Selkoe, D. Processing of beta-amyloid precursor protein in microglia and astrocytes favors an internal localization over constitutive secretion. J. Neurosci. 1991, 11, 3783–3793. [Google Scholar] [CrossRef] [PubMed]
- Nunan, J.; Small, D.H. Regulation of APP cleavage by alpha-, beta- and gamma-secretases. FEBS Lett. 2000, 483, 6–10. [Google Scholar] [CrossRef]
- Yang, M.; Teplow, D.B. Amyloid beta-protein monomer folding: Free-energy surfaces reveal alloform-specific differences. J. Mol. Biol. 2008, 384, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Sgourakis, N.G.; Yan, Y.; McCallum, S.A.; Wang, C.; Garcia, A.E. The Alzheimer’s peptides Abeta40 and 42 adopt distinct conformations in water: A combined MD/NMR study. J. Mol. Biol. 2007, 368, 1448–1457. [Google Scholar] [CrossRef]
- Pike, K.E.; Savage, G.; Villemagne, V.L.; Ng, S.; Moss, S.A.; Maruff, P.; Mathis, C.A.; Klunk, W.E.; Masters, C.L.; Rowe, C.C. Beta-amyloid imaging and memory in non-demented individuals: Evidence for preclinical Alzheimer’s disease. Brain 2007, 130 Pt 11, 2837–2844. [Google Scholar] [CrossRef]
- Martin, K.R.; Quigley, H.A.; Valenta, D.; Kielczewski, J.; Pease, M.E. Optic nerve dynein motor protein distribution changes with intraocular pressure elevation in a rat model of glaucoma. Exp. Eye Res. 2006, 83, 255–262. [Google Scholar] [CrossRef]
- Kipfer-Kauer, A.; McKinnon, S.J.; Frueh, B.E.; Goldblum, D. Distribution of amyloid precursor protein and amyloid-beta in ocular hypertensive C57BL/6 mouse eyes. Curr. Eye Res. 2010, 35, 828–834. [Google Scholar] [CrossRef] [PubMed]
- Minckler, D.S.; Bunt, A.H.; Johanson, G.W. Orthograde and retrograde axoplasmic transport during acute ocular hypertension in the monkey. Investig. Ophthalmol. Vis. Sci. 1977, 16, 426–441. [Google Scholar]
- Quigley, H.A.; Addicks, E.M.; Green, W.R.; Maumenee, A.E. Optic nerve damage in human glaucoma. II. The site of injury and susceptibility to damage. Arch. Ophthalmol. 1981, 99, 635–649. [Google Scholar] [CrossRef] [PubMed]
- Dias, M.S.; Luo, X.; Ribas, V.T.; Petrs-Silva, H.; Koch, J.C. The Role of Axonal Transport in Glaucoma. Int. J. Mol. Sci. 2022, 23, 3935. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.A.; Zhou, B.; Wernig, M.; Sudhof, T.C. ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Abeta Secretion. Cell 2017, 168, 427–441.e21. [Google Scholar] [CrossRef] [PubMed]
- Safieh, M.; Korczyn, A.D.; Michaelson, D.M. ApoE4: An emerging therapeutic target for Alzheimer’s disease. BMC Med. 2019, 17, 64. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Holm, M.L.; Liu, C.C.; Shinohara, M.; Aikawa, T.; Oue, H.; Yamazaki, Y.; Martens, Y.A.; Murray, M.E.; Sullivan, P.M.; et al. APOE4-mediated amyloid-beta pathology depends on its neuronal receptor LRP1. J. Clin. Investig. 2019, 129, 1272–1277. [Google Scholar] [CrossRef] [PubMed]
- Husain, M.A.; Laurent, B.; Plourde, M. APOE and Alzheimer’s Disease: From Lipid Transport to Physiopathology and Therapeutics. Front. Neurosci. 2021, 15, 630502. [Google Scholar] [CrossRef]
- Corbo, R.M.; Scacchi, R. Apolipoprotein E (APOE) allele distribution in the world. Is APOE*4 a ‘thrifty’ allele? Ann. Hum. Genet. 1999, 63, 301–310. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.P.; Singh, M.; Mastana, S.S. APOE distribution in world populations with new data from India and the UK. Ann. Hum. Biol. 2006, 33, 279–308. [Google Scholar] [CrossRef] [PubMed]
- Copin, B.; Brézin, A.P.; Valtot, F.; Dascotte, J.-C.; Béchetoille, A.; Garchon, H.-J. Apolipoprotein E–promoter single-nucleotide polymorphisms affect the phenotype of primary open-angle glaucoma and demonstrate interaction with the myocilin gene. Am. J. Hum. Genet. 2002, 70, 1575–1581. [Google Scholar] [CrossRef] [PubMed]
- Saadane, A.; Petrov, A.; Mast, N.; El-Darzi, N.; Dao, T.; Alnemri, A.; Song, Y.; Dunaief, J.L.; Pikuleva, I.A. Mechanisms that minimize retinal impact of apolipoprotein E absence. J. Lipid Res. 2018, 59, 2368–2382. [Google Scholar] [CrossRef] [PubMed]
- Margeta, M.A.; Letcher, S.M.; Igo, R.P.; Cooke Bailey, J.N.; Pasquale, L.R.; Haines, J.L.; Butovsky, O.; Wiggs, J.L. Association of APOE with Primary Open-Angle Glaucoma Suggests a Protective Effect for APOE epsilon4. Investig. Ophthalmol. Vis. Scence 2020, 61, 3. [Google Scholar] [CrossRef] [PubMed]
- Al-Dabbagh, N.M.; Al-Dohayan, N.; Arfin, M.; Tariq, M. Apolipoprotein E polymorphisms and primary glaucoma in Saudis. Mol. Vis. 2009, 15, 912–919. [Google Scholar]
- Vickers, J.C.; E Craig, J.; Stankovich, J.; McCormack, G.H.; West, A.K.; Dickinson, J.L.; McCartney, P.J.; A Coote, M.; Healey, D.L.; A Mackey, D. The apolipoprotein epsilon4 gene is associated with elevated risk of normal tension glaucoma. Mol. Vis. 2002, 8, 389–893. [Google Scholar]
- Mabuchi, F.; Tang, S.; Ando, D.; Yamakita, M.; Wang, J.; Kashiwagi, K.; Yamagata, Z.; Iijima, H.; Tsukahara, S. The apolipoprotein E gene polymorphism is associated with open angle glaucoma in the Japanese population. Mol. Vis. 2005, 11, 609–612. [Google Scholar] [PubMed]
- Saglar, E.; Yucel, D.; Bozkurt, B.; Ozgul, R.; Irkec, M.; Ogus, A. Association of polymorphisms in APOE, p53, and p21 with primary open-angle glaucoma in Turkish patients. Mol. Vis. 2009, 15, 1270–1276. [Google Scholar] [PubMed]
- Poirier, J. Apolipoprotein E in animal models of CNS injury and in Alzheimer’s disease. Trends Neurosci. 1994, 17, 525–530. [Google Scholar] [CrossRef]
- Rudajev, V.; Novotny, J. Cholesterol as a key player in amyloid beta-mediated toxicity in Alzheimer’s disease. Front. Mol. Neurosci. 2022, 15, 937056. [Google Scholar] [CrossRef]
- Jeong, W.; Lee, H.; Cho, S.; Seo, J. ApoE4-Induced Cholesterol Dysregulation and Its Brain Cell Type-Specific Implications in the Pathogenesis of Alzheimer’s Disease. Mol. Cells 2019, 42, 739–746. [Google Scholar] [PubMed]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Brionne, T.C.; Tesseur, I.; Masliah, E.; Wyss-Coray, T. Loss of TGF-beta 1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron 2003, 40, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Guillot-Sestier, M.-V.; Town, T. Innate immunity in Alzheimer’s disease: A complex affair. CNS Neurol. Disord.-Drug Targets 2013, 12, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Dalleau, S.; Baradat, M.; Guéraud, F.; Huc, L. Cell death and diseases related to oxidative stress:4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 2013, 20, 1615–1630. [Google Scholar] [CrossRef] [PubMed]
- Montine, K.S.; Reich, E.; Neely, M.D.; Sidell, K.R.; Olson, S.J.; Markesbery, W.R.; Montine, T.J. Distribution of reducible 4-hydroxynonenal adduct immunoreactivity in Alzheimer disease is associated with APOE genotype. J. Neuropathol. Exp. Neurol. 1998, 57, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Mattson, M.P. Apolipoprotein E and oxidative stress in brain with relevance to Alzheimer’s disease. Neurobiol. Dis. 2020, 138, 104795. [Google Scholar] [CrossRef] [PubMed]
- Shea, T.B.; Rogers, E.; Ashline, D.; Ortiz, D.; Sheu, M.-S. Apolipoprotein E deficiency promotes increased oxidative stress and compensatory increases in antioxidants in brain tissue. Free. Radic. Biol. Med. 2002, 33, 1115–1120. [Google Scholar] [CrossRef] [PubMed]
- Dose, J.; Huebbe, P.; Nebel, A.; Rimbach, G. APOE genotype and stress response—A mini review. Lipids Health Dis. 2016, 15, 121. [Google Scholar] [CrossRef]
- Miyata, M.; Smith, J.D. Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and beta-amyloid peptides. Nat. Genet. 1996, 14, 55–61. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Fernandes, M.A.; Proenca, M.T.; Nogueira, A.J.; Grazina, M.M.; Oliveira, L.M.; Fernandes, A.I.; Santiago, B.; Santana, I.; Oliveira, C.R. Influence of apolipoprotein E genotype on blood redox status of Alzheimer’s disease patients. Int. J. Mol. Med. 1999, 4, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Chico, L.; Simoncini, C.; Lo Gerfo, A.; Rocchi, A.; Petrozzi, L.; Carlesi, C.; Volpi, L.; Tognoni, G.; Siciliano, G.; Bonuccelli, U. Oxidative stress and APO E polymorphisms in Alzheimer’s disease and in mild cognitive impairment. Free Radic. Res. 2013, 47, 569–576. [Google Scholar] [CrossRef]
- Siegel, S.J.; Bieschke, J.; Powers, E.T.; Kelly, J.W. The oxidative stress metabolite 4-hydroxynonenal promotes Alzheimer protofibril formation. Biochemistry 2007, 46, 1503–1510. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Boyd-Kimball, D. Redox proteomics and amyloid beta-peptide: Insights into Alzheimer disease. J. Neurochem. 2019, 151, 459–487. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Drake, J.; Pocernich, C.; Castegna, A. Evidence of oxidative damage in Alzheimer’s disease brain: Central role for amyloid beta-peptide. Trends Mol. Med. 2001, 7, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.N.; Schmitt, F.A.; Scheff, S.W.; Ding, Q.; Chen, Q.; Butterfield, D.A.; Markesbery, W.R. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology 2005, 64, 1152–1156. [Google Scholar] [CrossRef]
- Hensley, K.; Hall, N.; Subramaniam, R.; Cole, P.; Harris, M.; Aksenov, M.; Aksenova, M.; Gabbita, S.P.; Wu, J.F.; Carney, J.M.; et al. Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J. Neurochem. 1995, 65, 2146–2156. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Chen, K.; Alexander, G.E.; Caselli, R.J.; Bandy, D.; Osborne, D.; Saunders, A.M.; Hardy, J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc. Natl. Acad. Sci. USA 2004, 101, 284–289. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Lauderback, C.M. Lipid peroxidation and protein oxidation in Alzheimer’ disease brain: Potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic. Biol. Med. 2002, 32, 1050–1060. [Google Scholar] [CrossRef]
- Scheff, S.W.; Ansari, M.A.; Mufson, E.J. Oxidative stress and hippocampal synaptic protein levels in elderly cognitively intact individuals with Alzheimer’s disease pathology. Neurobiol. Aging 2016, 42, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, N.; Coban, D.T.; Bayindir, A.; Erol, M.K.; Ellidag, H.Y.; Giray, O.; Sayrac, S.; Tekeli, S.O.; Eren, E. Higher serum lipids and oxidative stress in patients with normal tension glaucoma, but not pseudoexfoliative glaucoma. Bosn. J. Basic Med Sci. 2015, 16, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, C. Pseudoexfoliation syndrome and pseudoexfoliation glaucoma. J. Fr. Ophtalmol. 2018, 41, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Araujo, F.B.; Barbosa, D.S.; Hsin, C.Y.; Maranhão, R.C.; Abdalla, D.S. Evaluation of oxidative stress in patients with hyperlipidemia. Atherosclerosis 1995, 117, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.-L.; Shi, Y.-H.; Hao, G.; Li, W.; Le, G.-W. Increasing Oxidative Stress with Progressive Hyperlipidemia in Human: Relation between Malondialdehyde and Atherogenic Index. J. Clin. Biochem. Nutr. 2008, 43, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Roca-Agujetas, V.; Barbero-Camps, E.; de Dios, C.; Podlesniy, P.; Abadin, X.; Morales, A.; Marí, M.; Trullàs, R.; Colell, A. Cholesterol alters mitophagy by impairing optineurin recruitment and lysosomal clearance in Alzheimer’s disease. Mol. Neurodegener. 2021, 16, 15. [Google Scholar] [CrossRef] [PubMed]
- Morton, S.; Hesson, L.; Peggie, M.; Cohen, P. Enhanced binding of TBK1 by an optineurin mutant that causes a familial form of primary open angle glaucoma. FEBS Lett. 2008, 582, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, M.L.; Radha, V.; Gupta, V.; Agarwal, N.; Balasubramanian, D.; Swarup, G. A glaucoma-associated mutant of optineurin selectively induces death of retinal ganglion cells which is inhibited by antioxidants. Investig. Opthalmol. Vis. Sci. 2007, 48, 1607–1614. [Google Scholar] [CrossRef]
- Sun, J.-H.; Yu, J.-T.; Tan, L. The role of cholesterol metabolism in Alzheimer’s disease. Mol. Neurobiol. 2015, 51, 947–965. [Google Scholar] [CrossRef]
- Barbero-Camps, E.; Roca-Agujetas, V.; Bartolessis, I.; de Dios, C.; Fernández-Checa, J.C.; Marí, M.; Morales, A.; Hartmann, T.; Colell, A. Cholesterol impairs autophagy-mediated clearance of amyloid beta while promoting its secretion. Autophagy 2018, 14, 1129–1154. [Google Scholar] [CrossRef]
- Fernandez, A.; Llacuna, L.; Fernandez-Checa, J.C.; Colell, A. Mitochondrial cholesterol loading exacerbates amyloid beta peptide-induced inflammation and neurotoxicity. J. Neurosci. 2009, 29, 6394–6405. [Google Scholar] [CrossRef] [PubMed]
- de Dios, C.; Bartolessis, I.; Roca-Agujetas, V.; Barbero-Camps, E.; Mari, M.; Morales, A.; Colell, A. Oxidative inactivation of amyloid beta-degrading proteases by cholesterol-enhanced mitochondrial stress. Redox Biol. 2019, 26, 101283. [Google Scholar] [CrossRef] [PubMed]
- Barbero-Camps, E.; Fernandez, A.; Martinez, L.; Fernandez-Checa, J.C.; Colell, A. APP/PS1 mice overexpressing SREBP-2 exhibit combined Abeta accumulation and tau pathology underlying Alzheimer’s disease. Hum. Mol. Genet. 2013, 22, 3460–3476. [Google Scholar] [CrossRef] [PubMed]
- Haddad, M.; Herve, V.; Ben Khedher, M.R.; Rabanel, J.M.; Ramassamy, C. Glutathione: An Old and Small Molecule with Great Functions and New Applications in the Brain and in Alzheimer’s Disease. Antioxid Redox Signal 2021, 35, 270–292. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Saharan, S.; Tripathi, M.; Murari, G. Brain Glutathione Levels–A novel biomarker for mild cognitive impairment and Alzheimer’s disease. Biol. Psychiatry 2015, 78, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Colell, A.; Fernandez, A.; Fernandez-Checa, J.C. Mitochondria, cholesterol and amyloid beta peptide: A dangerous trio in Alzheimer disease. J. Bioenerg. Biomembr. 2009, 41, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Sano, H.; Namekata, K.; Kimura, A.; Shitara, H.; Guo, X.; Harada, C.; Mitamura, Y.; Harada, T. Differential effects of N-acetylcysteine on retinal degeneration in two mouse models of normal tension glaucoma. Cell Death Dis. 2019, 10, 75. [Google Scholar] [CrossRef] [PubMed]
- Harada, C.; Noro, T.; Kimura, A.; Guo, X.; Namekata, K.; Nakano, T.; Harada, T. Suppression of Oxidative Stress as Potential Therapeutic Approach for Normal Tension Glaucoma. Antioxidants 2020, 9, 874. [Google Scholar] [CrossRef]
- Aung, T.; Rezaie, T.; Okada, K.; Viswanathan, A.C.; Child, A.H.; Brice, G.; Bhattacharya, S.S.; Lehmann, O.J.; Sarfarazi, M.; Hitchings, R.A. Clinical features and course of patients with glaucoma with the E50K mutation in the optineurin gene. Investig. Opthalmol. Vis. Sci. 2005, 46, 2816–2822. [Google Scholar] [CrossRef]
- Zhang, S.; Shao, Z.; Liu, X.; Hou, M.; Cheng, F.; Lei, D.; Yuan, H. The E50K optineurin mutation impacts autophagy-mediated degradation of TDP-43 and leads to RGC apoptosis in vivo and in vitro. Cell Death Discov. 2021, 7, 49. [Google Scholar] [CrossRef]
- Ayala-Lugo, R.M.; Pawar, H.; Reed, D.M.; Lichter, P.R.; Moroi, S.E.; Page, M.; Eadie, J.; Azocar, V.; Maul, E.; Ntim-Amponsah, C.; et al. Variation in optineurin (OPTN) allele frequencies between and within populations. Mol. Vis. 2007, 13, 151–163. [Google Scholar]
- Umeda, T.; Matsuo, T.; Nagayama, M.; Tamura, N.; Tanabe, Y.; Ohtsuki, H. Clinical relevance of optineurin sequence alterations in Japanese glaucoma patients. Ophthalmic Genet. 2004, 25, 91–99. [Google Scholar] [CrossRef]
- Fuse, N.; Takahashi, K.; Akiyama, H.; Nakazawa, T.; Seimiya, M.; Kuwahara, S.; Tamai, M. Molecular genetic analysis of optineurin gene for primary open-angle and normal tension glaucoma in the Japanese population. Eur. J. Gastroenterol. Hepatol. 2004, 13, 299–303. [Google Scholar] [CrossRef]
- Chalasani, M.L.S.; Kumari, A.; Radha, V.; Swarup, G. E50K-OPTN-induced retinal cell death involves the rab GTPase-activating protein, TBC1D17 mediated block in autophagy. PLoS ONE 2014, 9, e95758. [Google Scholar] [CrossRef] [PubMed]
- VanderWall, K.B.; Huang, K.-C.; Pan, Y.; Lavekar, S.S.; Fligor, C.M.; Allsop, A.R.; Lentsch, K.A.; Dang, P.; Zhang, C.; Tseng, H.C.; et al. Retinal Ganglion Cells with a Glaucoma OPTN(E50K) Mutation Exhibit Neurodegenerative Phenotypes when Derived from Three-Dimensional Retinal Organoids. Stem Cell Rep. 2020, 15, 52–66. [Google Scholar] [CrossRef]
- Chi, Z.-L.; Akahori, M.; Obazawa, M.; Minami, M.; Noda, T.; Nakaya, N.; Tomarev, S.; Kawase, K.; Yamamoto, T.; Noda, S.; et al. Overexpression of optineurin E50K disrupts Rab8 interaction and leads to a progressive retinal degeneration in mice. Hum. Mol. Genet. 2010, 19, 2606–2615. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.; VanderWall, K.B.; Pan, Y.; Lu, X.; Lavekar, S.S.; Huang, K.-C.; Fligor, C.M.; Harkin, J.; Zhang, C.; Cummins, T.R.; et al. Astrocytes modulate neurodegenerative phenotypes associated with glaucoma in OPTN(E50K) human stem cell-derived retinal ganglion cells. Stem Cell Rep. 2022, 17, 1636–1649. [Google Scholar] [CrossRef]
- Minegishi, Y.; Iejima, D.; Kobayashi, H.; Chi, Z.-L.; Kawase, K.; Yamamoto, T.; Seki, T.; Yuasa, S.; Fukuda, K.; Iwata, T. Enhanced optineurin E50K–TBK1 interaction evokes protein insolubility and initiates familial primary open-angle glaucoma. Hum. Mol. Genet. 2013, 22, 3559–3567. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Kalamida, D.; Giatromanolaki, A.; Zois, C.E.; Sivridis, E.; Pouliliou, S.; Mitrakas, A.; Gatter, K.C.; Harris, A.L. Autophagosome Proteins LC3A, LC3B and LC3C Have Distinct Subcellular Distribution Kinetics and Expression in Cancer Cell Lines. PLoS ONE 2015, 10, e0137675. [Google Scholar] [CrossRef] [PubMed]
- Awadalla, M.S.; Fingert, J.H.; Roos, B.E.; Chen, S.; Holmes, R.; Graham, S.L.; Chehade, M.; Galanopolous, A.; Ridge, B.; Souzeau, E.; et al. Copy number variations of TBK1 in Australian patients with primary open-angle glaucoma. Am. J. Ophthalmol. 2015, 159, 124–130.e1. [Google Scholar] [CrossRef]
- Killer, H.; Pircher, A. Normal tension glaucoma: Review of current understanding and mechanisms of the pathogenesis. Eye 2018, 32, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Demailly, P.; Cambien, F.; Plouin, P.F.; Baron, P.; Chevallier, B. Do patients with low tension glaucoma have particular cardiovascular characteristics? Ophthalmologica 1984, 188, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Tielsch, J.M.; Katz, J.; Sommer, A.; Quigley, H.A.; Javitt, J.C. Hypertension, perfusion pressure, and primary open-angle glaucoma. A population-based assessment. Arch. Ophthalmol. 1995, 113, 216–221. [Google Scholar] [CrossRef]
- Jonas, J.B.; O Naumann, G. Parapapillary retinal vessel diameter in normal and glaucoma eyes. II. Correlations. Investig. Ophthalmol. Vis. Sci. 1989, 30, 1604–1611. [Google Scholar]
- Mitchell, P.; Leung, H.; Wang, J.J.; Rochtchina, E.; Lee, A.J.; Wong, T.Y.; Klein, R. Retinal vessel diameter and open-angle glaucoma: The Blue Mountains Eye Study. Ophthalmology 2005, 112, 245–250. [Google Scholar] [CrossRef]
- Xu, H.; Zhai, R.; Zong, Y.; Kong, X.; Jiang, C.; Sun, X.; He, Y.; Li, X. Comparison of retinal microvascular changes in eyes with high-tension glaucoma or normal-tension glaucoma: A quantitative optic coherence tomography angiographic study. Graefes Arch. Clin. Exp. Ophthalmol. 2018, 256, 1179–1186. [Google Scholar] [CrossRef]
- Berisha, F.; Feke, G.T.; Trempe, C.L.; McMeel, J.W.; Schepens, C.L. Retinal abnormalities in early Alzheimer’s disease. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2285–2289. [Google Scholar] [CrossRef]
- Kawasaki, R.; Cheung, N.; Mosley, T.; Islam, A.; Sharrett, A.R.; Klein, R.; Coker, L.; Knopman, D.; Shibata, D.; Catellier, D.; et al. Retinal microvascular signs and 10-year risk of cerebral atrophy: The Atherosclerosis Risk in Communities (ARIC) study. Stroke 2010, 41, 1826–1828. [Google Scholar] [CrossRef]
- Patton, N.; Pattie, A.; MacGillivray, T.; Aslam, T.; Dhillon, B.; Gow, A.; Starr, J.M.; Whalley, L.J.; Deary, I.J. The association between retinal vascular network geometry and cognitive ability in an elderly population. Investig. Opthalmol. Vis. Sci. 2007, 48, 1995–2000. [Google Scholar] [CrossRef]
- Folkman, J.; Shing, Y. Angiogenesis. J. Biol. Chem. 1992, 267, 10931–10934. [Google Scholar] [CrossRef]
- Vallon, M.; Chang, J.; Zhang, H.; Kuo, C.J. Developmental and pathological angiogenesis in the central nervous system. Cell. Mol. Life Sci. 2014, 71, 3489–3506. [Google Scholar] [CrossRef] [PubMed]
- Rosenstein, J.M.; Krum, J.M.; Ruhrberg, C. VEGF in the nervous system. Organogenesis 2010, 6, 107–114. [Google Scholar] [CrossRef]
- Grammas, P.; Sanchez, A.; Tripathy, D.; Luo, E.; Martinez, J. Vascular signaling abnormalities in Alzheimer disease. Cleve Clin. J. Med 2011, 78 (Suppl. S1), S50–S53. [Google Scholar] [CrossRef] [PubMed]
- Aiello, L.P.; Northrup, J.M.; Keyt, B.A.; Takagi, H.; Iwamoto, M.A. Hypoxic regulation of vascular endothelial growth factor in retinal cells. Arch. Ophthalmol. 1995, 113, 1538–1544. [Google Scholar] [CrossRef] [PubMed]
- Aiello, L.P.; Avery, R.L.; Arrigg, P.G.; Keyt, B.A.; Jampel, H.D.; Shah, S.T.; Pasquale, L.R.; Thieme, H.; Iwamoto, M.A.; Park, J.E.; et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N. Engl. J. Med. 1994, 331, 1480–1487. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, Z. Neovascular Age-Related Macular Degeneration and its Association with Alzheimer’s Disease. Curr. Aging Sci. 2020, 13, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.P.; Bae, D.G.; Kang, H.J.; Gwag, B.J.; Gho, Y.S.; Chae, C.B. Co-accumulation of vascular endothelial growth factor with beta-amyloid in the brain of patients with Alzheimer’s disease. Neurobiol. Aging 2004, 25, 283–290. [Google Scholar] [CrossRef]
- Alvarez, X.A.; Alvarez, I.; Aleixandre, M.; Linares, C.; Muresanu, D.; Winter, S.; Moessler, H. Severity-Related Increase and Cognitive Correlates of Serum VEGF Levels in Alzheimer’s Disease ApoE4 Carriers. J. Alzheimers Dis. 2018, 63, 1003–1013. [Google Scholar] [CrossRef]
- Ali, M.; Bracko, O. VEGF Paradoxically Reduces Cerebral Blood Flow in Alzheimer’s Disease Mice. Neurosci. Insights 2022, 17, 26331055221109254. [Google Scholar] [CrossRef]
- Harris, R.; Miners, J.S.; Allen, S.; Love, S. VEGFR1 and VEGFR2 in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 61, 741–752. [Google Scholar] [CrossRef]
- Ali, M.; Falkenhain, K.; Njiru, B.N.; Murtaza-Ali, M.; Ruiz-Uribe, N.E.; Haft-Javaherian, M.; Catchers, S.; Nishimura, N.; Schaffer, C.B.; Bracko, O. VEGF signalling causes stalls in brain capillaries and reduces cerebral blood flow in Alzheimer’s mice. Brain 2022, 145, 1449–1463. [Google Scholar] [CrossRef] [PubMed]
- Reeson, P.; Choi, K.; Brown, C.E. VEGF signaling regulates the fate of obstructed capillaries in mouse cortex. Elife 2018, 7, e33670. [Google Scholar] [CrossRef] [PubMed]
- Akiyode, O.; Dunkelly-Allen, N. Ranibizumab: A Review of Its Use in the Treatment of Diabetic Retinopathy in Patients with Diabetic Macular Edema. J. Pharm. Technol. 2016, 32, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Sin, B.H.; Song, B.J.; Park, S.P. Aqueous vascular endothelial growth factor and endothelin-1 levels in branch retinal vein occlusion associated with normal tension glaucoma. J. Glaucoma 2013, 22, 104–109. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, Z.; Li, H.; Xia, Y.; Xing, M.; Xiao, C.; Cai, W.; Bu, L.; Li, Y.; Park, T.E.; et al. Blockage of VEGF function by bevacizumab alleviates early-stage cerebrovascular dysfunction and improves cognitive function in a mouse model of Alzheimer’s disease. Transl. Neurodegener. 2024, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.U.; Park, I.W.; Suh, W. Long-term intraocular pressure changes after intravitreal injection of bevacizumab. Cutan. Ocul. Toxicol. 2016, 35, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Bressler, S.B.; Almukhtar, T.; Bhorade, A.; Bressler, N.M.; Glassman, A.R.; Huang, S.S.; Jampol, L.M.; Kim, J.E.; Melia, M.; Diabetic Retinopathy Clinical Research Network, I. Repeated intravitreous ranibizumab injections for diabetic macular edema and the risk of sustained elevation of intraocular pressure or the need for ocular hypotensive treatment. JAMA Ophthalmol. 2015, 133, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Pirinen, I.; Leinonen, S.; Helminen, M.; Hujanen, P.; Vaajanen, A.; Tuulonen, A.; Uusitalo-Jarvinen, H. Glaucoma progression in patients receiving intravitreal anti-VEGF treatment for neovascular age-related macular degeneration. Acta Ophthalmol. 2023, 101, 261–265. [Google Scholar] [CrossRef]
- Spini, A.; Giometto, S.; Donnini, S.; Posarelli, M.; Dotta, F.; Ziche, M.; Tosi, G.M.; Girardi, A.; Lucenteforte, E.; Gini, R.; et al. Risk of Intraocular Pressure Increase with Intravitreal Injections of Vascular Endothelial Growth Factor Inhibitors: A Cohort Study. Am. J. Ophthalmol. 2023, 248, 45–50. [Google Scholar] [CrossRef]
- Sultana, J.; Scondotto, G.; Cutroneo, P.M.; Morgante, F.; Trifiro, G. Intravitreal Anti-VEGF Drugs and Signals of Dementia and Parkinson-Like Events: Analysis of the VigiBase Database of Spontaneous Reports. Front. Pharmacol. 2020, 11, 315. [Google Scholar] [CrossRef]
- Yoshimoto, M.; Takeda, N.; Yoshimoto, T.; Matsumoto, S. Hypertensive cerebral hemorrhage with undetectable plasma vascular endothelial growth factor levels in a patient receiving intravitreal injection of aflibercept for bilateral diabetic macular edema: A case report. J. Med. Case Rep. 2021, 15, 403. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.K.; Manz, S.N. Brain health assessment in macular degeneration patients undergoing intravitreal anti-vascular endothelial growth factor injections (the bham study): An interim analysis. Retina 2021, 41, 1748–1753. [Google Scholar] [CrossRef] [PubMed]
- Bayhan, H.A.; Aslan Bayhan, S.; Celikbilek, A.; Tanik, N.; Gurdal, C. Evaluation of the chorioretinal thickness changes in Alzheimer’s disease using spectral-domain optical coherence tomography. Clin. Exp. Ophthalmol. 2015, 43, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Gharbiya, M.; Trebbastoni, A.; Parisi, F.; Manganiello, S.; Cruciani, F.; D’Antonio, F.; De Vico, U.; Imbriano, L.; Campanelli, A.; De Lena, C. Choroidal thinning as a new finding in Alzheimer’s disease: Evidence from enhanced depth imaging spectral domain optical coherence tomography. J. Alzheimers Dis. 2014, 40, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Trebbastoni, A.; Marcelli, M.; Mallone, F.; D’Antonio, F.; Imbriano, L.; Campanelli, A.; de Lena, C.; Gharbiya, M. Attenuation of Choroidal Thickness in Patients with Alzheimer Disease: Evidence From an Italian Prospective Study. Alzheimer Dis. Assoc. Disord. 2017, 31, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Cunha, J.P.; Proenca, R.; Dias-Santos, A.; Melancia, D.; Almeida, R.; Aguas, H.; Santos, B.O.; Alves, M.; Ferreira, J.; Papoila, A.L.; et al. Choroidal thinning: Alzheimer’s disease and aging. Alzheimers Dement. 2017, 8, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.M.; Hui, V.W.K.; Shi, J.; Wong, M.O.M.; Chan, P.P.; Chan, N.; Lai, I.; Cheung, C.Y.; Tham, C.C. Characterization of macular choroid in normal-tension glaucoma: A swept-source optical coherence tomography study. Acta Ophthalmol. 2021, 99, e1421–e1429. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Wu, X.; Wang, X.; Geng, Z.; Wang, L.; Xiao, G.; Wu, Y.; Zhou, S.; Liao, R.; Wei, L.; et al. The Retinal Vessel Density Can Reflect Cognitive Function in Patients with Alzheimer’s Disease: Evidence from Optical Coherence Tomography Angiography. J. Alzheimers Dis. 2021, 79, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.R. Glaucoma, capillaries and pericytes. 1. Blood flow regulation. Ophthalmologica 1996, 210, 257–262. [Google Scholar] [CrossRef]
- Venkataraman, S.T.; Flanagan, J.G.; Hudson, C. Vascular reactivity of optic nerve head and retinal blood vessels in glaucoma—A review. Microcirculation 2010, 17, 568–581. [Google Scholar] [CrossRef]
- Drance, S.M. Some factors in the production of low tension glaucoma. Br. J. Ophthalmol. 1972, 56, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Hayreh, S.S.; Podhajsky, P.; Zimmerman, M.B. Beta-blocker eyedrops and nocturnal arterial hypotension. Am. J. Ophthalmol. 1999, 128, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.W.; Jo, Y.H.; Song, M.K.; Won, H.J.; Kook, M.S. Nocturnal blood pressure dip and parapapillary choroidal microvasculature dropout in normal-tension glaucoma. Sci. Rep. 2021, 11, 206. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Jo, Y.H.; Jeong, D.; Shon, K.; Kook, M.S. Baseline Systolic versus Diastolic Blood Pressure Dip and Subsequent Visual Field Progression in Normal-Tension Glaucoma. Ophthalmology 2019, 126, 967–979. [Google Scholar] [CrossRef] [PubMed]
- Melgarejo, J.D.; Lee, J.H.; Petitto, M.; Yepez, J.B.; Murati, F.A.; Jin, Z.; Chavez, C.A.; Pirela, R.V.; Calmon, G.E.; Lee, W.; et al. Glaucomatous Optic Neuropathy Associated with Nocturnal Dip in Blood Pressure: Findings from the Maracaibo Aging Study. Ophthalmology 2018, 125, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Tabara, Y.; Igase, M.; Yamamoto, M.; Ochi, N.; Kido, T.; Uetani, E.; Taguchi, K.; Miki, T.; Kohara, K. Abnormal nocturnal blood pressure profile is associated with mild cognitive impairment in the elderly: The J-SHIPP study. Hypertens. Res. 2010, 33, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Su, W.W.; Cheng, S.T.; Hsu, T.S.; Ho, W.J. Abnormal flow-mediated vasodilation in normal-tension glaucoma using a noninvasive determination for peripheral endothelial dysfunction. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3390–3394. [Google Scholar] [CrossRef] [PubMed]
- Buckley, C.; Hadoke, P.W.; Henry, E.; O’Brien, C. Systemic vascular endothelial cell dysfunction in normal pressure glaucoma. Br. J. Ophthalmol. 2002, 86, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Galassi, F.; Giambene, B.; Varriale, R. Systemic vascular dysregulation and retrobulbar hemodynamics in normal-tension glaucoma. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4467–4471. [Google Scholar] [CrossRef]
- Sugiyama, T.; Moriya, S.; Oku, H.; Azuma, I. Association of endothelin-1 with normal tension glaucoma: Clinical and fundamental studies. Surv. Ophthalmol. 1995, 39 (Suppl. S1), S49–S56. [Google Scholar] [CrossRef]
- Cellini, M.; Possati, G.L.; Profazio, V.; Sbrocca, M.; Caramazza, N.; Caramazza, R. Color Doppler imaging and plasma levels of endothelin-1 in low-tension glaucoma. Acta Ophthalmol. Scand. Suppl. 1997, 75, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, A.; Kleniewska, P.; Kolodziejczyk, M.; Skibska, B.; Goraca, A. The role of endothelin-1 and endothelin receptor antagonists in inflammatory response and sepsis. Arch. Immunol. Ther. Exp. 2015, 63, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Kunimatsu, S.; Mayama, C.; Tomidokoro, A.; Araie, M. Plasma endothelin-1 level in Japanese normal tension glaucoma patients. Curr. Eye Res. 2006, 31, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.C.; Barker, R.; Kehoe, P.G.; Love, S. Endothelin-1 is elevated in Alzheimer’s disease and upregulated by amyloid-beta. J. Alzheimers Dis. 2012, 29, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Siaudvytyte, L.; Januleviciene, I.; Ragauskas, A.; Bartusis, L.; Meiliuniene, I.; Siesky, B.; Harris, A. The difference in translaminar pressure gradient and neuroretinal rim area in glaucoma and healthy subjects. J. Ophthalmol. 2014, 2014, 937360. [Google Scholar] [CrossRef] [PubMed]
- Quigley, H.A.; Hohman, R.M.; Addicks, E.M.; Massof, R.W.; Green, W.R. Morphologic changes in the lamina cribrosa correlated with neural loss in open-angle glaucoma. Am. J. Ophthalmol. 1983, 95, 673–691. [Google Scholar] [CrossRef]
- Berdahl, J.P.; Allingham, R.R.; Johnson, D.H. Cerebrospinal fluid pressure is decreased in primary open-angle glaucoma. Ophthalmology 2008, 115, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.; Jonas, J.B.; Tian, G.; Zhen, Y.; Ma, K.; Li, S.; Wang, H.; Li, B.; Zhang, X.; Wang, N. Cerebrospinal fluid pressure in glaucoma: A prospective study. Ophthalmology 2010, 117, 259–266. [Google Scholar] [CrossRef]
- Berdahl, J.P.; Fautsch, M.P.; Stinnett, S.S.; Allingham, R.R. Intracranial pressure in primary open angle glaucoma, normal tension glaucoma, and ocular hypertension: A case-control study. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5412–5418. [Google Scholar] [CrossRef]
- Wang, N.; Yang, D.; Jonas, J.B. Low cerebrospinal fluid pressure in the pathogenesis of primary open-angle glaucoma: Epiphenomenon or causal relationship? The Beijing Intracranial and Intraocular Pressure (iCOP) study. J. Glaucoma 2013, 22 (Suppl. S5), S11–S12. [Google Scholar] [CrossRef]
- Silverberg, G.; Mayo, M.; Saul, T.; Fellmann, J.; McGuire, D. Elevated cerebrospinal fluid pressure in patients with Alzheimer’s disease. Cerebrospinal Fluid Res. 2006, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Wostyn, P.; Audenaert, K.; De Deyn, P.P. Alzheimer’s disease and glaucoma: Is there a causal relationship? Br. J. Ophthalmol. 2009, 93, 1557–1559. [Google Scholar] [CrossRef] [PubMed]
- Killer, H.E.; Jaggi, G.P.; Flammer, J.; Miller, N.R.; Huber, A.R. The optic nerve: A new window into cerebrospinal fluid composition? Brain 2006, 129, 1027–1030. [Google Scholar] [CrossRef] [PubMed]
- Omatiga, A.G.; Onakpoya, O.H.; Idowu, B.M.; Asaleye, C.M.; Adegbehingbe, B.O.; Aderibigbe, A.S. B-mode sonographic evaluation of optic nerve sheath diameter and lens thickness in Nigerian adults with glaucoma. Afr. Health Sci. 2018, 18, 343–351. [Google Scholar] [CrossRef]
- Telano, L.N.; Baker, S. Physiology, Cerebral Spinal Fluid. In StatPearls; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Killer, H.E. Is stagnant cerebrospinal fluid involved in the pathophysiology of normal tension glaucoma. Prog. Brain Res. 2020, 256, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, E.; Gupta, N.; Ahari, A.; Zhou, X.; Hanna, J.; Yucel, Y.H. Evidence for Cerebrospinal Fluid Entry Into the Optic Nerve via a Glymphatic Pathway. Investig. Ophthalmol. Vis. Sci. 2017, 58, 4784–4791. [Google Scholar] [CrossRef] [PubMed]
- Wostyn, P.; Killer, H.E. Normal-Tension Glaucoma: A Glymphopathy? Eye Brain 2023, 15, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Killer, H.E.; Miller, N.R.; Flammer, J.; Meyer, P.; Weinreb, R.N.; Remonda, L.; Jaggi, G.P. Cerebrospinal fluid exchange in the optic nerve in normal-tension glaucoma. Br. J. Ophthalmol. 2012, 96, 544–548. [Google Scholar] [CrossRef]
- Silverberg, G.D.; Mayo, M.; Saul, T.; Rubenstein, E.; McGuire, D. Alzheimer’s disease, normal-pressure hydrocephalus, and senescent changes in CSF circulatory physiology: A hypothesis. Lancet Neurol. 2003, 2, 506–511. [Google Scholar] [CrossRef]
- Silverberg, G.D.; Heit, G.; Huhn, S.; Jaffe, R.A.; Chang, S.D.; Bronte-Stewart, H.; Rubenstein, E.; Possin, K.; Saul, T.A. The cerebrospinal fluid production rate is reduced in dementia of the Alzheimer’s type. Neurology 2001, 57, 1763–1766. [Google Scholar] [CrossRef]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, S.J.; Lehman, D.M.; Kerrigan-Baumrind, L.A.; Merges, C.A.; Pease, M.E.; Kerrigan, D.F.; Ransom, N.L.; Tahzib, N.G.; Reitsamer, H.A.; Levkovitch-Verbin, H.; et al. Caspase activation and amyloid precursor protein cleavage in rat ocular hypertension. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1077–1087. [Google Scholar] [PubMed]
- McKinnon, S.J. Glaucoma: Ocular Alzheimer’s disease? Front. Biosci. 2003, 8, s1140–s1156. [Google Scholar] [CrossRef]
- Goldblum, D.; Kipfer-Kauer, A.; Sarra, G.M.; Wolf, S.; Frueh, B.E. Distribution of amyloid precursor protein and amyloid-beta immunoreactivity in DBA/2J glaucomatous mouse retinas. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5085–5090. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Fong, J.; Ang, L.C.; Yucel, Y.H. Retinal tau pathology in human glaucomas. Can. J. Ophthalmol. 2008, 43, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Bolos, M.; Perea, J.R.; Avila, J. Alzheimer’s disease as an inflammatory disease. Biomol. Concepts 2017, 8, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.K.; Mestre, H.; Nedergaard, M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018, 17, 1016–1024. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, M. Neuroscience. Garbage truck of the brain. Science 2013, 340, 1529–1530. [Google Scholar] [CrossRef]
- Foglio, E.; Rodella, L.F. Aquaporins and neurodegenerative diseases. Curr. Neuropharmacol. 2010, 8, 112–121. [Google Scholar] [CrossRef]
- Kress, B.T.; Iliff, J.J.; Xia, M.; Wang, M.; Wei, H.S.; Zeppenfeld, D.; Xie, L.; Kang, H.; Xu, Q.; Liew, J.A.; et al. Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 2014, 76, 845–861. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Xiao, N.; Chen, Y.; Huang, H.; Marshall, C.; Gao, J.; Cai, Z.; Wu, T.; Hu, G.; Xiao, M. Deletion of aquaporin-4 in APP/PS1 mice exacerbates brain Abeta accumulation and memory deficits. Mol. Neurodegener. 2015, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Bothwell, S.W.; Janigro, D.; Patabendige, A. Cerebrospinal fluid dynamics and intracranial pressure elevation in neurological diseases. Fluids Barriers CNS 2019, 16, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lou, N.; Eberhardt, A.; Yang, Y.; Kusk, P.; Xu, Q.; Forstera, B.; Peng, S.; Shi, M.; Ladron-de-Guevara, A.; et al. An ocular glymphatic clearance system removes beta-amyloid from the rodent eye. Sci. Transl. Med. 2020, 12, eaaw3210. [Google Scholar] [CrossRef] [PubMed]
- Bidot, S.; Clough, L.; Saindane, A.M.; Newman, N.J.; Biousse, V.; Bruce, B.B. The Optic Canal Size Is Associated with the Severity of Papilledema and Poor Visual Function in Idiopathic Intracranial Hypertension. J. Neuroophthalmol. 2016, 36, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Pircher, A.; Montali, M.; Berberat, J.; Remonda, L.; Killer, H.E. The Optic Canal: A Bottleneck for Cerebrospinal Fluid Dynamics in Normal-Tension Glaucoma? Front. Neurol. 2017, 8, 47. [Google Scholar] [CrossRef]
- Soldatos, T.; Karakitsos, D.; Chatzimichail, K.; Papathanasiou, M.; Gouliamos, A.; Karabinis, A. Optic nerve sonography in the diagnostic evaluation of adult brain injury. Crit. Care 2008, 12, R67. [Google Scholar] [CrossRef] [PubMed]
- Geeraerts, T.; Merceron, S.; Benhamou, D.; Vigue, B.; Duranteau, J. Non-invasive assessment of intracranial pressure using ocular sonography in neurocritical care patients. Intensive Care Med. 2008, 34, 2062–2067. [Google Scholar] [CrossRef]
- Bachmann, G.; Achtelik, R.; Nekic, M.; Michel, O. Beta-trace protein in diagnosis of cerebrospinal fluid fistula. HNO 2000, 48, 496–500. [Google Scholar] [CrossRef]
- Hoffmann, A.; Conradt, H.S.; Gross, G.; Nimtz, M.; Lottspeich, F.; Wurster, U. Purification and chemical characterization of beta-trace protein from human cerebrospinal fluid: Its identification as prostaglandin D synthase. J. Neurochem. 1993, 61, 451–456. [Google Scholar] [CrossRef]
- Watanabe, K.; Urade, Y.; Mader, M.; Murphy, C.; Hayaishi, O. Identification of beta-trace as prostaglandin D synthase. Biochem. Biophys. Res. Commun. 1994, 203, 1110–1116. [Google Scholar] [CrossRef]
- Link, H.; Olsson, J.E. Beta-trace protein concentration in CSF in neurological disorders. Acta Neurol. Scand. 1972, 48, 57–68. [Google Scholar] [CrossRef]
- Urade, Y.; Hayaishi, O. Prostaglandin D synthase: Structure and function. Vitam. Horm. 2000, 58, 89–120. [Google Scholar] [CrossRef] [PubMed]
- Beuckmann, C.T.; Gordon, W.C.; Kanaoka, Y.; Eguchi, N.; Marcheselli, V.L.; Gerashchenko, D.Y.; Urade, Y.; Hayaishi, O.; Bazan, N.G. Lipocalin-type prostaglandin D synthase (beta-trace) is located in pigment epithelial cells of rat retina and accumulates within interphotoreceptor matrix. J. Neurosci. 1996, 16, 6119–6124. [Google Scholar] [CrossRef]
- Pircher, A.; Neutzner, A.; Montali, M.; Huber, A.; Scholl, H.P.N.; Berberat, J.; Remonda, L.; Killer, H.E. Lipocalin-type Prostaglandin D Synthase Concentration Gradients in the Cerebrospinal Fluid in Normal-tension Glaucoma Patients with Optic Nerve Sheath Compartmentation. Eye Brain 2021, 13, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Kannaian, B.; Sharma, B.; Phillips, M.; Chowdhury, A.; Manimekalai, M.S.S.; Adav, S.S.; Ng, J.T.Y.; Kumar, A.; Lim, S.; Mu, Y.; et al. Abundant neuroprotective chaperone Lipocalin-type prostaglandin D synthase (L-PGDS) disassembles the Amyloid-beta fibrils. Sci. Rep. 2019, 9, 12579. [Google Scholar] [CrossRef]
- Nishida, N.; Nagata, N.; Toda, H.; Jingami, N.; Uemura, K.; Ozaki, A.; Mase, M.; Urade, Y.; Matsumoto, S.; Iwasaki, K.; et al. Association of lipocalin-type prostaglandin D synthase with disproportionately enlarged subarachnoid-space in idiopathic normal pressure hydrocephalus. Fluids Barriers CNS 2014, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Jaggi, G.P.; Harlev, M.; Ziegler, U.; Dotan, S.; Miller, N.R.; Killer, H.E. Cerebrospinal fluid segregation optic neuropathy: An experimental model and a hypothesis. Br. J. Ophthalmol. 2010, 94, 1088–1093. [Google Scholar] [CrossRef]
- Xin, X.; Huber, A.; Meyer, P.; Flammer, J.; Neutzner, A.; Miller, N.R.; Killer, H.E. L-PGDS (betatrace protein) inhibits astrocyte proliferation and mitochondrial ATP production in vitro. J. Mol. Neurosci. 2009, 39, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Maesaka, J.K.; Sodam, B.; Palaia, T.; Ragolia, L.; Batuman, V.; Miyawaki, N.; Shastry, S.; Youmans, S.; El-Sabban, M. Prostaglandin D2 synthase: Apoptotic factor in alzheimer plasma, inducer of reactive oxygen species, inflammatory cytokines and dialysis dementia. J. Nephropathol. 2013, 2, 166–180. [Google Scholar] [CrossRef]
- Guan, P.P.; Liang, Y.Y.; Cao, L.L.; Yu, X.; Wang, P. Cyclooxygenase-2 Induced the beta-Amyloid Protein Deposition and Neuronal Apoptosis Via Upregulating the Synthesis of Prostaglandin E(2) and 15-Deoxy-Delta(12,14)-prostaglandin J(2). Neurotherapeutics 2019, 16, 1255–1268. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Peskind, E.R.; Millard, S.P.; Chi, P.; Sokal, I.; Yu, C.E.; Bekris, L.M.; Raskind, M.A.; Galasko, D.R.; Montine, T.J. Cerebrospinal fluid concentration of brain-derived neurotrophic factor and cognitive function in non-demented subjects. PLoS ONE 2009, 4, e5424. [Google Scholar] [CrossRef] [PubMed]
- Laske, C.; Stransky, E.; Leyhe, T.; Eschweiler, G.W.; Maetzler, W.; Wittorf, A.; Soekadar, S.; Richartz, E.; Koehler, N.; Bartels, M.; et al. BDNF serum and CSF concentrations in Alzheimer’s disease, normal pressure hydrocephalus and healthy controls. J. Psychiatr. Res. 2007, 41, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Zhang, Y.; Sterling, K.; Song, W. Brain-derived neurotrophic factor in Alzheimer’s disease and its pharmaceutical potential. Transl. Neurodegener. 2022, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Forlenza, O.V.; Diniz, B.S.; Teixeira, A.L.; Radanovic, M.; Talib, L.L.; Rocha, N.P.; Gattaz, W.F. Lower Cerebrospinal Fluid Concentration of Brain-Derived Neurotrophic Factor Predicts Progression from Mild Cognitive Impairment to Alzheimer’s Disease. Neuromol. Med. 2015, 17, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Pillai, A.; Bruno, D.; Sarreal, A.S.; Hernando, R.T.; Saint-Louis, L.A.; Nierenberg, J.; Ginsberg, S.D.; Pomara, N.; Mehta, P.D.; Zetterberg, H.; et al. Plasma BDNF levels vary in relation to body weight in females. PLoS ONE 2012, 7, e39358. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Takada, N.; Fujioka, A.; Himori, N.; Yokoyama, Y.; Tsuda, S.; Omodaka, K.; Kirihara, T.; Ishikawa, M.; Kunikata, H.; et al. Reduced Plasma BDNF Levels in Normal Tension Glaucoma Compared to Open Angle Glaucoma. J. Glaucoma 2023, 32, 734–737. [Google Scholar] [CrossRef]
- Klein, A.B.; Williamson, R.; Santini, M.A.; Clemmensen, C.; Ettrup, A.; Rios, M.; Knudsen, G.M.; Aznar, S. Blood BDNF concentrations reflect brain-tissue BDNF levels across species. Int. J. Neuropsychopharmacol. 2011, 14, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Conti, F.; Romano, G.L.; Eandi, C.M.; Toro, M.D.; Rejdak, R.; Di Benedetto, G.; Lazzara, F.; Bernardini, R.; Drago, F.; Cantarella, G.; et al. Brimonidine is Neuroprotective in Animal Paradigm of Retinal Ganglion Cell Damage. Front. Pharmacol. 2021, 12, 705405. [Google Scholar] [CrossRef]
- Yukita, M.; Omodaka, K.; Machida, S.; Yasuda, M.; Sato, K.; Maruyama, K.; Nishiguchi, K.M.; Nakazawa, T. Brimonidine Enhances the Electrophysiological Response of Retinal Ganglion Cells through the Trk-MAPK/ERK and PI3K Pathways in Axotomized Eyes. Curr. Eye Res. 2017, 42, 125–133. [Google Scholar] [CrossRef]
- Shen, T.; Gupta, V.K.; Klistorner, A.; Chitranshi, N.; Graham, S.L.; You, Y. Sex-Specific Effect of BDNF Val66Met Genotypes on the Progression of Open-Angle Glaucoma. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1069–1075. [Google Scholar] [CrossRef]
- Quigley, H.A.; McKinnon, S.J.; Zack, D.J.; Pease, M.E.; Kerrigan-Baumrind, L.A.; Kerrigan, D.F.; Mitchell, R.S. Retrograde axonal transport of BDNF in retinal ganglion cells is blocked by acute IOP elevation in rats. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3460–3466. [Google Scholar]
- Gonzalez, H.; Elgueta, D.; Montoya, A.; Pacheco, R. Neuroimmune regulation of microglial activity involved in neuroinflammation and neurodegenerative diseases. J. Neuroimmunol. 2014, 274, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Magni, P.; Ruscica, M.; Dozio, E.; Rizzi, E.; Beretta, G.; Maffei Facino, R. Parthenolide inhibits the LPS-induced secretion of IL-6 and TNF-alpha and NF-kappaB nuclear translocation in BV-2 microglia. Phytother. Res. 2012, 26, 1405–1409. [Google Scholar] [CrossRef]
- Ramirez, A.I.; de Hoz, R.; Salobrar-Garcia, E.; Salazar, J.J.; Rojas, B.; Ajoy, D.; Lopez-Cuenca, I.; Rojas, P.; Trivino, A.; Ramirez, J.M. The Role of Microglia in Retinal Neurodegeneration: Alzheimer’s Disease, Parkinson, and Glaucoma. Front. Aging Neurosci. 2017, 9, 214. [Google Scholar] [CrossRef] [PubMed]
- Polans, J.; Keller, B.; Carrasco-Zevallos, O.M.; LaRocca, F.; Cole, E.; Whitson, H.E.; Lad, E.M.; Farsiu, S.; Izatt, J.A. Wide-field retinal optical coherence tomography with wavefront sensorless adaptive optics for enhanced imaging of targeted regions. Biomed. Opt. Express 2017, 8, 16–37. [Google Scholar] [CrossRef]
- Kirbas, S.; Turkyilmaz, K.; Anlar, O.; Tufekci, A.; Durmus, M. Retinal nerve fiber layer thickness in patients with Alzheimer disease. J. Neuroophthalmol. 2013, 33, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Tepelus, T.C.; Song, S.; Borrelli, E.; Nittala, M.G.; Baghdasaryan, E.; Sadda, S.R.; Chopra, V. Quantitative Analysis of Retinal and Choroidal Vascular Parameters in Patients with Low Tension Glaucoma. J. Glaucoma 2019, 28, 557–562. [Google Scholar] [CrossRef]
- Parisi, V.; Restuccia, R.; Fattapposta, F.; Mina, C.; Bucci, M.G.; Pierelli, F. Morphological and functional retinal impairment in Alzheimer’s disease patients. Clin. Neurophysiol. 2001, 112, 1860–1867. [Google Scholar] [CrossRef]
- Tan, O.; Chopra, V.; Lu, A.T.; Schuman, J.S.; Ishikawa, H.; Wollstein, G.; Varma, R.; Huang, D. Detection of macular ganglion cell loss in glaucoma by Fourier-domain optical coherence tomography. Ophthalmology 2009, 116, 2305–2314. [Google Scholar] [CrossRef]
- Eraslan, M.; Cerman, E.; Cekic, O.; Balci, S.; Dericioglu, V.; Sahin, O.; Suer, D.; Chabou, B.; Tuncer Elmaci, E.N. Neurodegeneration in ocular and central nervous systems: Optical coherence tomography study in normal-tension glaucoma and Alzheimer disease. Turk. J. Med. Sci. 2015, 45, 1106–1114. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.R.; Lee, E.S.; Seong, G.J.; Kim, J.H.; An, H.G.; Kim, C.Y. Structure-function relationship and diagnostic value of macular ganglion cell complex measurement using Fourier-domain OCT in glaucoma. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4646–4651. [Google Scholar] [CrossRef] [PubMed]
- Asanad, S.; Ross-Cisneros, F.N.; Nassisi, M.; Barron, E.; Karanjia, R.; Sadun, A.A. The Retina in Alzheimer’s Disease: Histomorphometric Analysis of an Ophthalmologic Biomarker. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Huang, E.J.; Kuo, C.N.; Wu, P.L.; Chen, C.L.; Wu, P.C.; Wu, S.H.; King, Y.C.; Lai, C.H. The relationship between age, axial length and retinal nerve fiber layer thickness in the normal elderly population in Taiwan: The Chiayi eye study in Taiwan. PLoS ONE 2018, 13, e0194116. [Google Scholar] [CrossRef]
- Parisi, V. Correlation between morphological and functional retinal impairment in patients affected by ocular hypertension, glaucoma, demyelinating optic neuritis and Alzheimer’s disease. Semin. Ophthalmol. 2003, 18, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Iseri, P.K.; Altinas, O.; Tokay, T.; Yuksel, N. Relationship between cognitive impairment and retinal morphological and visual functional abnormalities in Alzheimer disease. J. Neuroophthalmol. 2006, 26, 18–24. [Google Scholar] [CrossRef]
- Kergoat, H.; Kergoat, M.J.; Justino, L.; Chertkow, H.; Robillard, A.; Bergman, H. An evaluation of the retinal nerve fiber layer thickness by scanning laser polarimetry in individuals with dementia of the Alzheimer type. Acta Ophthalmol. Scand. 2001, 79, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Paquet, C.; Boissonnot, M.; Roger, F.; Dighiero, P.; Gil, R.; Hugon, J. Abnormal retinal thickness in patients with mild cognitive impairment and Alzheimer’s disease. Neurosci. Lett. 2007, 420, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Moschos, M.M.; Markopoulos, I.; Chatziralli, I.; Rouvas, A.; Papageorgiou, S.G.; Ladas, I.; Vassilopoulos, D. Structural and functional impairment of the retina and optic nerve in Alzheimer’s disease. Curr. Alzheimer Res. 2012, 9, 782–788. [Google Scholar] [CrossRef]
- Marziani, E.; Pomati, S.; Ramolfo, P.; Cigada, M.; Giani, A.; Mariani, C.; Staurenghi, G. Evaluation of retinal nerve fiber layer and ganglion cell layer thickness in Alzheimer’s disease using spectral-domain optical coherence tomography. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5953–5958. [Google Scholar] [CrossRef]
- Moreno-Ramos, T.; Benito-Leon, J.; Villarejo, A.; Bermejo-Pareja, F. Retinal nerve fiber layer thinning in dementia associated with Parkinson’s disease, dementia with Lewy bodies, and Alzheimer’s disease. J. Alzheimers Dis. 2013, 34, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Kromer, R.; Serbecic, N.; Hausner, L.; Froelich, L.; Aboul-Enein, F.; Beutelspacher, S.C. Detection of Retinal Nerve Fiber Layer Defects in Alzheimer’s Disease Using SD-OCT. Front. Psychiatry 2014, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Wu, Y.; Wang, M.; Cao, J.; Feng, W.; Cheng, Y.; Li, C.; Shen, Y. Greater attenuation of retinal nerve fiber layer thickness in Alzheimer’s disease patients. J. Alzheimers Dis. 2014, 40, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Coppola, G.; Di Renzo, A.; Ziccardi, L.; Martelli, F.; Fadda, A.; Manni, G.; Barboni, P.; Pierelli, F.; Sadun, A.A.; Parisi, V. Optical Coherence Tomography in Alzheimer’s Disease: A Meta-Analysis. PLoS ONE 2015, 10, e0134750. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Liu, Y.; Li, X.; Bai, Q.; Liu, P. Abnormal retinal nerve fiber layer thickness and macula lutea in patients with mild cognitive impairment and Alzheimer’s disease. Arch. Gerontol. Geriatr. 2015, 60, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhang, L.; Li, Z.; Zhang, X.; Wu, Y.; Yang, H.; Min, B.; Zhang, X.; Ma, D.; Lu, Y. Thinner changes of the retinal nerve fiber layer in patients with mild cognitive impairment and Alzheimer’s disease. BMC Neurol. 2015, 15, 14. [Google Scholar] [CrossRef] [PubMed]
- La Morgia, C.; Ross-Cisneros, F.N.; Koronyo, Y.; Hannibal, J.; Gallassi, R.; Cantalupo, G.; Sambati, L.; Pan, B.X.; Tozer, K.R.; Barboni, P.; et al. Melanopsin retinal ganglion cell loss in Alzheimer disease. Ann. Neurol. 2016, 79, 90–109. [Google Scholar] [CrossRef] [PubMed]
- Cunha, J.P.; Moura-Coelho, N.; Proenca, R.P.; Dias-Santos, A.; Ferreira, J.; Louro, C.; Castanheira-Dinis, A. Alzheimer’s disease: A review of its visual system neuropathology. Optical coherence tomography—A potential role as a study tool in vivo. Graefes Arch. Clin. Exp. Ophthalmol. 2016, 254, 2079–2092. [Google Scholar] [CrossRef] [PubMed]
- Doustar, J.; Torbati, T.; Black, K.L.; Koronyo, Y.; Koronyo-Hamaoui, M. Optical Coherence Tomography in Alzheimer’s Disease and Other Neurodegenerative Diseases. Front. Neurol. 2017, 8, 701. [Google Scholar] [CrossRef]
- Ferrari, L.; Huang, S.C.; Magnani, G.; Ambrosi, A.; Comi, G.; Leocani, L. Optical Coherence Tomography Reveals Retinal Neuroaxonal Thinning in Frontotemporal Dementia as in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 56, 1101–1107. [Google Scholar] [CrossRef]
- Polo, V.; Rodrigo, M.J.; Garcia-Martin, E.; Otin, S.; Larrosa, J.M.; Fuertes, M.I.; Bambo, M.P.; Pablo, L.E.; Satue, M. Visual dysfunction and its correlation with retinal changes in patients with Alzheimer’s disease. Eye 2017, 31, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Bulut, M.; Yaman, A.; Erol, M.K.; Kurtulus, F.; Toslak, D.; Dogan, B.; Turgut Coban, D.; Kaya Basar, E. Choroidal Thickness in Patients with Mild Cognitive Impairment and Alzheimer’s Type Dementia. J. Ophthalmol. 2016, 2016, 7291257. [Google Scholar] [CrossRef] [PubMed]
- Janez-Escalada, L.; Janez-Garcia, L.; Salobrar-Garcia, E.; Santos-Mayo, A.; de Hoz, R.; Yubero, R.; Gil, P.; Ramirez, J.M. Spatial analysis of thickness changes in ten retinal layers of Alzheimer’s disease patients based on optical coherence tomography. Sci. Rep. 2019, 9, 13000. [Google Scholar] [CrossRef] [PubMed]
- Czako, C.; Kovacs, T.; Ungvari, Z.; Csiszar, A.; Yabluchanskiy, A.; Conley, S.; Csipo, T.; Lipecz, A.; Horvath, H.; Sandor, G.L.; et al. Retinal biomarkers for Alzheimer’s disease and vascular cognitive impairment and dementia (VCID): Implication for early diagnosis and prognosis. Geroscience 2020, 42, 1499–1525. [Google Scholar] [CrossRef] [PubMed]
- Dumitrascu, O.M.; Koronyo-Hamaoui, M. Retinal vessel changes in cerebrovascular disease. Curr. Opin. Neurol. 2020, 33, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Mejia-Vergara, A.J.; Karanjia, R.; Sadun, A.A. OCT parameters of the optic nerve head and the retina as surrogate markers of brain volume in a normal population, a pilot study. J. Neurol. Sci. 2021, 420, 117213. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, D.; Votruba, M. Can the retina be used to diagnose and plot the progression of Alzheimer’s disease? Acta Ophthalmol. 2017, 95, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Chan, V.T.T.; Sun, Z.; Tang, S.; Chen, L.J.; Wong, A.; Tham, C.C.; Wong, T.Y.; Chen, C.; Ikram, M.K.; Whitson, H.E.; et al. Spectral-Domain OCT Measurements in Alzheimer’s Disease: A Systematic Review and Meta-analysis. Ophthalmology 2019, 126, 497–510. [Google Scholar] [CrossRef] [PubMed]
- den Haan, J.; Verbraak, F.D.; Visser, P.J.; Bouwman, F.H. Retinal thickness in Alzheimer’s disease: A systematic review and meta-analysis. Alzheimers Dement. 2017, 6, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Hinton, D.R.; Sadun, A.A.; Blanks, J.C.; Miller, C.A. Optic-nerve degeneration in Alzheimer’s disease. N. Engl. J. Med. 1986, 315, 485–487. [Google Scholar] [CrossRef]
- Blanks, J.C.; Schmidt, S.Y.; Torigoe, Y.; Porrello, K.V.; Hinton, D.R.; Blanks, R.H. Retinal pathology in Alzheimer’s disease. II. Regional neuron loss and glial changes in GCL. Neurobiol. Aging 1996, 17, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Blanks, J.C.; Hinton, D.R.; Sadun, A.A.; Miller, C.A. Retinal ganglion cell degeneration in Alzheimer’s disease. Brain Res. 1989, 501, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Koronyo, Y.; Biggs, D.; Barron, E.; Boyer, D.S.; Pearlman, J.A.; Au, W.J.; Kile, S.J.; Blanco, A.; Fuchs, D.T.; Ashfaq, A.; et al. Retinal amyloid pathology and proof-of-concept imaging trial in Alzheimer’s disease. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Sadun, A.A.; Bassi, C.J. Optic nerve damage in Alzheimer’s disease. Ophthalmology 1990, 97, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.A.; Syed, A.B. Alzheimer’s disease and the eye. Ophthalmic Physiol. Opt. 1996, 16 (Suppl. S1), S2–S8. [Google Scholar] [CrossRef] [PubMed]
- Risacher, S.L.; WuDunn, D.; Tallman, E.F.; West, J.D.; Gao, S.; Farlow, M.R.; Brosch, J.R.; Apostolova, L.G.; Saykin, A.J. Visual contrast sensitivity is associated with the presence of cerebral amyloid and tau deposition. Brain Commun. 2020, 2, fcaa019. [Google Scholar] [CrossRef] [PubMed]
- Trick, G.L.; Barris, M.C.; Bickler-Bluth, M. Abnormal pattern electroretinograms in patients with senile dementia of the Alzheimer type. Ann. Neurol. 1989, 26, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Pilorz, V.; Tam, S.K.; Hughes, S.; Pothecary, C.A.; Jagannath, A.; Hankins, M.W.; Bannerman, D.M.; Lightman, S.L.; Vyazovskiy, V.V.; Nolan, P.M.; et al. Melanopsin Regulates Both Sleep-Promoting and Arousal-Promoting Responses to Light. PLoS Biol. 2016, 14, e1002482. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Holtzman, D.M. Bidirectional relationship between sleep and Alzheimer’s disease: Role of amyloid, tau, and other factors. Neuropsychopharmacology 2020, 45, 104–120. [Google Scholar] [CrossRef] [PubMed]
- Ciulla, L.; Moorthy, M.; Mathew, S.; Siesky, B.; Verticchio Vercellin, A.C.; Price, D.; Januleviciene, I.; Harris, A. Circadian Rhythm and Glaucoma: What do We Know? J. Glaucoma 2020, 29, 127–132. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, Y.; Ding, J.; Wang, N. Changes in the circadian rhythm in patients with primary glaucoma. PLoS ONE 2013, 8, e62841. [Google Scholar] [CrossRef] [PubMed]
- Drouyer, E.; Dkhissi-Benyahya, O.; Chiquet, C.; WoldeMussie, E.; Ruiz, G.; Wheeler, L.A.; Denis, P.; Cooper, H.M. Glaucoma alters the circadian timing system. PLoS ONE 2008, 3, e3931. [Google Scholar] [CrossRef] [PubMed]
- Obara, E.A.; Hannibal, J.; Heegaard, S.; Fahrenkrug, J. Loss of Melanopsin-Expressing Retinal Ganglion Cells in Severely Staged Glaucoma Patients. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4661–4667. [Google Scholar] [CrossRef] [PubMed]
- Lanzani, M.F.; de Zavalia, N.; Fontana, H.; Sarmiento, M.I.; Golombek, D.; Rosenstein, R.E. Alterations of locomotor activity rhythm and sleep parameters in patients with advanced glaucoma. Chronobiol. Int. 2012, 29, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.S.; Ritch, R.; Schwartz, B.; Lee, S.S.; Miller, N.R.; Chi, T.; Hsieh, F.Y. Optic nerve head and nerve fiber layer in Alzheimer’s disease. Arch. Ophthalmol. 1991, 109, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.R.; Xu, L.; Yang, Y. A comparative study of optic nerve damage between primary open angle glaucoma and normal tension glaucoma. Zhonghua Yan Ke Za Zhi 2005, 41, 136–140. [Google Scholar] [PubMed]
- Frezzotti, P.; Giorgio, A.; Toto, F.; De Leucio, A.; De Stefano, N. Early changes of brain connectivity in primary open angle glaucoma. Hum. Brain Mapp. 2016, 37, 4581–4596. [Google Scholar] [CrossRef]
- Jiang, M.M.; Zhou, Q.; Liu, X.Y.; Shi, C.Z.; Chen, J.; Huang, X.H. Structural and functional brain changes in early- and mid-stage primary open-angle glaucoma using voxel-based morphometry and functional magnetic resonance imaging. Medicine 2017, 96, e6139. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Morelli, J.N.; Ai, F.; Yin, D.; Hu, C.; Xu, D.; Li, Y. Resting-state functional MRI: Functional connectivity analysis of the visual cortex in primary open-angle glaucoma patients. Hum. Brain Mapp. 2013, 34, 2455–2463. [Google Scholar] [CrossRef]
- Wang, J.; Li, T.; Wang, N.; Xian, J.; He, H. Graph theoretical analysis reveals the reorganization of the brain network pattern in primary open angle glaucoma patients. Eur. Radiol. 2016, 26, 3957–3967. [Google Scholar] [CrossRef]
- Song, S.K.; Sun, S.W.; Ramsbottom, M.J.; Chang, C.; Russell, J.; Cross, A.H. Dysmyelination revealed through MRI as increased radial (but unchanged axial) diffusion of water. Neuroimage 2002, 17, 1429–1436. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.J.; Fisman, M.; Hachinski, V.; Blume, W.; Fox, A.; Kral, V.A.; Kirshen, A.J.; Fox, H.; Merskey, H. A new definition of Alzheimer’s disease: A hippocampal dementia. Lancet 1985, 1, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Price, J.L.; Morris, J.C. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann. Neurol. 1999, 45, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Mintun, M.A.; Larossa, G.N.; Sheline, Y.I.; Dence, C.S.; Lee, S.Y.; Mach, R.H.; Klunk, W.E.; Mathis, C.A.; DeKosky, S.T.; Morris, J.C. [11C]PIB in a nondemented population: Potential antecedent marker of Alzheimer disease. Neurology 2006, 67, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Frisoni, G.B.; Fox, N.C.; Jack, C.R., Jr.; Scheltens, P.; Thompson, P.M. The clinical use of structural MRI in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Leow, A.D.; Parikshak, N.; Lee, S.; Chiang, M.C.; Toga, A.W.; Jack, C.R., Jr.; Weiner, M.W.; Thompson, P.M.; Alzheimer’s Disease Neuroimaging, I. Tensor-based morphometry as a neuroimaging biomarker for Alzheimer’s disease: An MRI study of 676 AD, MCI, and normal subjects. Neuroimage 2008, 43, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Bartzokis, G. Alzheimer’s disease as homeostatic responses to age-related myelin breakdown. Neurobiol. Aging 2011, 32, 1341–1371. [Google Scholar] [CrossRef]
- Braskie, M.N.; Jahanshad, N.; Stein, J.L.; Barysheva, M.; Johnson, K.; McMahon, K.L.; de Zubicaray, G.I.; Martin, N.G.; Wright, M.J.; Ringman, J.M.; et al. Relationship of a variant in the NTRK1 gene to white matter microstructure in young adults. J. Neurosci. 2012, 32, 5964–5972. [Google Scholar] [CrossRef] [PubMed]
- Karas, G.B.; Scheltens, P.; Rombouts, S.A.; Visser, P.J.; van Schijndel, R.A.; Fox, N.C.; Barkhof, F. Global and local gray matter loss in mild cognitive impairment and Alzheimer’s disease. Neuroimage 2004, 23, 708–716. [Google Scholar] [CrossRef]
- Grady, C.L.; Furey, M.L.; Pietrini, P.; Horwitz, B.; Rapoport, S.I. Altered brain functional connectivity and impaired short-term memory in Alzheimer’s disease. Brain 2001, 124, 739–756. [Google Scholar] [CrossRef]
- Bozzali, M.; Falini, A.; Franceschi, M.; Cercignani, M.; Zuffi, M.; Scotti, G.; Comi, G.; Filippi, M. White matter damage in Alzheimer’s disease assessed in vivo using diffusion tensor magnetic resonance imaging. J. Neurol. Neurosurg. Psychiatry 2002, 72, 742–746. [Google Scholar] [CrossRef]
- Greicius, M.D.; Srivastava, G.; Reiss, A.L.; Menon, V. Default-mode network activity distinguishes Alzheimer’s disease from healthy aging: Evidence from functional MRI. Proc. Natl. Acad. Sci. USA 2004, 101, 4637–4642. [Google Scholar] [CrossRef] [PubMed]
- Naggara, O.; Oppenheim, C.; Rieu, D.; Raoux, N.; Rodrigo, S.; Dalla Barba, G.; Meder, J.F. Diffusion tensor imaging in early Alzheimer’s disease. Psychiatry Res. 2006, 146, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Stam, C.J.; Jones, B.F.; Nolte, G.; Breakspear, M.; Scheltens, P. Small-world networks and functional connectivity in Alzheimer’s disease. Cereb. Cortex 2007, 17, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Lowe, M.J.; Dzemidzic, M.; Lurito, J.T.; Mathews, V.P.; Phillips, M.D. Correlations in low-frequency BOLD fluctuations reflect cortico-cortical connections. NeuroImage 2000, 12, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Cordes, D.; Haughton, V.M.; Arfanakis, K.; Wendt, G.J.; Turski, P.A.; Moritz, C.H.; Quigley, M.A.; Meyerand, M.E. Mapping functionally related regions of brain with functional connectivity MR imaging. AJNR Am. J. Neuroradiol. 2000, 21, 1636–1644. [Google Scholar] [PubMed]
- Golden, H.L.; Agustus, J.L.; Nicholas, J.M.; Schott, J.M.; Crutch, S.J.; Mancini, L.; Warren, J.D. Functional neuroanatomy of spatial sound processing in Alzheimer’s disease. Neurobiol. Aging 2016, 39, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.A.; Akrofi, K.; Carpenter-Thompson, J.R.; Husain, F.T. Default mode, dorsal attention and auditory resting state networks exhibit differential functional connectivity in tinnitus and hearing loss. PLoS ONE 2013, 8, e76488. [Google Scholar] [CrossRef]
- Zhao, J.; Du, Y.H.; Ding, X.T.; Wang, X.H.; Men, G.Z. Alteration of functional connectivity in patients with Alzheimer’s disease revealed by resting-state functional magnetic resonance imaging. Neural Regen. Res. 2020, 15, 285–292. [Google Scholar] [CrossRef]
Figure 1.
Schematic depicting the connections between Alzheimer’s Disease and normal-tension glaucoma. APOE = Apolipoprotein e4 allele, TBK1 = TANK Binding Kinase 1, OPTN = Optineurin, RNFL = retinal nerve fiber layer, CSF = cerebrospinal fluid, and TLPG = translaminar pressure gradient. Created with BioRender.com.
Figure 1.
Schematic depicting the connections between Alzheimer’s Disease and normal-tension glaucoma. APOE = Apolipoprotein e4 allele, TBK1 = TANK Binding Kinase 1, OPTN = Optineurin, RNFL = retinal nerve fiber layer, CSF = cerebrospinal fluid, and TLPG = translaminar pressure gradient. Created with BioRender.com.

Figure 2.
Altered CSF fluid dynamics and bio-active molecules in NTG and AD pathology. L-PGDS = lipocalin-type prostaglandin D synthases, IOP = intraocular pressure, ICP = intracranial pressure, CSF = cerebrospinal fluid, NTG = normal-tension glaucoma, AD = Alzheimer’s Disease, and Aβ = Amyloid β. Created with BioRender.com.
Figure 2.
Altered CSF fluid dynamics and bio-active molecules in NTG and AD pathology. L-PGDS = lipocalin-type prostaglandin D synthases, IOP = intraocular pressure, ICP = intracranial pressure, CSF = cerebrospinal fluid, NTG = normal-tension glaucoma, AD = Alzheimer’s Disease, and Aβ = Amyloid β. Created with BioRender.com.

Figure 3.
Ocular and cerebral manifestations in NTG and AD. CSF = cerebrospinal fluid and Aβ = Amyloid β. Created with BioRender.com.
Figure 3.
Ocular and cerebral manifestations in NTG and AD. CSF = cerebrospinal fluid and Aβ = Amyloid β. Created with BioRender.com.

Figure 4.
Mechanistic pathogenic linkage between Alzheimer’s and normal-tension glaucoma. CSF = cerebrospinal fluid. Created with BioRender.com.
Figure 4.
Mechanistic pathogenic linkage between Alzheimer’s and normal-tension glaucoma. CSF = cerebrospinal fluid. Created with BioRender.com.
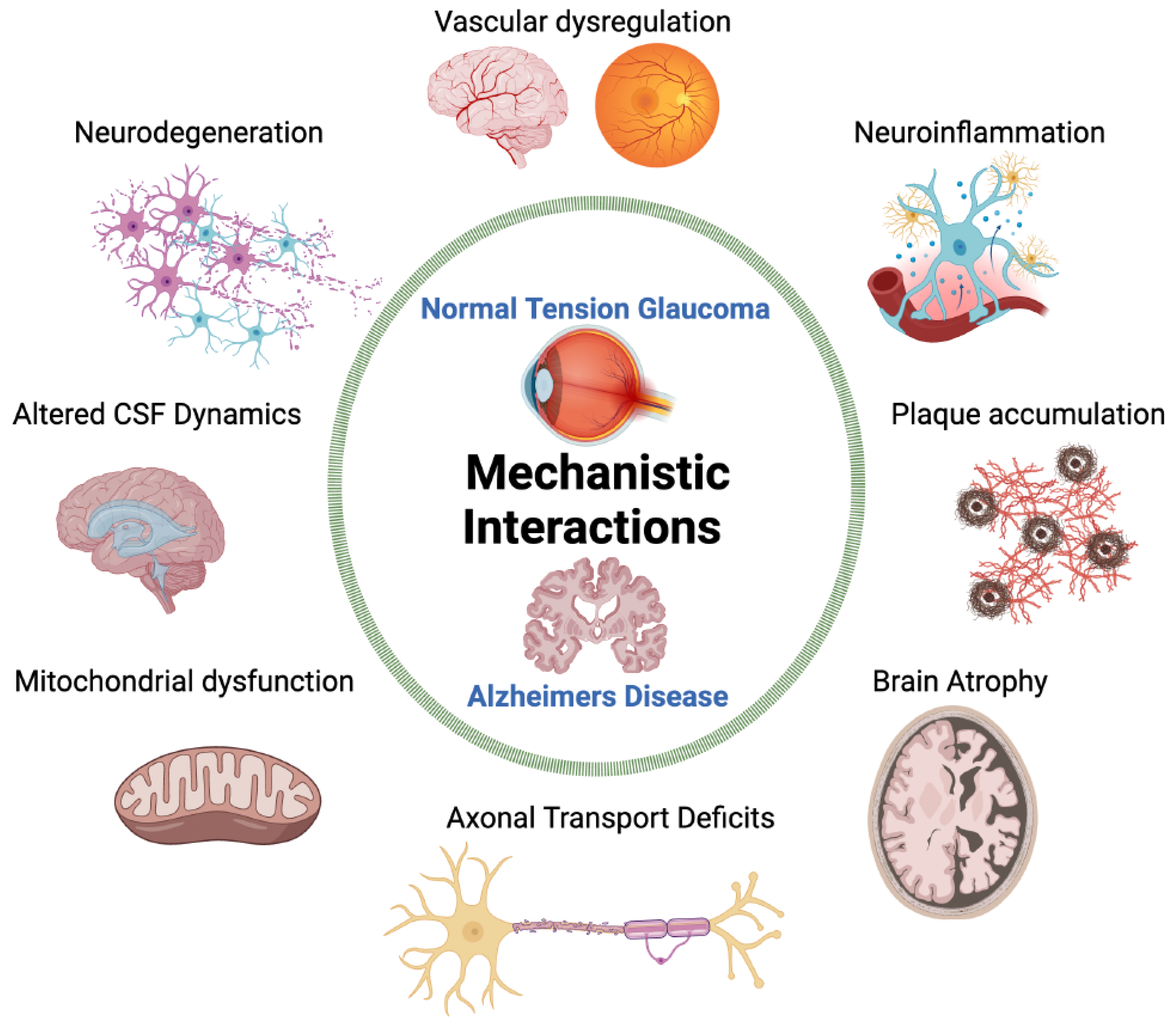
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ho, K.; Bodi, N.E.; Sharma, T.P. Normal-Tension Glaucoma and Potential Clinical Links to Alzheimer’s Disease. J. Clin. Med. 2024, 13, 1948. https://doi.org/10.3390/jcm13071948
AMA Style
Ho K, Bodi NE, Sharma TP. Normal-Tension Glaucoma and Potential Clinical Links to Alzheimer’s Disease. Journal of Clinical Medicine. 2024; 13(7):1948. https://doi.org/10.3390/jcm13071948
Chicago/Turabian StyleHo, Kathleen, Nicole E. Bodi, and Tasneem P. Sharma. 2024. "Normal-Tension Glaucoma and Potential Clinical Links to Alzheimer’s Disease" Journal of Clinical Medicine 13, no. 7: 1948. https://doi.org/10.3390/jcm13071948
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.