
Endothelial Function in Pulmonary Arterial Hypertension: From Bench to Bedside
 , , , ,
, , , ,
Abstract
:1. Introduction
2. What Is Endothelial Function and What Regulates It?
3. Why Is Endothelial Function Relevant in PAH?
Genetics
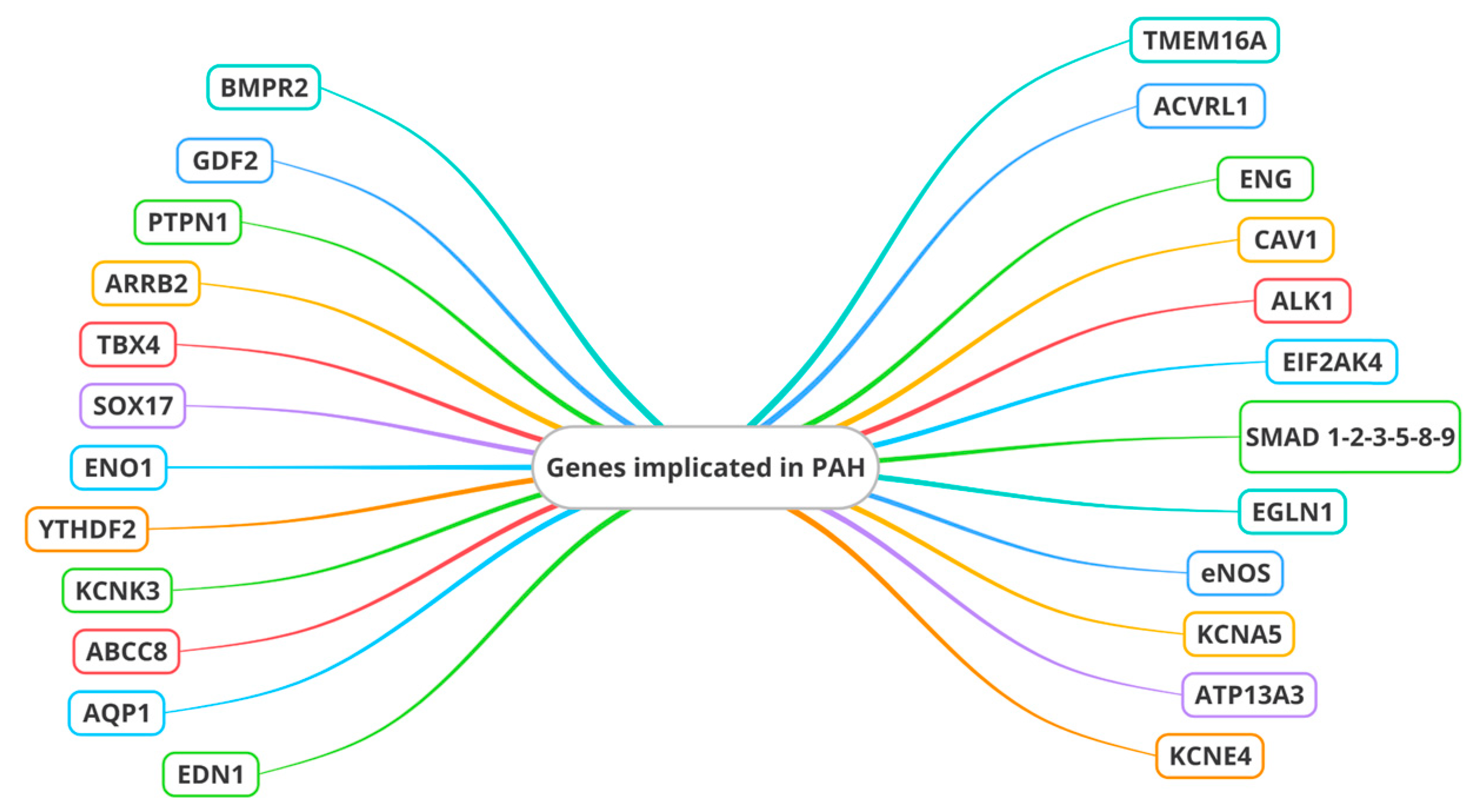
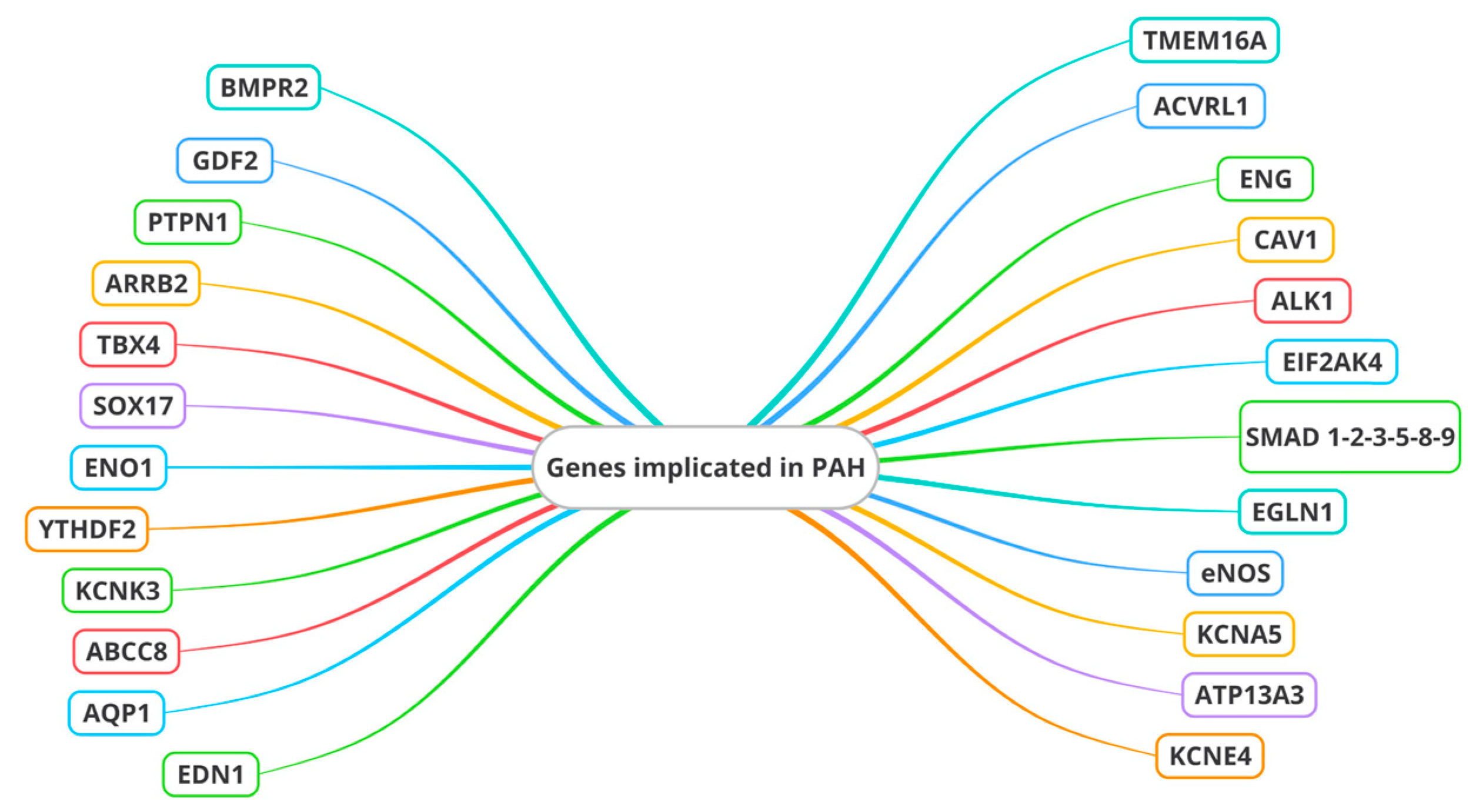
4. Genes in PAH
- Alpha-enolase 1 (ENO1), which influences genes associated with mitochondria and the PI3K-Akt signaling pathway [64];
- YTH N6-methyladenosine RNA binding protein 2 (YTHDF2), which increased pulmonary vascular resistance, right ventricular hypertrophy, macrophage polarization, and oxidative stress [65].
- Endothelial nitric oxide synthase (eNOS), crucial for NO synthesis. Impaired eNOS activity is a hallmark of ED in PAH, leading to reduced NO bioavailability and enhanced vasoconstriction. Genetic variations affecting eNOS function have an important impact on endothelial homeostasis and the development of PAH [72].
- Channel genes. Mutations in the Potassium Two Pore Domain Channel Subfamily K Member 3 (KCNK3) gene, encoding an outward-rectifying potassium channel, are related to ED and, thus, to pulmonary vascular remodeling. Another member of the potassium channel family, namely potassium voltage-gated channel subfamily A member 5 (KCNA5), plays a role in the ED of PH due to its regulatory function in pulmonary vascular tone, cell proliferation, apoptosis, and oxygen sensitivity [73,74]. Two other channel genes are involved: ATP-binding cassette subfamily member 8 (ABCC8) and ATPase 13A3 (ATP13A3). Another channel gene, aquaporin 1 (AQP1), was reported in one study conducted by Stefan Graf et al. [41]. ABCC8 is expressed in PAECs and PASMCs [75,76], influencing vasoreactivity and cell proliferation, thereby impacting pulmonary vascular tone and remodeling. KCNE4 (Potassium Voltage-Gated Channel Subfamily E Regulatory Subunit 4) also seems to be involved in vascular tone regulation in pulmonary arteries [77]. In the pathogenesis of PAH, Cl-activated Ca2+ channels (CaCCs) also appear to be implicated. It has been demonstrated that the most significant member of the CaCCs, Transmembrane Protein 16A (TMEM16A), contributes to the pathogenesis of IPAH in PASMCs. It was proposed that the extracellular signal-regulated kinase ½ (ERK1/2) pathway is specifically influenced by elevated TMEM16A activity, leading to functional consequences such as reduced NO production, alterations in Ca2+ dynamics and eNOS activity, the proliferation of PAECs, wound healing, tube formation, and the acetylcholine-mediated relaxation of human pulmonary arteries [78].
- ET-1 (endothelin 1) is a potent vasoconstrictor. Genetic variations in the EDN1 gene contribute to ED by promoting excessive vasoconstriction and smooth muscle cell proliferation. So, EDN1 has an important impact on the delicate balance of endothelial homeostasis [79].
- Eukaryotic translation initiation factor 2-alpha kinase 4 (EIF2AK4). This gene is involved in the regulation of protein synthesis in eukaryotic cells. Its specific functions can vary depending on the cellular context and environmental signals. EIF2AK4 plays an important role in the cellular stress response, particularly in endoplasmic reticulum (ER) stress. When cells undergo stress, such as hypoxia or the accumulation of misfolded proteins in the endoplasmic reticulum, EIF2AK4 can be activated. Once activated, it phosphorylates a subunit of the protein translation initiator, called eIF2α. This phosphorylation prevents the initiation of global protein translation, allowing cells to adapt to stress by activating survival or repair programs [80,81] (Table 1).
5. Aspects of Vascular Biology
How to Evaluate Peripheral Endothelial Function in Patients with Pulmonary Hypertension
6. Endothelial Function and Pulmonary Therapy
7. Conclusions
Funding
Conflicts of Interest
References
- Vonk-Noordegraaf, A.; Haddad, F.; Chin, K.M.; Forfia, P.R.; Kawut, S.M.; Lumens, J.; Naeije, R.; Newman, J.; Oudiz, R.J.; Provencher, S.; et al. Right heart adaptation to pulmonary arterial hypertension: Physiology and pathobiology. J. Am. Coll. Cardiol. 2013, 62 (Suppl. 25), D22–D33. [Google Scholar] [CrossRef] [PubMed]
- Stacher, E.; Graham, B.B.; Hunt, J.M.; Gandjeva, A.; Groshong, S.D.; McLaughlin, V.V.; Jessup, M.; Grizzle, W.E.; Aldred, M.A.; Cool, C.D.; et al. Modern age pathology of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 261–272. [Google Scholar] [CrossRef]
- Zolty, R. Novel Experimental Therapies for Treatment of Pulmonary Arterial Hypertension. J. Exp. Pharmacol. 2021, 13, 817–857. [Google Scholar] [CrossRef]
- Hassoun, P.M. Pulmonary Arterial Hypertension. N. Engl. J. Med. 2021, 385, 2361–2376. [Google Scholar] [CrossRef] [PubMed]
- Gorelova, A.; Berman, M.; Al Ghouleh, I. Endothelial-to-Mesenchymal Transition in Pulmonary Arterial Hypertension. Antioxid. Redox Signal. 2021, 34, 891–914. [Google Scholar] [CrossRef] [PubMed]
- Haensel, M.; Wojciak-Stothard, B. The role of endothelial cells in pulmonary hypertension: Old concepts and new science. Curr. Opin. Physiol. 2023, 34, 100667. [Google Scholar] [CrossRef]
- Amraoui, F.; Olde Engberink, R.H.; van Gorp, J.; Ramdani, A.; Vogt, L.; van den Born, B.J. Microvascular glycocalyx dimension estimated by automated SDF imaging is not related to cardiovascular disease. Microcirculation 2014, 21, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Yang, Z.C.; Liu, Y. Attenuating Pulmonary Hypertension by Protecting the Integrity of Glycocalyx in Rats Model of Pulmonary Artery Hypertension. Inflammation 2019, 42, 1951–1956. [Google Scholar] [CrossRef]
- Sumpio, B.E.; Riley, J.T.; Dardik, A. Cells in focus: Endothelial cell. Int. J. Biochem. Cell Biol. 2002, 34, 1508–1512. [Google Scholar] [CrossRef]
- Godo, S.; Shimokawa, H. Divergent roles of endothelial nitric oxide synthases system in maintaining cardiovascular homeostasis. Free Radic. Biol. Med. 2017, 109, 4–10. [Google Scholar] [CrossRef]
- Shimokawa, H.; Satoh, K. Vascular function. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2359–2362. [Google Scholar] [CrossRef]
- Zhou, J.; Li, Y.S.; Chien, S. Shear stress-initiated signaling and its regulation of endothelial function. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2191–2198. [Google Scholar] [CrossRef]
- Godo, S.; Shimokawa, H. Endothelial Functions. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e108–e114. [Google Scholar] [CrossRef]
- Bonetti, P.O.; Lerman, L.O.; Lerman, A. Endothelial dysfunction: A marker of atherosclerotic risk. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 168–175. [Google Scholar] [CrossRef]
- Evans, C.E.; Cober, N.D.; Dai, Z.; Stewart, D.J.; Zhao, Y.Y. Endothelial cells in the pathogenesis of pulmonary arterial hypertension. Eur. Respir. J. 2021, 58, 2003957. [Google Scholar] [CrossRef]
- Humbert, M.; Morrell, N.W.; Archer, S.L.; Stenmark, K.R.; MacLean, M.R.; Lang, I.M.; Christman, B.W.; Weir, E.K.; Eickelberg, O.; Voelkel, N.F.; et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, 13S–24S. [Google Scholar] [CrossRef]
- Tuder, R.M.; Groves, B.; Badesch, D.B.; Voelkel, N.F. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am. J. Pathol. 1994, 144, 275–285. [Google Scholar]
- Ranchoux, B.; Harvey, L.D.; Ayon, R.J.; Babicheva, A.; Bonnet, S.; Chan, S.Y.; Yuan, J.X.; Perez, V.J. Endothelial dysfunction in pulmonary arterial hypertension: An evolving landscape (2017 Grover Conference Series). Pulm. Circ. 2018, 8, 2045893217752912. [Google Scholar] [CrossRef]
- Cober, N.D.; VandenBroek, M.M.; Ormiston, M.L.; Stewart, D.J. Evolving Concepts in Endothelial Pathobiology of Pulmonary Arterial Hypertension. Hypertension 2022, 79, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.J.; Levy, R.D.; Cernacek, P.; Langleben, D. Increased plasma endothelin-1 in pulmonary hypertension: Marker or mediator of disease? Ann. Intern. Med. 1991, 114, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Christman, B.W.; McPherson, C.D.; Newman, J.H.; King, G.A.; Bernard, G.R.; Groves, B.M.; Loyd, J.E. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N. Engl. J. Med. 1992, 327, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.; Dewachter, L.; Gore, B.; Fadel, E.; Dartevelle, P.; Simonneau, G.; Humbert, M.; Eddahibi, S.; Guignabert, C. Autocrine fibroblast growth factor-2 signaling contributes to altered endothelial phenotype in pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 2011, 45, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Zhu, M.M.; Peng, Y.; Jin, H.; Machireddy, N.; Qian, Z.; Zhang, X.; Zhao, Y.Y. Endothelial and Smooth Muscle Cell Interaction via FoxM1 Signaling Mediates Vascular Remodeling and Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 788–802. [Google Scholar] [CrossRef] [PubMed]
- Budhiraja, R.; Tuder, R.M.; Hassoun, P.M. Endothelial dysfunction in pulmonary hypertension. Circulation 2004, 109, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, M.R. Pulmonary hypertension: The science behind the disease spectrum. Eur. Respir. Rev. 2012, 21, 19–26. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hanna, V.S.; Hafez, E.A.A. Synopsis of arachidonic acid metabolism: A review. J. Adv. Res. 2018, 11, 23–32. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hinton, M.; Gutsol, A.; Dakshinamurti, S. Thromboxane hypersensitivity in hypoxic pulmonary artery myocytes: Altered TP receptor localization and kinetics. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L654–L663. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Cool, C.D.; Geraci, M.W.; Wang, J.; Abman, S.H.; Wright, L.; Badesch, D.; Voelkel, N.F. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1999, 159, 1925–1932. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.A.; Loscalzo, J. Pulmonary hypertension: Pathophysiology and signaling pathways. Handb. Exp. Pharmacol. 2013, 218, 31–58. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaïci, A.; Weitzenblum, E.; Cordier, J.F.; Chabot, F.; et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 2010, 122, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Chung, W.K. The role of genetics in pulmonary arterial hypertension. J. Pathol. 2017, 241, 273–280. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Good, R.B.; Gilbane, A.J.; Trinder, S.L.; Denton, C.P.; Coghlan, G.; Abraham, D.J.; Holmes, A.M. Endothelial to Mesenchymal Transition Contributes to Endothelial Dysfunction in Pulmonary Arterial Hypertension. Am. J. Pathol. 2015, 185, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Aschner, Y.; Downey, G.P. Transforming Growth Factor-β: Master Regulator of the Respiratory System in Health and Disease. Am. J. Respir. Cell Mol. Biol. 2016, 54, 647–655. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, F.; Ventura, F.; Doody, J.; Massagué, J. Human type II receptor for bone morphogenic proteins (BMPs): Extension of the two-kinase receptor model to the BMPs. Mol. Cell. Biol. 1995, 15, 3479–3486. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Zwijsen, A.; Ten Dijke, P.; Bailly, S. Bone Morphogenetic Proteins in Vascular Homeostasis and Disease. Cold Spring Harb. Perspect. Biol. 2018, 10, a031989. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Duffhues, G.; Williams, E.; Goumans, M.J.; Heldin, C.H.; Ten Dijke, P. Bone morphogenetic protein receptors: Structure, function and targeting by selective small molecule kinase inhibitors. Bone 2020, 138, 115472. [Google Scholar] [CrossRef]
- Kurakula, K.; Goumans, M.J.; Ten Dijke, P. Regulatory RNAs controlling vascular (dys)function by affecting TGF-ß family signalling. EXCLI J. 2015, 14, 832–850. [Google Scholar]
- Yang, X.; Long, L.; Southwood, M.; Rudarakanchana, N.; Upton, P.D.; Jeffery, T.K.; Atkinson, C.; Chen, H.; Trembath, R.C.; Morrell, N.W. Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ. Res. 2005, 96, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Teichert-Kuliszewska, K.; Kutryk, M.J.B.; Kuliszewski, M.A.; Karoubi, G.; Courtman, D.W.; Zucco, L.; Granton, J.; Stewart, D.J. Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: Implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ. Res. 2006, 98, 209–217. [Google Scholar] [CrossRef]
- Zhang, S.; Fantozzi, I.; Tigno, D.D.; Yi, E.S.; Platoshyn, O.; Thistlethwaite, P.A.; Kriett, J.M.; Yung, G.; Rubin, L.J.; Yuan, J.X. Bone morphogenetic proteins induce apoptosis in human pulmonary vascular smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L740–L754. [Google Scholar] [CrossRef]
- Gräf, S.; Haimel, M.; Bleda, M.; Hadinnapola, C.; Southgate, L.; Li, W.; Hodgson, J.; Liu, B.; Salmon, R.M.; Southwood, M.; et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat. Commun. 2018, 9, 1416. [Google Scholar] [CrossRef] [PubMed]
- Frump, A.; Prewitt, A.; de Caestecker, M.P. BMPR2 mutations and endothelial dysfunction in pulmonary arterial hypertension (2017 Grover Conference Series). Pulm. Circ. 2018, 8, 2045894018765840. [Google Scholar] [CrossRef]
- Soon, E.; Crosby, A.; Southwood, M.; Yang, P.; Tajsic, T.; Toshner, M.; Appleby, S.; Shanahan, C.M.; Bloch, K.D.; Pepke-Zaba, J.; et al. Bone morphogenetic protein receptor type II deficiency and increased inflammatory cytokine production: A gateway to pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wang, J.; Kinzel, B.; Müeller, M.; Mao, X.; Valdez, R.; Liu, Y.; Li, E. Dosage-dependent requirement of BMP type II receptor for maintenance of vascular integrity. Blood 2007, 110, 1502–1510. [Google Scholar] [CrossRef]
- Long, L.; MacLean, M.R.; Jeffery, T.K.; Morecroft, I.; Yang, X.; Rudarakanchana, N.; Southwood, M.; James, V.; Trembath, R.C.; Morrell, N.W. Serotonin increases susceptibility to pulmonary hypertension in BMPR2-deficient mice. Circ. Res. 2006, 98, 818–827. [Google Scholar] [CrossRef]
- Atkinson, C.; Stewart, S.; Upton, P.D.; Machado, R.; Thomson, J.R.; Trembath, R.C.; Morrell, N.W. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation 2002, 105, 1672–1678. [Google Scholar] [CrossRef]
- Brock, M.; Trenkmann, M.; Gay, R.E.; Michel, B.A.; Gay, S.; Fischler, M.; Ulrich, S.; Speich, R.; Huber, L.C. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ. Res. 2009, 104, 1184–1191. [Google Scholar] [CrossRef]
- Andruska, A.; Spiekerkoetter, E. Consequences of BMPR2 Deficiency in the Pulmonary Vasculature and Beyond: Contributions to Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2018, 19, 2499. [Google Scholar] [CrossRef] [PubMed]
- Happé, C.; Kurakula, K.; Sun, X.Q.; da Silva Goncalves Bos, D.; Rol, N.; Guignabert, C.; Tu, L.; Schalij, I.; Wiesmeijer, K.C.; Tura-Ceide, O.; et al. The BMP Receptor 2 in Pulmonary Arterial Hypertension: When and Where the Animal Model Matches the Patient. Cells 2020, 9, 1422. [Google Scholar] [CrossRef]
- Hong, K.H.; Lee, Y.J.; Lee, E.; Park, S.O.; Han, C.; Beppu, H.; Li, E.; Raizada, M.K.; Bloch, K.D.; Oh, S.P. Genetic ablation of the BMPR2 gene in pulmonary endothelium is sufficient to predispose to pulmonary arterial hypertension. Circulation 2008, 118, 722–730. [Google Scholar] [CrossRef]
- Majka, S.; Hagen, M.; Blackwell, T.; Harral, J.; Johnson, J.A.; Gendron, R.; Paradis, H.; Crona, D.; Loyd, J.E.; Nozik-Grayck, E.; et al. Physiologic and molecular consequences of endothelial Bmpr2 mutation. Respir. Res. 2011, 12, 84. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Ormiston, M.L.; Yang, X.; Southwood, M.; Gräf, S.; Machado, R.D.; Mueller, M.; Kinzel, B.; Yung, L.M.; Wilkinson, J.M.; et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat. Med. 2015, 21, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.K.; Tian, X.; Zhao, L.; Schimmel, K.; Rhodes, C.J.; Wilkins, M.R.; Nicolls, M.R.; Spiekerkoetter, E.F. PTPN1 Deficiency Modulates BMPR2 Signaling and Induces Endothelial Dysfunction in Pulmonary Arterial Hypertension. Cells 2023, 12, 316. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, L.; Moonen, J.R.; Cao, A.; Isobe, S.; Li, C.G.; Tojais, N.F.; Taylor, S.; Marciano, D.P.; Chen, P.I.; Gu, M.; et al. Dysregulated Smooth Muscle Cell BMPR2-ARRB2 Axis Causes Pulmonary Hypertension. Circ. Res. 2023, 132, 545–564. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Southgate, L.; Machado, R.D.; Gräf, S.; Morrell, N.W. Molecular genetic framework underlying pulmonary arterial hypertension. Nat. Rev. Cardiol. 2020, 17, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Katseff, A.; Alhawaj, R.; Wolin, M.S. Redox and Inflammatory Signaling, the Unfolded Protein Response, and the Pathogenesis of Pulmonary Hypertension. Adv. Exp. Med. Biol. 2021, 1304, 333–373. [Google Scholar] [CrossRef] [PubMed]
- Upton, P.D.; Davies, R.J.; Tajsic, T.; Morrell, N.W. Transforming growth factor-β(1) represses bone morphogenetic protein-mediated Smad signaling in pulmonary artery smooth muscle cells via Smad3. Am. J. Respir. Cell Mol. Biol. 2013, 49, 1135–1145. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Austin, E.D.; Ma, L.; LeDuc, C.; Berman Rosenzweig, E.; Borczuk, A.; Phillips, J.A., 3rd; Palomero, T.; Sumazin, P.; Kim, H.R.; Talati, M.H.; et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ. Cardiovasc. Genet. 2012, 5, 336–343. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wertz, J.W.; Bauer, P.M. Caveolin-1 regulates BMPRII localization and signaling in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2008, 375, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Hartung, A.; Bitton-Worms, K.; Rechtman, M.M.; Wenzel, V.; Boergermann, J.H.; Hassel, S.; Henis, Y.I.; Knaus, P. Different routes of bone morphogenic protein (BMP) receptor endocytosis influence BMP signaling. Mol. Cell Biol. 2006, 26, 7791–7805. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yoshida, Y.; Uchida, K.; Kodo, K.; Shibata, H.; Furutani, Y.; Nakayama, T.; Sakai, S.; Nakanishi, T.; Takahashi, T.; Yamagishi, H. Genetic and functional analyses of TBX4 reveal novel mechanisms underlying pulmonary arterial hypertension. J. Mol. Cell Cardiol. 2022, 171, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Sangam, S.; Sun, X.; Schwantes-An, T.H.; Yegambaram, M.; Lu, Q.; Shi, Y.; Cook, T.; Fisher, A.; Frump, A.L.; Coleman, A.; et al. Desai AA. SOX17 Deficiency Mediates Pulmonary Hypertension: At the Crossroads of Sex, Metabolism, and Genetics. Am. J. Respir. Crit. Care Med. 2023, 207, 1055–1069. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shi, Y.; Liu, J.; Zhang, R.; Zhang, M.; Cui, H.; Wang, L.; Cui, Y.; Wang, W.; Sun, Y.; Wang, C. Targeting Endothelial ENO1 (Alpha-Enolase)-PI3K-Akt-mTOR Axis Alleviates Hypoxic Pulmonary Hypertension. Hypertension 2023, 80, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Yu, Y.; Shen, Y.; Huang, H.; Lin, D.; Wang, K.; Yu, Y.; Li, K.; Cao, Y.; Wang, Q.; et al. Ythdf2 promotes pulmonary hypertension by suppressing Hmox1-dependent anti-inflammatory and antioxidant function in alveolar macrophages. Redox Biol. 2023, 61, 102638. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gong, H.; Rehman, J.; Tang, H.; Wary, K.; Mittal, M.; Chaturvedi, P.; Zhao, Y.Y.; Komarova, Y.A.; Vogel, S.M.; Malik, A.B. HIF2α signaling inhibits adherens junctional disruption in acute lung injury. J. Clin. Investig. 2015, 125, 652–664, Erratum in: J. Clin. Investig. 2015, 125, 1364. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dai, Z.; Li, M.; Wharton, J.; Zhu, M.M.; Zhao, Y.Y. Prolyl-4 Hydroxylase 2 (PHD2) Deficiency in Endothelial Cells and Hematopoietic Cells Induces Obliterative Vascular Remodeling and Severe Pulmonary Arterial Hypertension in Mice and Humans through Hypoxia-Inducible Factor-2α. Circulation 2016, 133, 2447–2458. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kapitsinou, P.P.; Rajendran, G.; Astleford, L.; Michael, M.; Schonfeld, M.P.; Fields, T.; Shay, S.; French, J.L.; West, J.; Haase, V.H. The Endothelial Prolyl-4-Hydroxylase Domain 2/Hypoxia-Inducible Factor 2 Axis Regulates Pulmonary Artery Pressure in Mice. Mol. Cell Biol. 2016, 36, 1584–1594. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, S.; Zeng, H.; Xie, X.J.; Tao, Y.K.; He, X.; Roman, R.J.; Aschner, J.L.; Chen, J.X. Loss of prolyl hydroxylase domain protein 2 in vascular endothelium increases pericyte coverage and promotes pulmonary arterial remodeling. Oncotarget 2016, 7, 58848–58861. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Kumar, S.M.; Martin, J.S.; Yang, R.; Xu, X. Snail1 mediates hypoxia-induced melanoma progression. Am. J. Pathol. 2011, 179, 3020–3031. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, X.; Tan, X.; Tampe, B.; Sanchez, E.; Zeisberg, M.; Zeisberg, E.M. Snail Is a Direct Target of Hypoxia-inducible Factor 1α (HIF1α) in Hypoxia-induced Endothelial to Mesenchymal Transition of Human Coronary Endothelial Cells. J. Biol. Chem. 2015, 290, 16653–16664. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tang, H.; Babicheva, A.; McDermott, K.M.; Gu, Y.; Ayon, R.J.; Song, S.; Wang, Z.; Gupta, A.; Zhou, T.; Sun, X.; et al. Endothelial HIF-2α contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L256–L275. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tran, N.; Garcia, T.; Aniqa, M.; Ali, S.; Ally, A.; Nauli, S.M. Endothelial Nitric Oxide Synthase (eNOS) and the Cardiovascular System: In Physiology and in Disease States. Am. J. Biomed. Sci. Res. 2022, 15, 153–177. [Google Scholar] [PubMed] [PubMed Central]
- Remillard, C.V.; Tigno, D.D.; Platoshyn, O.; Burg, E.D.; Brevnova, E.E.; Conger, D.; Nicholson, A.; Rana, B.K.; Channick, R.N.; Rubin, L.J.; et al. Function of Kv1.5 channels and genetic variations of KCNA5 in patients with idiopathic pulmonary arterial hypertension. Am. J. Physiol. Cell Physiol. 2007, 292, C1837–C1853. [Google Scholar] [CrossRef] [PubMed]
- Moudgil, R.; Michelakis, E.D.; Archer, S.L. The role of k+ channels in determining pulmonary vascular tone, oxygen sensing, cell proliferation, and apoptosis: Implications in hypoxic pulmonary vasoconstriction and pulmonary arterial hypertension. Microcirculation 2006, 13, 615–632. [Google Scholar] [CrossRef] [PubMed]
- Bohnen, M.S.; Ma, L.; Zhu, N.; Qi, H.; McClenaghan, C.; Gonzaga-Jauregui, C.; Dewey, F.E.; Overton, J.D.; Reid, J.G.; Shuldiner, A.R.; et al. Loss-of-Function ABCC8 Mutations in Pulmonary Arterial Hypertension. Circ. Genom. Precis. Med. 2018, 11, e002087. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Le Ribeuz, H.; Capuano, V.; Girerd, B.; Humbert, M.; Montani, D.; Antigny, F. Implication of Potassium Channels in the Pathophysiology of Pulmonary Arterial Hypertension. Biomolecules 2020, 10, 1261. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Belin de Chantemèle, E.J.; Muta, K.; Mintz, J.; Tremblay, M.L.; Marrero, M.B.; Fulton, D.J.; Stepp, D.W. Protein tyrosine phosphatase 1B, a major regulator of leptin-mediated control of cardiovascular function. Circulation 2009, 120, 753–763. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Skofic Maurer, D.; Zabini, D.; Nagaraj, C.; Sharma, N.; Lengyel, M.; Nagy, B.M.; Frank, S.; Klepetko, W.; Gschwandtner, E.; Enyedi, P.; et al. Endothelial Dysfunction Following Enhanced TMEM16A Activity in Human Pulmonary Arteries. Cells 2020, 9, 1984. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chester, A.H.; Yacoub, M.H. The role of endothelin-1 in pulmonary arterial hypertension. Glob. Cardiol. Sci. Pract. 2014, 2014, 62–78. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jonas, K.; Żuławińska, B.; Borys, A.; Wołkow, P.; Małecki, M.; Kopeć, G. A new mutation in the EIF2AK4 gene in familial pulmonary veno-occlusive disease. Pol. Arch. Intern. Med. 2023, 133, 16413. [Google Scholar] [CrossRef] [PubMed]
- Bignard, J.; Atassi, F.; Claude, O.; Ghigna, M.R.; Mougenot, N.; Abdoulkarim, B.S.; Deknuydt, F.; Gestin, A.; Monceau, V.; Montani, D.; et al. T-cell dysregulation and inflammatory process in Gcn2 (Eif2ak4-/-)-deficient rats in basal and stress conditions. Am. J. Physiol. Lung Cell Mol. Physiol. 2023, 324, L609–L624. [Google Scholar] [CrossRef] [PubMed]
- Eyries, M.; Montani, D.; Girerd, B.; Perret, C.; Leroy, A.; Lonjou, C.; Chelghoum, N.; Coulet, F.; Bonnet, D.; Dorfmuller, P.; et al. EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nat. Genet. 2014, 46, 65–69. [Google Scholar] [CrossRef]
- Galiè, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk, A.; Noordegraaf, A.V.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Respir. J. 2015, 46, 903–975. [Google Scholar] [PubMed]
- Tillet, E.; Bailly, S. Emerging roles of BMP9 and BMP10 in hereditary hemorrhagic telangiectasia. Front. Genet. 2015, 5, 456. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kurakula, K.; Smolders, V.F.E.D.; Tura-Ceide, O.; Jukema, J.W.; Quax, P.H.A.; Goumans, M.J. Endothelial Dysfunction in Pulmonary Hypertension: Cause or Consequence? Biomedicines 2021, 9, 57. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Christou, H.; Khalil, R.A. Mechanisms of pulmonary vascular dysfunction in pulmonary hypertension and implications for novel therapies. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H702–H724. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmüller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Galambos, C.; Sims-Lucas, S.; Abman, S.H.; Cool, C.D. Intrapulmonary Bronchopulmonary Anastomoses and Plexiform Lesions in Idiopathic Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2016, 193, 574–576. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wagenvoort, C.A.; Wagenvoort, N. Primary Pulmonary Hypertension A Pathologic Study of the Lung Vessels in 156 Clinically Diagnosed Cases. Circulation 1970, 42, 1163–1184. [Google Scholar] [CrossRef]
- Ghigna, M.R.; Guignabert, C.; Montani, D.; Girerd, B.; Jaïs, X.; Savale, L.; Hervé, P.; Thomas de Montpréville, V.; Mercier, O.; Sitbon, O.; et al. BMPR2 mutation status influences bronchial vascular changes in pulmonary arterial hypertension. Eur. Respir. J. 2016, 48, 1668–1681. [Google Scholar] [CrossRef] [PubMed]
- Pu, X.; Du, L.; Hu, Y.; Fan, Y.; Xu, Q. Stem/Progenitor Cells and Pulmonary Arterial Hypertension. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Ni, Z.; Tan, Y.Q.; Deng, J.; Zhang, S.J.; Lv, Z.C.; Wang, X.J.; Chen, T.; Zhang, Z.; Hu, Y.; et al. Adventitial cell atlas of wt (Wild Type) and ApoE (Apolipoprotein E)-deficient mice defined by single-cell RNA sequencing. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1055–1071. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.F.; Liang, H.Y.; Yuan, B.; Li, L.F.; Wang, T.; Kan, Q.C.; Wang, L.X.; Sun, T.W. Efficacy of stem cell therapy for pulmonary arterial hypertension: A systematic review and meta-analysis of preclinical studies. Stem Cell Res. Ther. 2019, 10, 55. [Google Scholar] [CrossRef]
- Xu, J.; Linneman, J.; Zhong, Y.; Yin, H.; Xia, Q.; Kang, K.; Gou, D. MicroRNAs in Pulmonary Hypertension, from Pathogenesis to Diagnosis and Treatment. Biomolecules 2022, 12, 496. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Parikh, V.N.; Jin, R.C.; Rabello, S.; Gulbahce, N.; White, K.; Hale, A.; Cottrill, K.A.; Shaik, R.S.; Waxman, A.B.; Zhang, Y.Y.; et al. MicroRNA-21 integrates pathogenic signaling to control pulmonary hypertension: Results of a network bioinformatics approach. Circulation 2012, 125, 1520–1532. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Galiè, N.; Manes, A.; Branzi, A. Prostanoids for pulmonary arterial hypertension. Am. J. Respir. Med. 2003, 2, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Sitbon, O.; Channick, R.; Chin, K.M.; Frey, A.; Gaine, S.; Galiè, N.; Ghofrani, H.A.; Hoeper, M.M.; Lang, I.M.; Preiss, R.; et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2015, 373, 2522–2533. [Google Scholar] [CrossRef] [PubMed]
- Correale, M.; Ferraretti, A.; Monaco, I.; Grazioli, D.; Di Biase, M.; Brunetti, N.D. Endothelin-receptor antagonists in the management of pulmonary arterial hypertension: Where do we stand? Vasc. Health Risk Manag. 2018, 14, 253–264. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tettey, A.; Jiang, Y.; Li, X.; Li, Y. Therapy for Pulmonary Arterial Hypertension: Glance on Nitric Oxide Pathway. Front. Pharmacol. 2021, 12, 767002. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nicolls, M.R.; Mizuno, S.; Taraseviciene-Stewart, L.; Farkas, L.; Drake, J.I.; Al Husseini, A.; Gomez-Arroyo, J.G.; Voelkel, N.F.; Bogaard, H.J. New models of pulmonary hypertension based on VEGF receptor blockade-induced endothelial cell apoptosis. Pulm. Circ. 2012, 2, 434–442. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Frump, A.L.; Bonnet, S.; de Jesus Perez, V.A.; Lahm, T. Emerging role of angiogenesis in adaptive and maladaptive right ventricular remodeling in pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L443–L460. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liang, S.; Yegambaram, M.; Wang, T.; Wang, J.; Black, S.M.; Tang, H. Mitochondrial Metabolism, Redox, and Calcium Homeostasis in Pulmonary Arterial Hypertension. Biomedicines 2022, 10, 341. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Culley, M.K.; Chan, S.Y. Mitochondrial metabolism in pulmonary hypertension: Beyond mountains there are mountains. J. Clin. Investig. 2018, 128, 3704–3715. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhou, G.; Chen, T.; Raj, J.U. MicroRNAs in pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 2015, 52, 139–151. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Thijssen, D.H.; Black, M.A.; Pyke, K.E.; Padilla, J.; Atkinson, G.; Harris, R.A.; Parker, B.; Widlansky, M.E.; Tschakovsky, M.E.; Green, D.J. Assessment of flow-mediated dilation in humans: A methodological and physiological guideline. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H2–H12. [Google Scholar] [CrossRef] [PubMed]
- Hudlická, O. Effect of training on macro- and microcirculatory changes in exercise. Exerc. Sport. Sci. Rev. 1977, 5, 181–230. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, I.B.; Webb, D.J. Venous occlusion plethysmography in cardiovascular research: Methodology and clinical applications. Br. J. Clin. Pharmacol. 2001, 52, 631–646. [Google Scholar] [CrossRef]
- Lind, L. Impact of ageing on the measurement of endothelium-dependent vasodilation. Pharmacol. Rep. 2006, 58, 41–46. [Google Scholar]
- Higashi, Y.; Yoshizumi, M. New methods to evaluate endothelial function: Method for assessing endothelial function in humans using a strain-gauge plethysmography: Nitric oxide-dependent and -independent vasodilation. J. Pharmacol. Sci. 2003, 93, 399–404. [Google Scholar] [CrossRef]
- Silva, A.M.; Schaan, B.D.; Signori, L.U.; Plentz, R.D.; Moreno, H., Jr.; Bertoluci, M.C.; Irigoyen, M.C. Microalbuminuria is associated with impaired arterial and venous endothelium-dependent vasodilation in patients with Type 2 diabetes. J. Endocrinol. Investig. 2010, 33, 696–700. [Google Scholar] [CrossRef]
- Aellig, W.H. A new technique for recording compliance of human hand veins. Br. J. Clin. Pharmacol. 1981, 11, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Leeson, C.P.; Robinson, M.; Francis, J.M.; Robson, M.D.; Channon, K.M.; Neubauer, S.; Wiesmann, F. Cardiovascular magnetic resonance imaging for non-invasive assessment of vascular function: Validation against ultrasound. J. Cardiovasc. Magn. Reson. 2006, 8, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Böhm, B.; Oberhoffer, R. Vascular health determinants in children. Cardiovasc. Diagn. Ther. 2019, 9 (Suppl. 2), S269–S280. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, E.A. Cell biology of endothelial cells. Hum. Pathol. 1987, 18, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Corretti, M.C.; Anderson, T.J.; Benjamin, E.J.; Celermajer, D.; Charbonneau, F.; Creager, M.A.; Deanfield, J.; Drexler, H.; Gerhard-Herman, M.; Herrington, D.; et al. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: A report of the International Brachial Artery Reactivity Task Force. J. Am. Coll. Cardiol. 2002, 39, 257–265, Erratum in: J. Am. Coll. Cardiol. 2002, 39, 1082. [Google Scholar] [CrossRef] [PubMed]
- Mućka, S.; Miodońska, M.; Jakubiak, G.K.; Starzak, M.; Cieślar, G.; Stanek, A. Endothelial Function Assessment by Flow-Mediated Dilation Method: A Valuable Tool in the Evaluation of the Cardiovascular System. Int. J. Environ. Res. Public. Health 2022, 19, 11242. [Google Scholar] [CrossRef] [PubMed]
- Gori, T.; Dragoni, S.; Lisi, M.; Di Stolfo, G.; Sonnati, S.; Fineschi, M.; Parker, J.D. Conduit artery constriction mediated by low flow a novel noninvasive method for the assessment of vascular function. J. Am. Coll. Cardiol. 2008, 51, 1953–1958. [Google Scholar] [CrossRef] [PubMed]
- Gori, T.; Grotti, S.; Dragoni, S.; Lisi, M.; Di Stolfo, G.; Sonnati, S.; Fineschi, M.; Parker, J.D. Assessment of vascular function: Flow-mediated constriction complements the information of flow-mediated dilatation. Heart 2010, 96, 141–147. [Google Scholar] [CrossRef]
- Kuvin, J.T.; Patel, A.R.; Sliney, K.A.; Pandian, N.G.; Sheffy, J.; Schnall, R.P.; Karas, R.H.; Udelson, J.E. Assessment of peripheral vascular endothelial function with finger arterial pulse wave amplitude. Am. Heart J. 2003, 146, 168–174. [Google Scholar] [CrossRef]
- Axtell, A.L.; Gomari, F.A.; Cooke, J.P. Assessing endothelial vasodilator function with the Endo-PAT 2000. J. Vis. Exp. 2010, 15, 2167. [Google Scholar] [CrossRef]
- Matsuzawa, Y.; Kwon, T.G.; Lennon, R.J.; Lerman, L.O.; Lerman, A. Prognostic Value of Flow-Mediated Vasodilation in Brachial Artery and Fingertip Artery for Cardiovascular Events: A Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2015, 4, e002270. [Google Scholar] [CrossRef]
- Lewis, T.; Grant, R. Observations upon reactive hyperaemia in man. Heart 1925, 12, 120. [Google Scholar]
- Whitney, R.J. The measurement of volume changes in human limbs. J. Physiol. 1953, 121, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, A.D.; Whitney, R.J.; Mowbray, J.F. Methods for the investigation of peripheral blood flow. Br. Med. Bull. 1963, 19, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Schraibman, I.G.; Mott, D.; Naylor, G.P.; Charlesworth, D. Impedance plethysmography: Evaluation of a simplified system of electrodes for the measurement of blood flow in the lower limb. Br. J. Surg. 1976, 63, 413–416. [Google Scholar] [CrossRef]
- Junejo, R.T.; Ray, C.J.; Marshall, J.M. Cuff inflation time significantly affects blood flow recorded with venous occlusion plethysmography. Eur. J. Appl. Physiol. 2019, 119, 665–674. [Google Scholar] [CrossRef]
- Gamble, J.; Gartside, I.B.; Christ, F. A reassessment of mercury in silastic strain gauge plethysmography for microvascular permeability assessment in man. J. Physiol. 1993, 464, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Roztocil, K.; Prerovský, I.; Studnicka, J.; Oliva, I.; Hálová, J. Capillary filtration during postischaemic hyperaemia in human limbs. Physiol. Bohemoslov. 1978, 27, 31–35. [Google Scholar]
- Ando, S.; Imaizumi, T.; Harada, S.; Hirooka, Y.; Takeshita, A. Atrial natriuretic peptide increases human capillary filtration and venous distensibility. J. Hypertens. 1992, 10, 451–457. [Google Scholar] [CrossRef]
- Jaap, A.J.; Shore, A.C.; Gartside, I.B.; Gamble, J.; Tooke, J.E. Increased microvascular fluid permeability in young type 1 (insulin-dependent) diabetic patients. Diabetologia 1993, 36, 648–652. [Google Scholar] [CrossRef]
- Mahy, I.R.; Lewis, D.M.; Tooke, J.E. Limb capillary filtration coefficient in human subjects: The importance of the site of measurement. Physiol. Meas. 1998, 19, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.; Bruegger, D.; Gamble, J.; Christ, F. Influence of different cuff inflation protocols on capillary filtration capacity in human calves—A congestion plethysmography study. J. Physiol. 2002, 543 Pt 3, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Hekman, C.M.; Loskutoff, D.J. Fibrinolytic pathways and the endothelium. Semin. Thromb. Hemost. 1987, 13, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Newby, D.E.; Wright, R.A.; Ludlam, C.A.; Fox, K.A.; Boon, N.A.; Webb, D.J. An in vivo model for the assessment of acute fibrinolytic capacity of the endothelium. Thromb. Haemost. 1997, 78, 1242–1248. [Google Scholar] [CrossRef] [PubMed]
- Hunter, A.L.; Shah, A.S.; Langrish, J.P.; Raftis, J.B.; Lucking, A.J.; Brittan, M.; Venkatasubramanian, S.; Stables, C.L.; Stelzle, D.; Marshall, J.; et al. Fire Simulation and Cardiovascular Health in Firefighters. Circulation 2017, 135, 1284–1295. [Google Scholar] [CrossRef] [PubMed]
- Noh, R.M.; Venkatasubramanian, S.; Daga, S.; Langrish, J.; Mills, N.L.; Lang, N.N.; Hoffmann, E.; Waterhouse, B.; Newby, D.E.; Frier, B.M. Cardiometabolic effects of a novel SIRT1 activator, SRT2104, in people with type 2 diabetes mellitus. Open Heart 2017, 4, e000647. [Google Scholar] [CrossRef] [PubMed]
- Hirano, H.; Takama, R.; Matsumoto, R.; Tanaka, H.; Hirano, H.; Soh, Z.; Ukawa, T.; Takayanagi, T.; Morimoto, H.; Nakamura, R.; et al. Assessment of Lower-limb Vascular Endothelial Function Based on Enclosed Zone Flow-mediated Dilation. Sci. Rep. 2018, 8, 9263. [Google Scholar] [CrossRef] [PubMed]
- Roustit, M.; Cracowski, J.L. Non-invasive assessment of skin microvascular function in humans: An insight into methods. Microcirculation 2012, 19, 47–64. [Google Scholar] [CrossRef]
- Stern, M.D. In vivo evaluation of microcirculation by coherent light scattering. Nature 1975, 254, 56–58. [Google Scholar] [CrossRef]
- Cracowski, J.L.; Minson, C.T.; Salvat-Melis, M.; Halliwill, J.R. Methodological issues in the assessment of skin microvascular endothelial function in humans. Trends Pharmacol. Sci. 2006, 27, 503–508. [Google Scholar] [CrossRef]
- Babos, L.; Járai, Z.; Nemcsik, J. Evaluation of microvascular reactivity with laser Doppler flowmetry in chronic kidney disease. World J. Nephrol. 2013, 2, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Greaney, J.L.; Saunders, E.F.H.; Santhanam, L.; Alexander, L.M. Oxidative Stress Contributes to Microvascular Endothelial Dysfunction in Men and Women with Major Depressive Disorder. Circ. Res. 2019, 124, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Heeman, W.; Steenbergen, W.; van Dam, G.; Boerma, E.C. Clinical applications of laser speckle contrast imaging: A review. J. Biomed. Opt. 2019, 24, 080901. [Google Scholar] [CrossRef] [PubMed]
- Farrero, M.; Blanco, I.; Batlle, M.; Santiago, E.; Cardona, M.; Vidal, B.; Castel, M.A.; Sitges, M.; Barbera, J.A.; Perez-Villa, F. Pulmonary hypertension is related to peripheral endothelial dysfunction in heart failure with preserved ejection fraction. Circ. Heart Fail. 2014, 7, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Weber, T.; Auer, J.; O‘Rourke, M.F.; Kvas, E.; Lassnig, E.; Berent, R.; Eber, B. Arterial stiffness, wave reflections, and the risk of coronary artery disease. Circulation 2004, 109, 184–189. [Google Scholar] [CrossRef]
- Van Bortel, L.M.; Laurent, S.; Boutouyrie, P.; Chowienczyk, P.; Cruickshank, J.K.; De Backer, T.; Filipovsky, J.; Huybrechts, S.; Mattace-Raso, F.U.; Protogerou, A.D.; et al. 1on Vascular Structure and Function; European Network for Noninvasive Investigation of Large Arteries. Expert consensus document on the measurement of aortic stiffness in daily practice using carotid-femoral pulse wave velocity. J. Hypertens. 2012, 30, 445–448. [Google Scholar] [CrossRef]
- Wassertheurer, S.; Kropf, J.; Weber, T.; van der Giet, M.; Baulmann, J.; Ammer, M.; Hametner, B.; Mayer, C.C.; Eber, B.; Magometschnigg, D. A new oscillometric method for pulse wave analysis: Comparison with a common tonometric method. J. Hum. Hypertens. 2010, 24, 498–504. [Google Scholar] [CrossRef]
- Vriz, O.; Driussi, C.; La Carrubba, S.; Di Bello, V.; Zito, C.; Carerj, S.; Antonini-Canterin, F. Comparison of sequentially measured Aloka echo-tracking one-point pulse wave velocity with SphygmoCor carotid-femoral pulse wave velocity. SAGE Open Med. 2013, 1, 2050312113507563. [Google Scholar] [CrossRef]
- Giannattasio, C.; Mancia, G. Arterial distensibility in humans. Modulating mechanisms, alterations in diseases and effects of treatment. J. Hypertens. 2002, 20, 1889–1899. [Google Scholar] [CrossRef]
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.; Coca, A.; De Simone, G.; Dominiczak, A.; et al. 2018 Practice Guidelines for the management of arterial hypertension of the European Society of Hypertension and the European Society of Cardiology: ESH/ESC Task Force for the Management of Arterial Hypertension. J. Hypertens. 2018, 36, 2284–2309, Erratum in J. Hypertens. 2019, 37, 456. [Google Scholar] [CrossRef]
- Shahin, Y.; Khan, J.A.; Chetter, I. Angiotensin converting enzyme inhibitors effect on arterial stiffness and wave reflections: A meta-analysis and meta-regression of randomised controlled trials. Atherosclerosis 2012, 221, 18–33. [Google Scholar] [CrossRef]
- Guerin, A.P.; Blacher, J.; Pannier, B.; Marchais, S.J.; Safar, M.E.; London, G.M. Impact of aortic stiffness attenuation on survival of patients in end-stage renal failure. Circulation 2001, 103, 987–992. [Google Scholar] [CrossRef]
- Bin-Nun, A.; Schreiber, M.D. Role of iNO in the modulation of pulmonary vascular resistance. J. Perinatol. 2008, 28 (Suppl. 3), S84–S92. [Google Scholar] [CrossRef] [PubMed]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef]
- Lan, N.S.H.; Massam, B.D.; Kulkarni, S.S.; Lang, C.C. Pulmonary Arterial Hypertension: Pathophysiology and Treatment. Diseases 2018, 6, 38. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fattouch, K.; Sbraga, F.; Bianco, G.; Speziale, G.; Gucciardo, M.; Sampognaro, R.; Ruvolo, G. Inhaled prostacyclin, nitric oxide, and nitroprusside in pulmonary hypertension after mitral valve replacement. J. Card. Surg. 2005, 20, 171–176. [Google Scholar] [CrossRef]
- Burger, C.D. Pulmonary hypertension in COPD: A review and consideration of the role of arterial vasodilators. COPD 2009, 6, 137–144. [Google Scholar] [CrossRef]
- Lee, J.E.; Hillier, S.C.; Knoderer, C.A. Use of sildenafil to facilitate weaning from inhaled nitric oxide in children with pulmonary hypertension following surgery for congenital heart disease. J. Intensive Care Med. 2008, 23, 329–334. [Google Scholar] [CrossRef]
- Stehlik, J.; Movsesian, M.A. Combined use of PDE5 inhibitors and nitrates in the treatment of pulmonary arterial hypertension in patients with heart failure. J. Card. Fail. 2009, 15, 31–34. [Google Scholar] [CrossRef]
- Yin, N.; Kaestle, S.; Yin, J.; Hentschel, T.; Pries, A.R.; Kuppe, H.; Kuebler, W.M. Inhaled nitric oxide versus aerosolized iloprost for the treatment of pulmonary hypertension with left heart disease. Crit. Care Med. 2009, 37, 980–986. [Google Scholar] [CrossRef]
- Zolty, R. Pulmonary arterial hypertension specific therapy: The old and the new. Pharmacol. Ther. 2020, 214, 107576. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, V.V.; Benza, R.L.; Rubin, L.J.; Channick, R.N.; Voswinckel, R.; Tapson, V.F.; Robbins, I.M.; Olschewski, H.; Rubenfire, M.; Seeger, W. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: A randomized controlled clinical trial. J. Am. Coll. Cardiol. 2010, 55, 1915–1922. [Google Scholar] [CrossRef]
- Hoeper, M.M.; Markevych, I.; Spiekerkoetter, E.; Welte, T.; Niedermeyer, J. Goal-oriented treatment and combination therapy for pulmonary arterial hypertension. Eur. Respir. J. 2005, 26, 858–863. [Google Scholar] [CrossRef]
- Boucly, A.; Savale, L.; Jaïs, X.; Bauer, F.; Bergot, E.; Bertoletti, L.; Beurnier, A.; Bourdin, A.; Bouvaist, H.; Bulifon, S.; et al. Association between initial treatment strategy and long-term survival in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2021, 204, 842–854. [Google Scholar] [CrossRef]
- “Pah, Prostacyclin Analogs: Drug Class, Uses, Side Effects, Drug Names”. RxList, RxList, 21 October 2021. Available online: www.rxlist.com/how_do_pah_prostacyclin_analogs_work/drug-class.htm (accessed on 18 April 2024).
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Heart J. 2022, 43, 3618–3731, Erratum in: Eur. Heart J. 2023, 44, 1312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, X.; Huang, J.; Li, H.; Su, Z.; Wang, J. Comparative Efficacy and Safety of Prostacyclin Ana-logs for Pulmonary Arterial Hypertension: A Network Meta-Analysis. Medicine 2016, 95, e2575. [Google Scholar] [CrossRef]
- Barnes, H.; Yeoh, H.L.; Fothergill, T.; Burns, A.; Humbert, M.; Williams, T. Prostacyclin for pulmonary arterial hypertension. Cochrane Database Syst. Rev. 2019, 5, CD012785. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hye, T.; Hossain, M.R.; Saha, D.; Foyez, T. Emerging biologics for the treatment of pulmonary arterial hypertension. J. Drug Target. 2023, 31, 471–485. [Google Scholar] [CrossRef]
- Lázár, Z.; Mészáros, M.; Bikov, A. The Nitric Oxide Pathway in Pulmonary Arterial Hypertension: Pathomechanism, Biomarkers and Drug Targets. Curr. Med. Chem. 2020, 27, 7168–7188. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Brundage, B.H.; Ghofrani, H.A.; Oudiz, R.J.; Simonneau, G.; Safdar, Z.; Shapiro, S.; White, R.J.; Chan, M.; Beardsworth, A.; et al. Pulmonary Arterial Hypertension and Response to Tadalafil (PHIRST) Study Group. Tadalafil therapy for pulmonary arterial hypertension. Circulation 2009, 119, 2894–2903, Erratum in Circulation 2011, 124, e279. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.J.; Beckmann, T.; Vorla, M.; Kalra, D.K. New Drugs and Therapies in Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2023, 24, 5850. [Google Scholar] [CrossRef]
- Tielemans, B.; Delcroix, M.; Belge, C.; Quarck, R. TGFβ and BMPRII signalling pathways in the pathogenesis of pulmonary arterial hypertension. Drug Discov. Today 2019, 24, 703–716. [Google Scholar] [CrossRef]
- Naeije, R.; Chin, K. Differentiating Precapillary from Postcapillary Pulmonary Hypertension. Circulation 2019, 140, 712–714. [Google Scholar] [CrossRef] [PubMed]
- Wolff, B.; Lodziewski, S.; Bollmann, T.; Opitz, C.F.; Ewert, R. Impaired peripheral endothelial function in severe idiopathic pulmonary hypertension correlates with the pulmonary vascular response to inhaled iloprost. Am. Heart J. 2007, 153, e1–e7. [Google Scholar] [CrossRef]
- Sfikakis, P.P.; Papamichael, C.; Stamatelopoulos, K.S.; Tousoulis, D.; Fragiadaki, K.G.; Katsichti, P.; Stefanadis, C.; Mavrikakis, M. Improvement of vascular endothelial function using the oral endothelin receptor antagonist bosentan in patients with systemic sclerosis. Arthritis Rheum. 2007, 56, 1985–1993. [Google Scholar] [CrossRef] [PubMed]
- Hirashiki, A.; Adachi, S.; Nakano, Y.; Kamimura, Y.; Shimokata, S.; Takeshita, K.; Murohara, T.; Kondo, T. Effects of bosentan on peripheral endothelial function in patients with pulmonary arterial hypertension or chronic thromboembolic pulmonary hypertension. Pulm. Circ. 2016, 6, 168–173. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schwartz, B.G.; Jackson, G.; Stecher, V.J.; Campoli-Richards, D.M.; Kloner, R.A. Phosphodiesterase type 5 inhibitors improve endothelial function and may benefit cardiovascular conditions. Am. J. Med. 2013, 126, 192–199. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Genes associated with the superfamily of TGF-β receptors | BMPR2 is the main gene implicated | ACVRL1 |
| ENG | ||
| CAV1 | ||
| ALK1 | ||
| SMAD 1,2,3, 4, 5, 8, 9 | ||
| EDN1 | ||
| ARRB2 | ||
| PTN1 | ||
| GDF2 | ||
| Other genes | Rarer | Transcription factors (TBX4 and SOX17) |
| ENO1 | ||
| YTHDF2 | ||
| EGLN1 | ||
| eNOS | ||
| Channel genes (KCNK3, KCNA5, ABCC8, ATP13A3, AQP1, TMEM16) | ||
| ET-1 | ||
| EIF2AK4 |
| Cellular changes | Proliferation of pulmonary artery smooth muscle cells (PASMCs) and pulmonary artery endothelial cells (PAECs) contributes to PAH [83]. |
| Typical Lesions in PAH | Plexiform lesions (including onion-skin, plexiform core, and dilation lesions) play a complex role in PAH pathology [85]. |
| Vascular Shunting | Systemic blood vessels (e.g., vasa vasorum, bronchial arteries) may contribute to plexiform vasculopathy [86]. |
| Bronchial-Pulmonary Shunts | Morphometric analysis reveals vascular shunts between bronchial and pulmonary blood vessels in PAH patients [87]. |
| SiMFis Structures | Large fibrovascular singular millimetric fibrovascular lesion (SiMFis) structures connect pulmonary arteries and veins to the systemic circulation [85]. |
| Stem/Progenitor Cells | Stem/progenitor cells may impact PAH progression and repair damaged endothelia [89]. |
| miRNA Dysregulation | Aberrant miRNA expression is linked to apoptosis resistance, inflammation, and vascular proliferation in PAH [92]. |
| miR-204 and miR-21 | MiR-204 down-regulation and MiR-21 up-regulation impact PAH pathophysiology [92,93]. |
| Other miRNAs | MiR-22, miR-17/92 cluster, and more contribute to the complex signaling network in PAH. |
| Endothelin-1 (ET-1) | ET-1 contributes to cellular proliferation and pulmonary artery constriction in PAH [96]. |
| RNA Manipulation | miRNA mimics, antagomirs, and siRNAs aim to precisely regulate gene expression [98,99]. |
| Angiogenesis Modulation | Angiogenic growth factors and inhibitors may restore vascular balance in PAH [98,99]. |
| Stem Cells and Regenerative Medicine | Repairing or replacing damaged pulmonary vascular tissue is now possible [98,99]. |
| Mitochondrial Dysfunction | Mitochondrial metabolism modulators offer potential therapeutic targets [100,101]. |
| NON-INVASIVE | FMD [110] |
| L-FMC [116,117] | |
| ezFMD [136] | |
| LDF [137] | |
| RHI [118] | |
| Doppler Echocardiography [143] | |
| Magnetic Resonance Imaging [143] | |
| Positron Emission Tomography [143] | |
| Pulse Wave Velocity [144] | |
| Nailfold Videocapillaroscopy [143] | |
| Near-Infrared Spectroscopy [143] | |
| INVASIVE | Venous Occlusion Plethysmography [38,115,116,117,118,119,120,121,122,123,124] |
| EDD using intra-arterial or intravenous infusion [106,107,108,109] | |
| Angiography [153] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Correale, M.; Chirivì, F.; Bevere, E.M.L.; Tricarico, L.; D’Alto, M.; Badagliacca, R.; Brunetti, N.D.; Vizza, C.D.; Ghio, S. Endothelial Function in Pulmonary Arterial Hypertension: From Bench to Bedside. J. Clin. Med. 2024, 13, 2444. https://doi.org/10.3390/jcm13082444
Correale M, Chirivì F, Bevere EML, Tricarico L, D’Alto M, Badagliacca R, Brunetti ND, Vizza CD, Ghio S. Endothelial Function in Pulmonary Arterial Hypertension: From Bench to Bedside. Journal of Clinical Medicine. 2024; 13(8):2444. https://doi.org/10.3390/jcm13082444
Chicago/Turabian StyleCorreale, Michele, Francesco Chirivì, Ester Maria Lucia Bevere, Lucia Tricarico, Michele D’Alto, Roberto Badagliacca, Natale D. Brunetti, Carmine Dario Vizza, and Stefano Ghio. 2024. "Endothelial Function in Pulmonary Arterial Hypertension: From Bench to Bedside" Journal of Clinical Medicine 13, no. 8: 2444. https://doi.org/10.3390/jcm13082444