
Immune Dysregulation in Patients Persistently Infected with Human Papillomaviruses 6 and 11

Abstract
: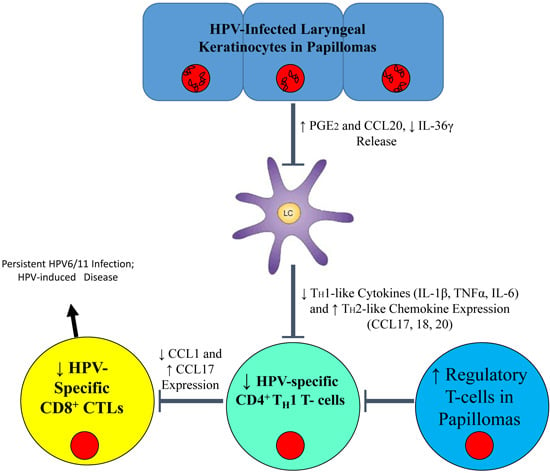
{kind=link}
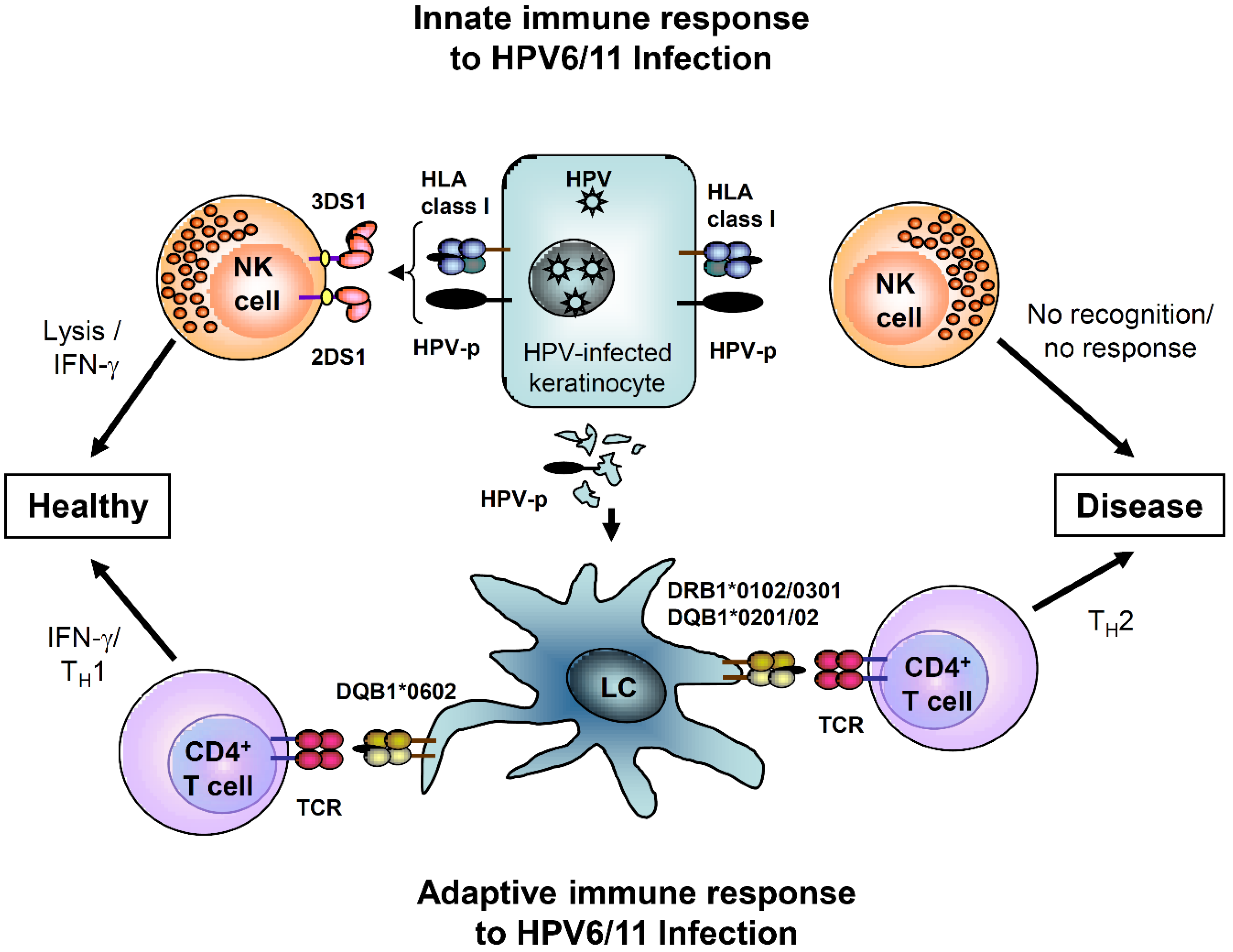{kind=link}
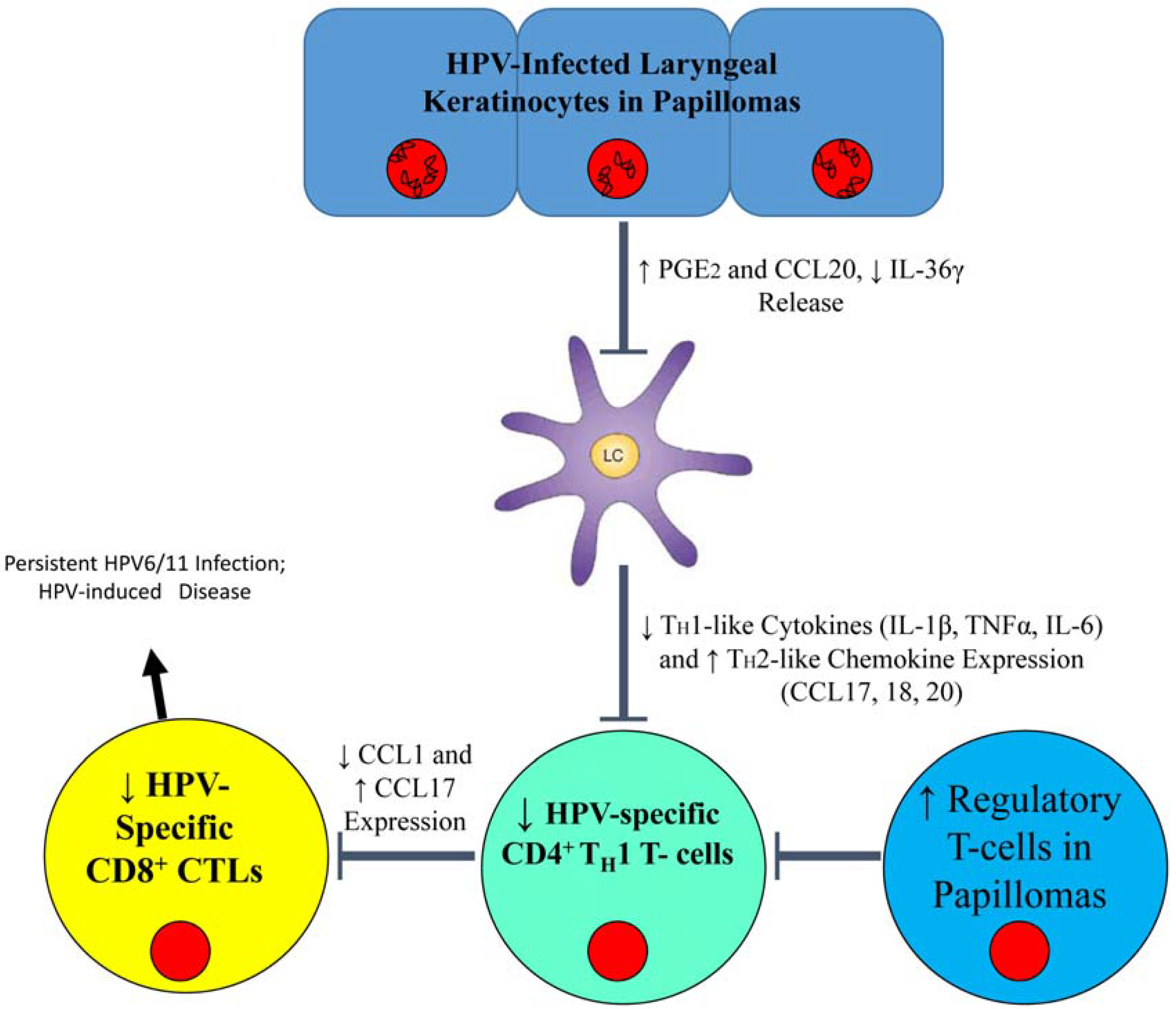{kind=link}
1. Introduction
2. Langerhans Cells


3. T Cells
4. Natural Killer Cells
5. Discussion
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Patel, H.; Wagner, M.; Singhal, P.; Kothari, S. Systematic review of the incidence and prevalence of genital warts. BMC Infect. Dis. 2013, 13. [Google Scholar] [CrossRef] [PubMed]
- Derkay, C.S.; Wiatrak, B. Recurrent respiratory papillomatosis: A review. Laryngoscope 2008, 118, 1236–1247. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Zhao, J.; Lei, Z.; Shen, S.; Liu, C.; Li, D.; Liu, J.; Shen, G.X.; Zhang, G.M.; Feng, Z.H.; et al. Local accumulation of FOXP3+ regulatory T cells: Evidence for an immune evasion mechanism in patients with large condylomata acuminata. J. Immunol. 2008, 180, 7681–7686. [Google Scholar] [CrossRef]
- Bonagura, V.R.; Siegal, F.P.; Abramson, A.L.; Santiagoschwarz, F.; Oreilly, M.E.; Shah, K.; Drake, D.; Steinberg, B.M. Enriched Hla-Dq3 phenotype and decreased class-I major histocompatibility complex antigen expression in recurrent respiratory papillomatosis. Clin. Diagn. Lab. Immunol. 1994, 1, 357–360. [Google Scholar] [PubMed]
- Stanley, M. Immunobiology of HPV and HPV vaccines. Gynecol. Oncol. 2008, 109 (Suppl. S2), 15–21. [Google Scholar] [CrossRef]
- Amador-Molina, A.; Hernández-Valencia, J.F.; Lamoyi, E.; Contreras-Paredes, A.; Lizano, M. Role of innate immunity against human papillomavirus (HPV) infections and effect of adjuvants in promoting specific immune response. Viruses 2013, 5, 2624–2642. [Google Scholar] [CrossRef] [PubMed]
- Sasagawa, T.; Takagi, H.; Makinoda, S. Immune responses against human papillomavirus (HPV) infection and evasion of host defense in cervical cancer. J. Infect. Chemother. 2012, 18, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Bonagura, V.R.; Hatam, L.; DeVoti, J.; Zeng, F.F.; Steinberg, B.M. Recurrent respiratory papillomatosis: Altered CD8(+) T-cell subsets and T(h)1/T(h)2 cytokine imbalance. Clin. Immunol. 1999, 93, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Bonagura, V.R.; Vambutas, A.; DeVoti, J.A.; Rosenthal, D.W.; Steinberg, B.M.; Abramson, A.L.; Shikowitz, M.J.; Gjertson, D.W.; Reed, E.F. Hla alleles, IFN-gamma responses to HPV-11 E6, and disease severity in patients with recurrent respiratory papillomatosis. Hum. Immunol. 2004, 65, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, D.W.; DeVoti, J.A.; Steinberg, B.M.; Abramson, A.L.; Bonagura, V.R. T(h)2-like chemokine patterns correlate with disease severity in patients with recurrent respiratory papillomatosis. Mol. Med. 2012, 18, 1338–1345. [Google Scholar] [CrossRef] [PubMed]
- Hatam, L.J.; Rosenthal, D.W.; DeVoti, J.A.; Lam, F.; Abramson, A.; Steinberg, B.M.; Bonagura, V.R. CD4(+)FOXP3(+)CD127(+low) T-regulatory cells are increased in HPV infected papillomas in patients with recurrent respiratory papillomatosis (RRP). J. Allergy Clin. Immunol. 2008, 121. [Google Scholar] [CrossRef] [PubMed]
- Hatam, L.J.; DeVoti, J.A.; Rosenthal, D.W.; Lam, F.; Abramson, A.L.; Steinberg, B.M.; Bonagura, V.R. Immune suppression in premalignant respiratory papillomas: Enriched functional CD4(+)FOXP3(+) regulatory T cells and PD-1/PD-L1/L2 expression. Clin. Cancer Res. 2012, 18, 1925–1935. [Google Scholar] [CrossRef] [PubMed]
- Bonagura, V.R.; Hatam, L.J.; Rosenthal, D.W.; De Voti, J.A.; Lam, F.; Steinberg, B.M.; Abramson, A.L. Recurrent respiratory papillomatosis: A complex defect in immune responsiveness to human papillomavirus-6 and -11. Apmis 2010, 118, 455–470. [Google Scholar] [CrossRef] [PubMed]
- Bonagura, V.R.; Du, Z.; Ashouri, E.; Luo, L.; Hatam, L.J.; DeVoti, J.A.; Rosenthal, D.W.; Steinberg, B.M.; Abramson, A.L.; Gjertson, D.W.; et al. Activating killer cell immunoglobulin-like receptors 3DS1 and 2DS1 protect against developing the severe form of recurrent respiratory papillomatosis. Hum. Immunol. 2010, 71, 212–219. [Google Scholar] [CrossRef]
- Steinman, R.M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 1991, 9, 271–296. [Google Scholar] [CrossRef] [PubMed]
- Hart, D.N. Dendritic cells: Unique leukocyte populations which control the primary immune response. Blood 1997, 90, 3245–3287. [Google Scholar] [PubMed]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef] [PubMed]
- Noel, W.; Raes, G.; Hassanzadeh Ghassabeh, G.; De Baetselier, P.; Beschin, A. Alternatively activated macrophages during parasite infections. Trends Parasitol. 2004, 20, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Lutz, M.B.; Suri, R.M.; Niimi, M.; Ogilvie, A.L.; Kukutsch, N.A.; Rossner, S.; Schuler, G.; Austyn, J.M. Immature dendritic cells generated with low doses of GM-CSF in the absence of IL-4 are maturation resistant and prolong allograft survival in vivo. Eur. J. Immunol. 2000, 30, 1813–1822. [Google Scholar] [CrossRef] [PubMed]
- McGuirk, P.; McCann, C.; Mills, K.H. Pathogen-specific T regulatory 1 cells induced in the respiratory tract by a bacterial molecule that stimulates interleukin 10 production by dendritic cells: A novel strategy for evasion of protective T helper type 1 responses by bordetella pertussis. J. Exp. Med. 2002, 195, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Shortman, K.; Liu, Y.J. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2002, 2, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, A.L.; Carbone, F.; Geijtenbeek, T.B. Langerhans cells and viral immunity. Eur J Immunol 2008, 38, 2377–2385. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, D.M.; Movius, C.A.; Raff, A.B.; Brand, H.E.; Skeate, J.G.; Wong, M.K.; Kast, W.M. Suppression of langerhans cell activation is conserved amongst human papillomavirus α and β genotypes, but not a µ genotype. Virology 2014, 452–453, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Leong, C.M.; Doorbar, J.; Nindl, I.; Yoon, H.S.; Hibma, M.H. Deregulation of e-cadherin by human papillomavirus is not confined to high-risk, cancer-causing types. Br. J. Dermatol. 2010, 163, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Bhawan, J.; Dayal, Y.; Bhan, A.K. Langerhans cells in molluscum contagiosum, verruca vulgaris, plantar wart, and condyloma acuminatum. J. Am. Acad. Dermatol. 1986, 15, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Viac, J.; Chardonnet, Y.; Euvrard, S.; Chignol, M.C.; Thivolet, J. Langerhans cells, inflammation markers and human papillomavirus infections in benign and malignant epithelial tumors from transplant recipients. J. Dermatol. 1992, 19, 67–77. [Google Scholar] [PubMed]
- Coleman, N.; Birley, H.D.; Renton, A.M.; Hanna, N.F.; Ryait, B.K.; Byrne, M.; Taylor-Robinson, D.; Stanley, M.A. Immunological events in regressing genital warts. Am. J. Clin. Pathol. 1994, 102, 768–774. [Google Scholar] [PubMed]
- McMillan, A.; Bishop, P.E.; Fletcher, S. An immunohistological study of condylomata acuminata. Histopathology 1990, 17, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Arany, I.; Tyring, S.K. Status of local cellular immunity in interferon-responsive and -nonresponsive human papillomavirus-associated lesions. Sex. Transm. Dis. 1996, 23, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Morelli, A.E.; Belardi, G.; DiPaola, G.; Paredes, A.; Fainboim, L. Cellular subsets and epithelial ICAM-1 and HLA-DR expression in human papillomavirus infection of the vulva. Acta Derm. Venereol. 1994, 74, 45–50. [Google Scholar] [PubMed]
- Feng, J.Y.; Peng, Z.H.; Tang, X.P.; Geng, S.M.; Liu, Y.P. Immunohistochemical and ultrastructural features of langerhans cells in condyloma acuminatum. J. Cutan. Pathol. 2008, 35, 15–20. [Google Scholar] [PubMed]
- Devoti, J.; Hatam, L.; Lucs, A.; Afzal, A.; Abramson, A.; Steinberg, B.; Bonagura, V. Decreased langerhans cell responses to IL-36γ: Altered innate immunity in patients with recurrent respiratory papillomatosis. Mol. Med. 2014, 20, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Arany, I.; Tyring, S.K.; Stanley, M.A.; Tomai, M.A.; Miller, R.L.; Smith, M.H.; McDermott, D.J.; Slade, H.B. Enhancement of the innate and cellular immune response in patients with genital warts treated with topical imiquimod cream 5%. Antivir. Res. 1999, 43, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Wang, B.; Shivji, G.M.; Toto, P.; Amerio, P.; Tomai, M.A.; Miller, R.L.; Sauder, D.N. Imiquimod, a topical immune response modifier, induces migration of langerhans cells. J. Investig. Dermatol. 2000, 114, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Shirakata, Y. Regulation of epidermal keratinocytes by growth factors. J. Dermatol. Sci. 2010, 59, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Pastore, S.; Mascia, F.; Girolomoni, G. The contribution of keratinocytes to the pathogenesis of atopic dermatitis. Eur. J. Dermatol. 2006, 16, 125–131. [Google Scholar] [PubMed]
- Werner, S.; Krieg, T.; Smola, H. Keratinocyte-fibroblast interactions in wound healing. J. Investig. Dermatol. 2007, 127, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Amerio, P.; Sauder, D.N. Role of cytokines in epidermal langerhans cell migration. J. Leukoc. Biol. 1999, 66, 33–39. [Google Scholar] [PubMed]
- Kel, J.M.; Girard-Madoux, M.J.; Reizis, B.; Clausen, B.E. TGF-beta is required to maintain the pool of immature langerhans cells in the epidermis. J. Immunol. 2010, 185, 3248–3255. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef] [PubMed]
- Del Hoyo, G.M.; Martin, P.; Vargas, H.H.; Ruiz, S.; Arias, C.F.; Ardavin, C. Characterization of a common precursor population for dendritic cells. Nature 2002, 415, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Paczesny, S.; Blanco, P.; Bennett, L.; Pascual, V.; Fay, J.; Palucka, A.K. Dendritic cells: Controllers of the immune system and a new promise for immunotherapy. Ann. N. Y. Acad. Sci. 2003, 987, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Caux, C.; Massacrier, C.; Vanbervliet, B.; Dubois, B.; Durand, I.; Cella, M.; Lanzavecchia, A.; Banchereau, J. CD34+ hematopoietic progenitors from human cord blood differentiate along two independent dendritic cell pathways in response to granulocyte-macrophage colony-stimulating factor plus tumor necrosis factor alpha: II. Functional analysis. Blood 1997, 90, 1458–1470. [Google Scholar] [PubMed]
- Steinman, R.M.; Turley, S.; Mellman, I.; Inaba, K. The induction of tolerance by dendritic cells that have captured apoptotic cells. J. Exp. Med. 2000, 191, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Kelsall, B.L. Freshly isolated peyer’s patch, but not spleen, dendritic cells produce interleukin 10 and induce the differentiation of T helper type 2 cells. J. Exp. Med. 1999, 190, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Enk, A.H.; Angeloni, V.L.; Udey, M.C.; Katz, S.I. Inhibition of langerhans cell antigen-presenting function by IL-10. A role for il-10 in induction of tolerance. J. Immunol. 1993, 151, 2390–2398. [Google Scholar] [PubMed]
- Beissert, S.; Hosoi, J.; Grabbe, S.; Asahina, A.; Granstein, R.D. IL-10 inhibits tumor antigen presentation by epidermal antigen-presenting cells. J. Immunol. 1995, 154, 1280–1286. [Google Scholar] [PubMed]
- Cottrez, F.; Groux, H. Specialization in tolerance: Innate CD(4+)CD(25+) versus acquired TR1 and TH3 regulatory T cells. Transplantation 2004, 77 (Suppl. S1), 12–15. [Google Scholar] [CrossRef]
- Johnston, A.; Xing, X.; Guzman, A.M.; Riblett, M.; Loyd, C.M.; Ward, N.L.; Wohn, C.; Prens, E.P.; Wang, F.; Maier, L.E.; et al. IL-1F5, -F6, -F8, and -F9: A novel IL-1 family signaling system that is active in psoriasis and promotes keratinocyte antimicrobial peptide expression. J. Immunol. 2011, 186, 2613–2622. [Google Scholar] [CrossRef] [PubMed]
- Romani, N.; Ebner, S.; Tripp, C.H.; Flacher, V.; Koch, F.; Stoitzner, P. Epidermal langerhans cells—Changing views on their function in vivo. Immunol. Lett. 2006, 106, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Elias, A.; Nanda, V.; Barr, R. CD1A expression in psoriatic skin following treatment with propylthiouracil, an antithyroid thioureylene. BMC Dermatol. 2003, 3. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, D.H.; Jenison, M.C.; Saeland, S.; Shlomchik, W.D.; Shlomchik, M.J. Epidermal langerhans cell-deficient mice develop enhanced contact hypersensitivity. Immunity 2005, 23, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Klechevsky, E.; Morita, R.; Liu, M.; Cao, Y.; Coquery, S.; Thompson-Snipes, L.; Briere, F.; Chaussabel, D.; Zurawski, G.; Palucka, A.K.; et al. Functional specializations of human epidermal langerhans cells and CD14+ dermal dendritic cells. Immunity 2008, 29, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.L.; Fallah-Arani, F.; Conlan, T.; Trouillet, C.; Goold, H.; Chorro, L.; Flutter, B.; Means, T.K.; Geissmann, F.; Chakraverty, R. Langerhans cells regulate cutaneous injury by licensing CD8 effector cells recruited to the skin. Blood 2011, 117, 7063–7069. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.M.; Adurthi, S.; Ramachandran, S.; Mukherjee, G.; Joy, O.; Krishnamurthy, H.; Krishna, S.; Bafna, U.D.; Uma, D.K.; Jayshree, R.S. Toll-like receptors 7, 8, and 9 expression and function in primary human cervical cancer langerhans cells: Evidence of anergy. Int. J. Gynecol. Cancer 2013, 23, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Sather, B.D.; Treuting, P.; Perdue, N.; Miazgowicz, M.; Fontenot, J.D.; Rudensky, A.Y.; Campbell, D.J. Altering the distribution of FOXP3(+) regulatory T cells results in tissue-specific inflammatory disease. J. Exp. Med. 2007, 204, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Zhao, J.; Yang, Z.; Cai, Z.; Zhang, B.; Zhou, Y.; Shen, G.X.; Chen, X.; Li, S.; Huang, B. CD4+FOXP3+ regulatory T cell depletion by low-dose cyclophosphamide prevents recurrence in patients with large condylomata acuminata after laser therapy. Clin. Immunol. 2010, 136, 21–29. [Google Scholar] [CrossRef] [PubMed]
- DeVoti, J.A.; Steinberg, B.M.; Rosenthal, D.W.; Hatam, L.; Vambutas, A.; Abramson, A.L.; Shikowitz, M.J.; Bonagura, V.R. Failure of gamma interferon but not interleukin-10 expression in response to human papillomavirus type 11 e6 protein in respiratory papillomatosis. Clin. Diagn. Lab. Immunol. 2004, 11, 538–547. [Google Scholar] [PubMed]
- Xu, Y.; Zhu, K.J.; Zhu, N.; Jiang, D.H.; Chen, X.Z.; Cheng, H. Expression of FOXP3+CD4+CD25+ regulatory T cells and TH1/TH2, TC1/Tc2 profiles in the peripheral blood of patients with condyloma acuminatum. Clin. Exp. Dermatol. 2009, 34, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Pedroza-Pacheco, I.; Madrigal, A.; Saudemont, A. Interaction between natural killer cells and regulatory T cells: Perspectives for immunotherapy. Cell. Mol. Immunol. 2013, 10, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Cauda, R.; Tyring, S.K.; Grossi, C.E.; Tilden, A.B.; Hatch, K.D.; Sams, W.M.; Baron, S.; Whitley, R.J. Patients with condyloma acuminatum exhibit decreased interleukin-2 and interferon gamma production and depressed natural killer activity. J. Clin. Immunol. 1987, 7, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Subbaramaiah, K.; Dannenberg, A.J. Cyclooxygenase-2 transcription is regulated by human papillomavirus 16 e6 and e7 oncoproteins: Evidence of a corepressor/coactivator exchange. Cancer Res 2007, 67, 3976–3985. [Google Scholar] [CrossRef] [PubMed]
- Vambutas, A.; DeVoti, J.; Pinn, W.; Steinberg, B.M.; Bonagura, V.R. Tap-1 down-regulation in laryngeal papillomas: Contribution of HPV 6/11 E7 protein. FASEB J. 2001, 15, A1009. [Google Scholar]
- Li, H.; Zhan, T.; Li, C.; Liu, M.; Wang, Q.K. Repression of MHC class I transcription by HPV16E7 through interaction with a putative rxrbeta motif and NF-kappaB cytoplasmic sequestration. Biochem. Biophys. Res. Commun. 2009, 388, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.J.; Yang, J.; Yang, W. Mechanistic investigation of immunosuppression in patients with condyloma acuminata. Mol. Med. Rep. 2013, 8, 480–486. [Google Scholar] [PubMed]
- DeVoti, J.A.; Rosenthal, D.W.; Wu, R.; Abramson, A.L.; Steinberg, B.M.; Bonagura, V.R. Immune dysregulation and tumor-associated gene changes in recurrent respiratory papillomatosis: A paired microarray analysis. Mol. Med. 2008, 14, 608–617. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucs, A.V.; DeVoti, J.A.; Hatam, L.; Afzal, A.; Abramson, A.L.; Steinberg, B.M.; Bonagura, V.R. Immune Dysregulation in Patients Persistently Infected with Human Papillomaviruses 6 and 11. J. Clin. Med. 2015, 4, 375-388. https://doi.org/10.3390/jcm4030375
Lucs AV, DeVoti JA, Hatam L, Afzal A, Abramson AL, Steinberg BM, Bonagura VR. Immune Dysregulation in Patients Persistently Infected with Human Papillomaviruses 6 and 11. Journal of Clinical Medicine. 2015; 4(3):375-388. https://doi.org/10.3390/jcm4030375
Chicago/Turabian StyleLucs, Alexandra V., James A. DeVoti, Lynda Hatam, Ali Afzal, Allan L. Abramson, Bettie M. Steinberg, and Vincent R. Bonagura. 2015. "Immune Dysregulation in Patients Persistently Infected with Human Papillomaviruses 6 and 11" Journal of Clinical Medicine 4, no. 3: 375-388. https://doi.org/10.3390/jcm4030375
APA StyleLucs, A. V., DeVoti, J. A., Hatam, L., Afzal, A., Abramson, A. L., Steinberg, B. M., & Bonagura, V. R. (2015). Immune Dysregulation in Patients Persistently Infected with Human Papillomaviruses 6 and 11. Journal of Clinical Medicine, 4(3), 375-388. https://doi.org/10.3390/jcm4030375