
Mechanisms by Which B Cells and Regulatory T Cells Influence Development of Murine Organ-Specific Autoimmune Diseases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
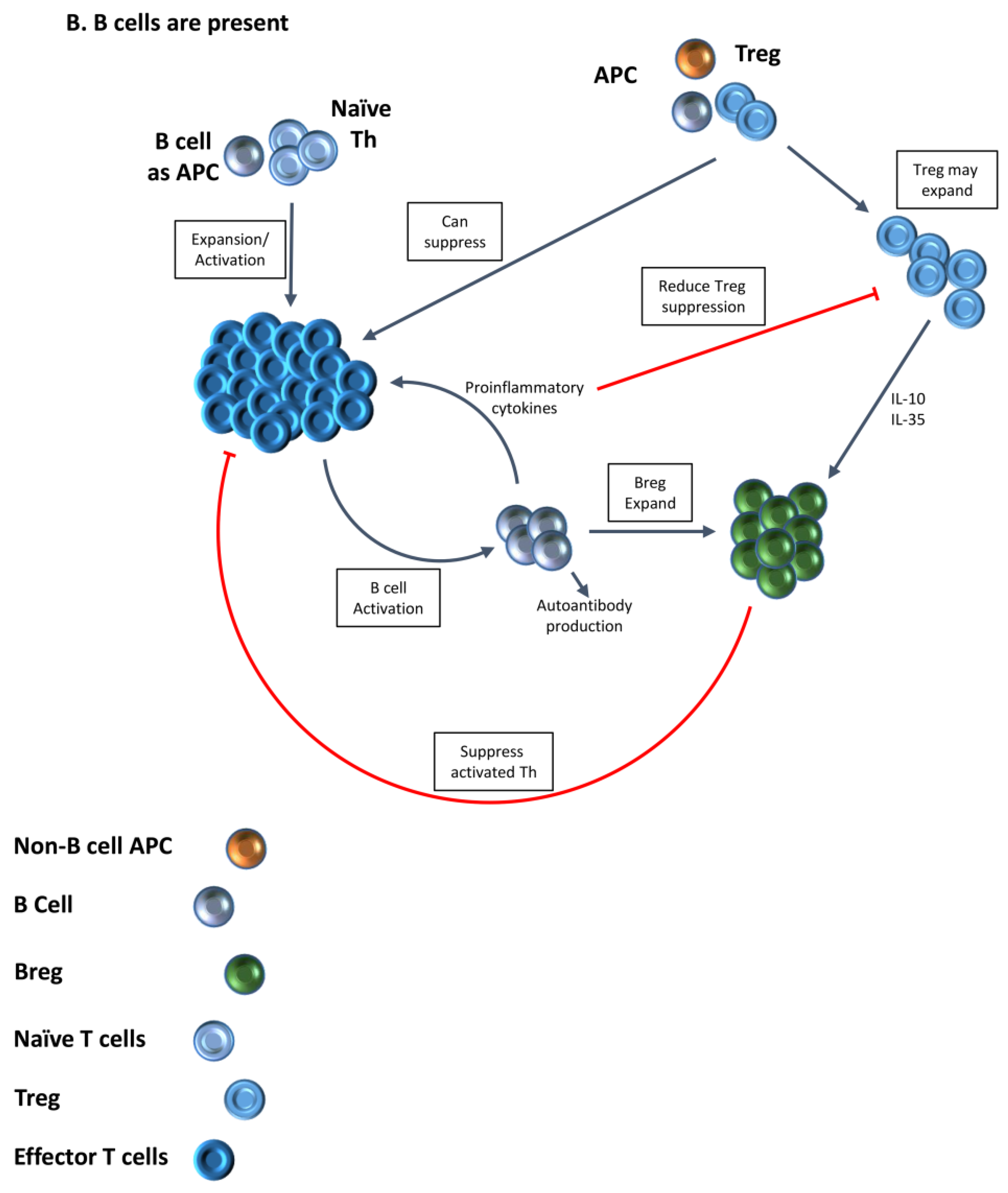
2. B Cells Are Important APC for Activation of Autoreactive T Cells
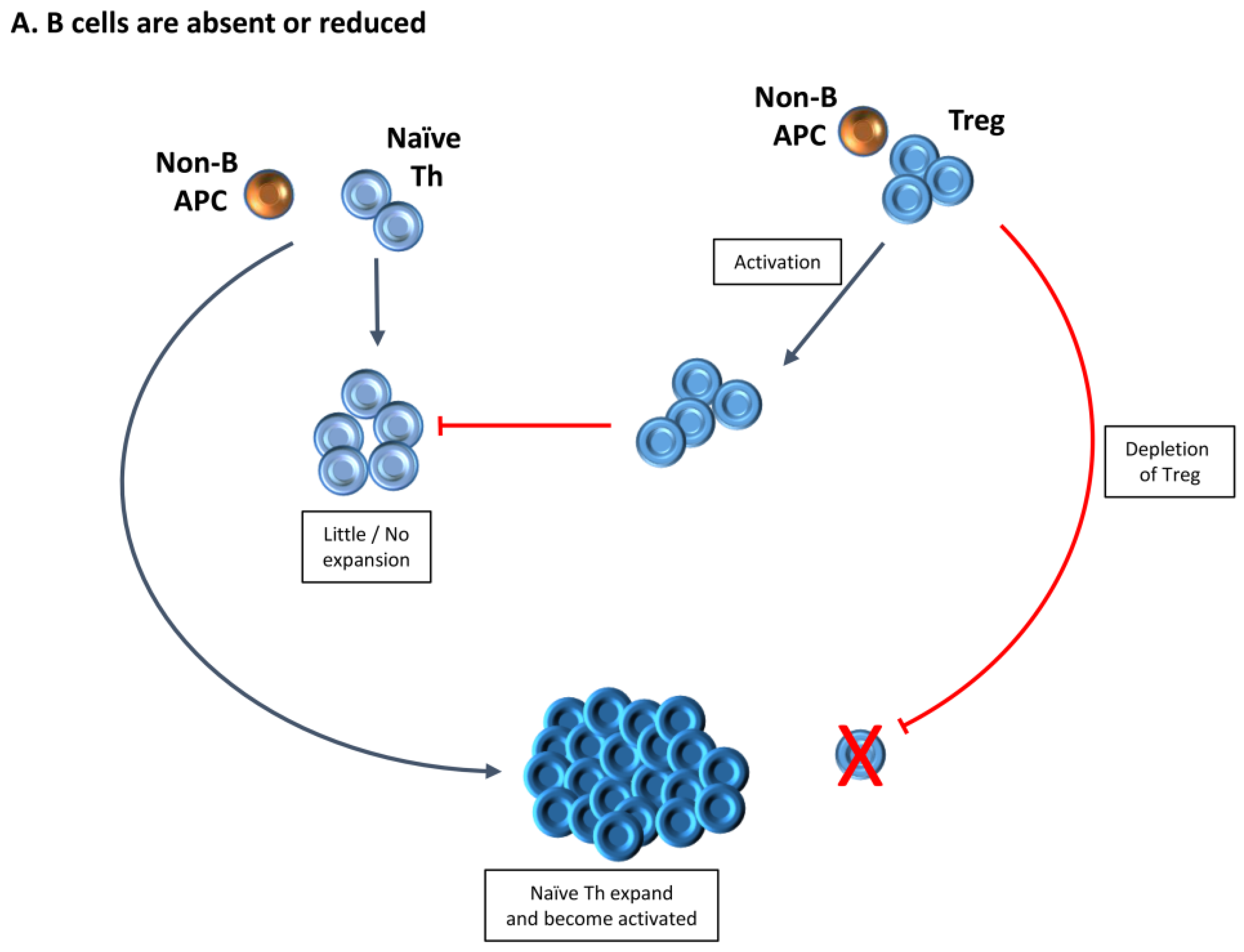
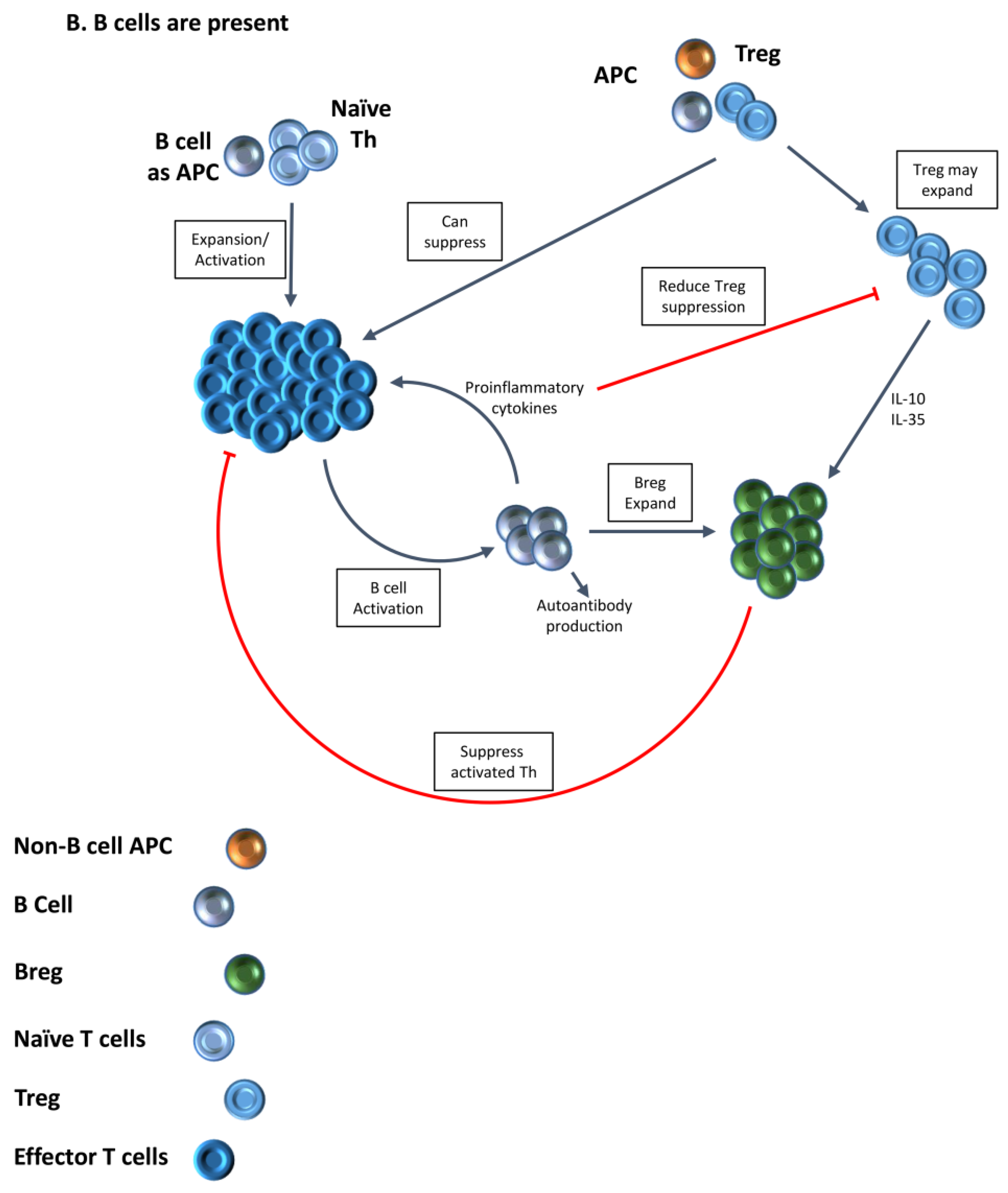
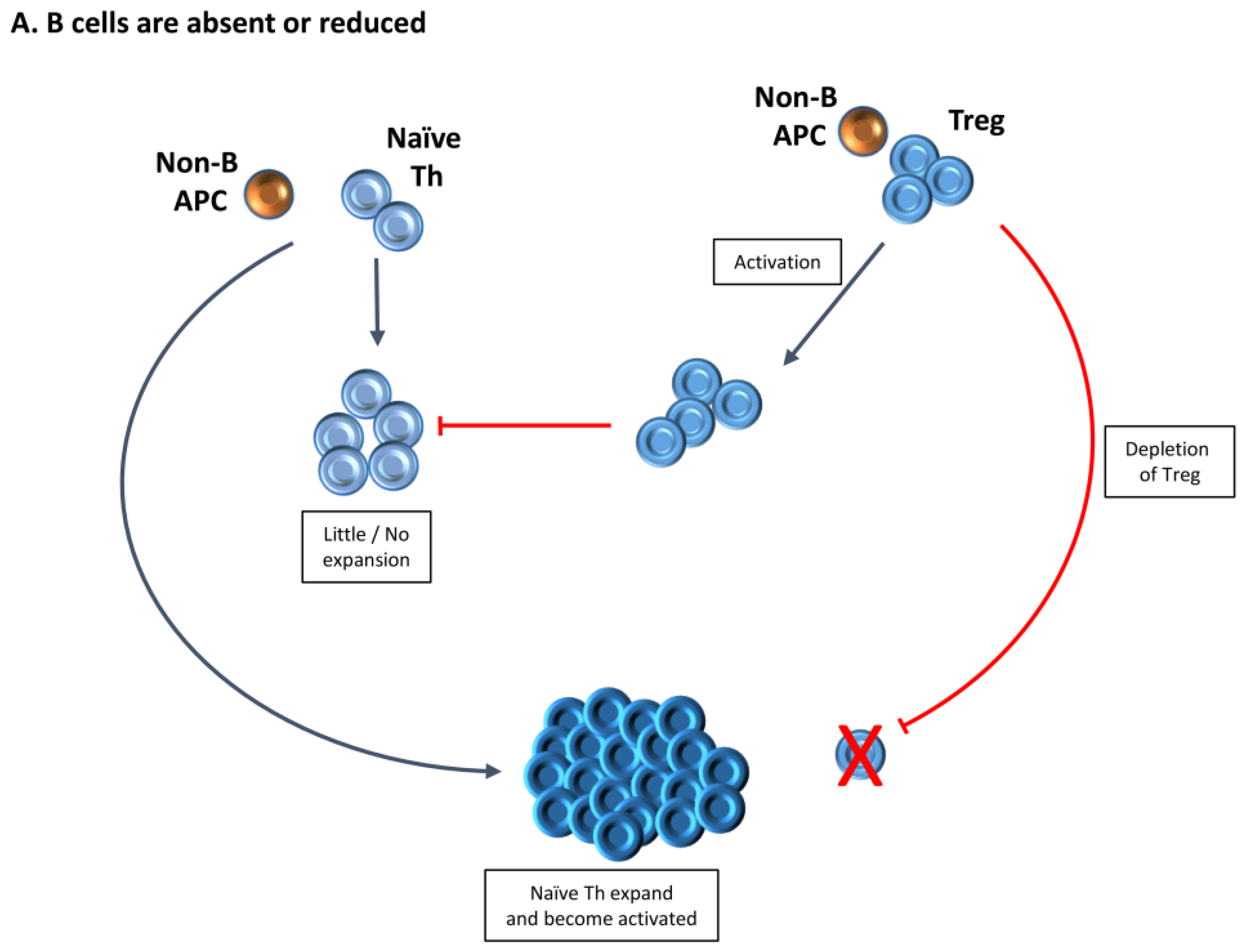
3. Increased Function of Tregs in the Absence of B Cells Provides One Explanation for the Requirement for B Cells for Development of Autoimmune Diseases
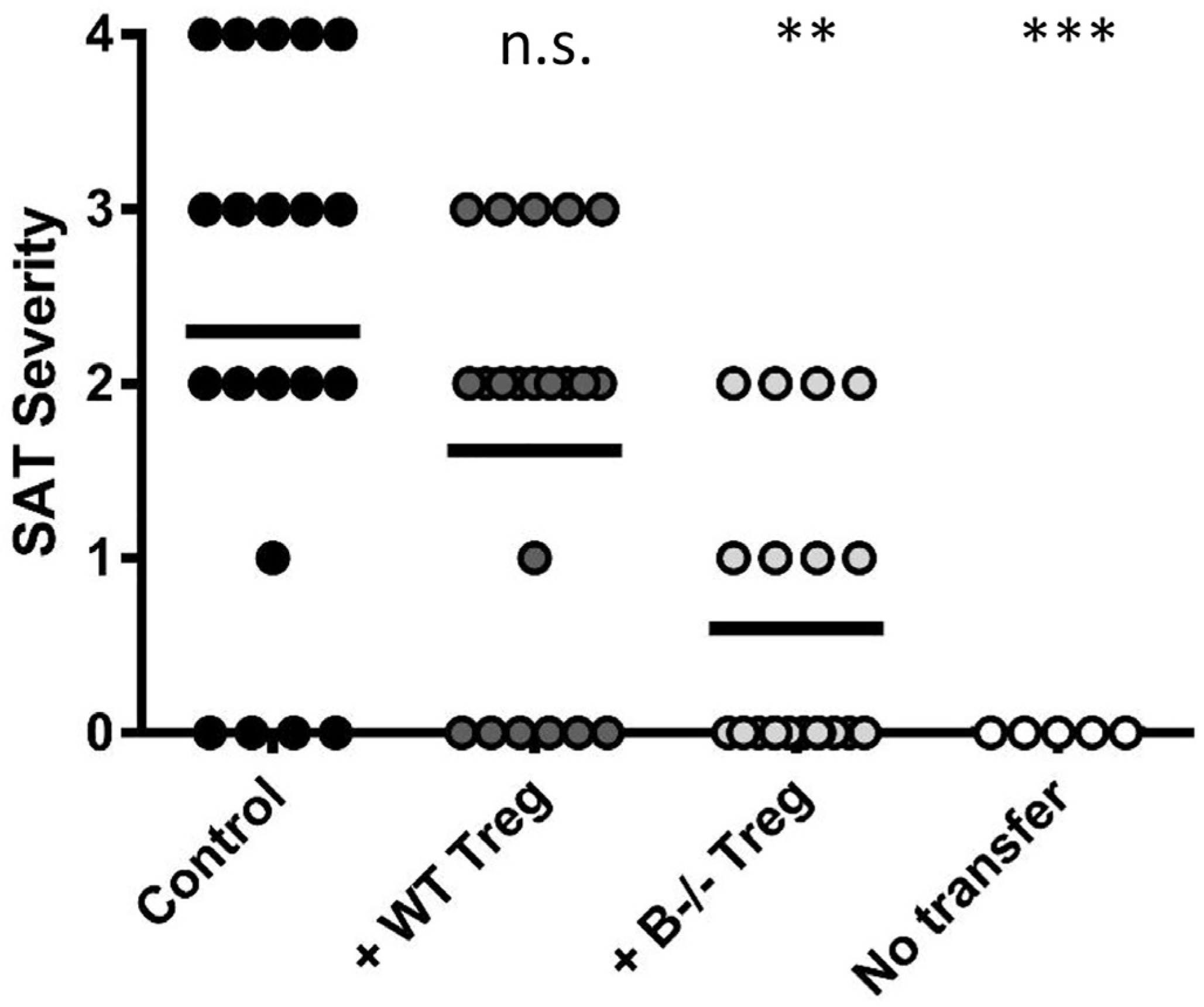
4. Treg Numbers and Function Differ in the Presence or Absence of B Cells
5. Treg in Mice Lacking B Cells Differ Functionally from Those in B Cell-Positive Mice
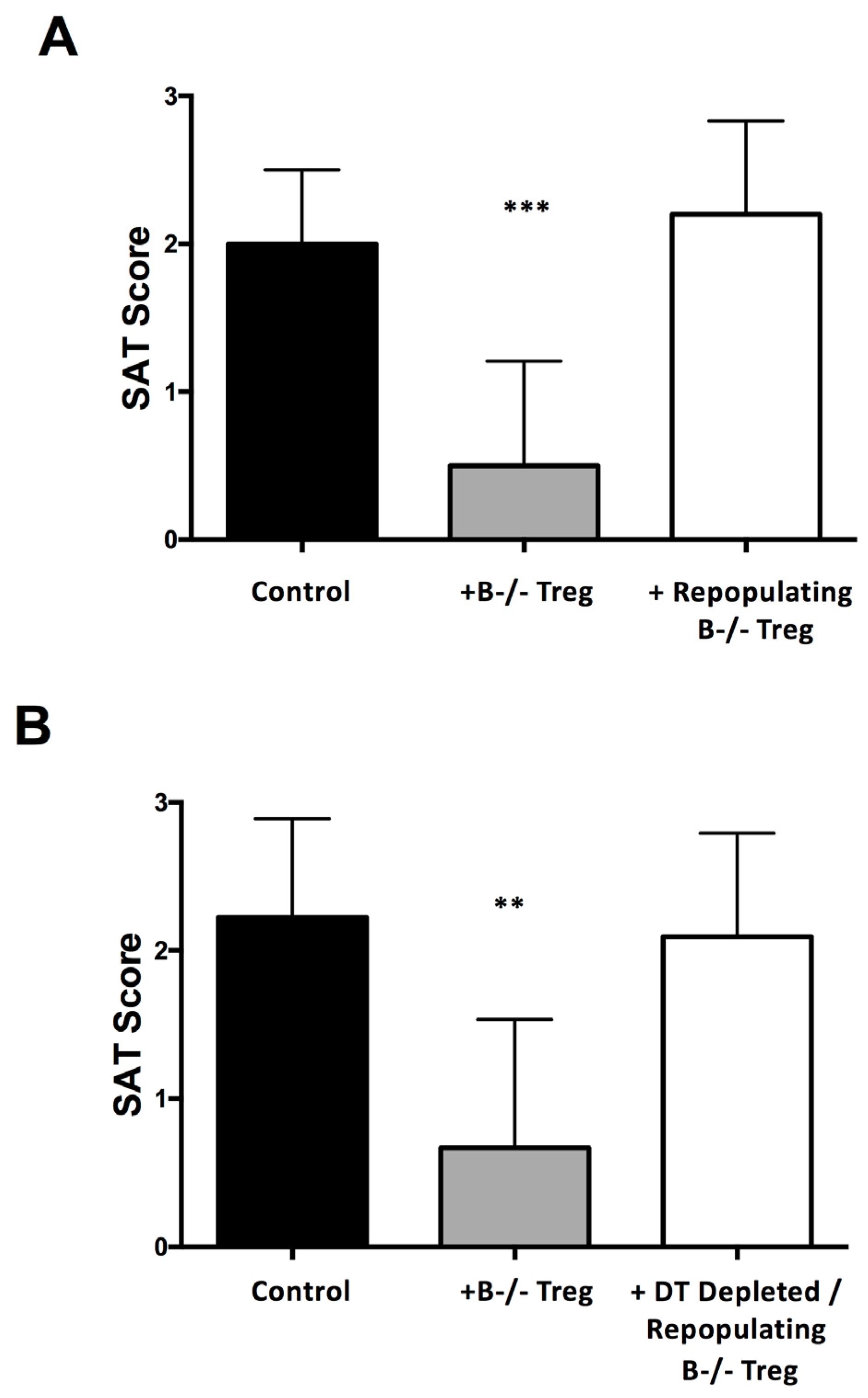
6. Transient Depletion of Treg Is Sufficient to Result in Autoimmune Disease in B−/− Mice Because Tregs That Repopulate Following Depletion Have Reduced Function
7. Multiple Factors Influence the Effectiveness of B Cell Depletion by Anti-CD20
8. Summary/Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Braley-Mullen, H.; Yu, S. Early requirement for B cells for development of spontaneous autoimmune thyroiditis in NOD.H-2h4 mice. J. Immunol. 2000, 165, 7262–7269. [Google Scholar] [CrossRef] [PubMed]
- Serreze, D.V.; Fleming, S.A.; Chapman, H.D.; Richard, S.D.; Leiter, E.H.; Tisch, R.M. B lymphocytes are critical antigen-presenting cells for the initiation of T cell-mediated autoimmune diabetes in nonobese diabetic mice. J. Immunol. 1998, 161, 3912–3918. [Google Scholar] [PubMed]
- Wong, F.S.; Wen, L.; Tang, M.; Ramanathan, M.; Visintin, I.; Daugherty, J.; Hannum, L.G.; Janeway, C.A., Jr.; Shlomchik, M.J. Investigation of the role of B-cells in type 1 diabetes in the NOD mouse. Diabetes 2004, 53, 2581–2587. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, I.; Tedder, T.F.; Zhuang, Y. B-lymphocyte depletion ameliorates Sjogren’s syndrome in ID3 knockout mice. Immunology 2007, 122, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.S.; Wan, X.; Braley-Mullen, H. Transient depletion of CD4+ CD25+ regulatory T cells results in multiple autoimmune diseases in wild-type and B-cell-deficient NOD mice. Immunology 2013, 139, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Chan, O.T.; Madaio, M.P.; Shlomchik, M.J. The central and multiple roles of B cells in lupus pathogenesis. Immunol. Rev. 1999, 169, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Hamel, K.; Doodes, P.; Cao, Y.; Wang, Y.; Martinson, J.; Dunn, R.; Kehry, M.R.; Farkas, B.; Finnegan, A. Suppression of proteoglycan-induced arthritis by anti-CD20 B cell depletion therapy is mediated by reduction in autoantibodies and CD4+ T cell reactivity. J. Immunol. 2008, 180, 4994–5003. [Google Scholar] [CrossRef] [PubMed]
- Svensson, L.; Jirholt, J.; Holmdahl, R.; Jansson, L. B cell-deficient mice do not develop type II collagen-induced arthritis (CIA). Clin. Exp. Immunol. 1998, 111, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Quan, S.; Sheng, J.R.; Abraham, P.M.; Soliven, B. Regulatory T and B lymphocytes in a spontaneous autoimmune polyneuropathy. Clin. Exp. Immunol. 2016, 184, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.C.; Cambridge, G. B-cell targeting in rheumatoid arthritis and other autoimmune diseases. Nat. Rev. Immunol. 2006, 6, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Erdei, A.; Paragh, G.; Kovacs, P.; Karanyi, Z.; Berenyi, E.; Galuska, L.; Lenkey, A.; Szabados, L.; Gyory, F.; Ujhelyi, B.; et al. Rapid response to and long-term effectiveness of anti-CD20 antibody in conventional therapy resistant graves’ orbitopathy: A five-year follow-up study. Autoimmunity 2014, 47, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Hampe, C.S. B Cell in Autoimmune Diseases. Scientifica 2012, 2012, 215308. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Dunn, R.; Kehry, M.R.; Braley-Mullen, H. B cell depletion inhibits spontaneous autoimmune thyroiditis in NOD.H-2h4 mice. J. Immunol. 2008, 180, 7706–7713. [Google Scholar] [CrossRef] [PubMed]
- Serreze, D.V.; Chapman, H.D.; Niens, M.; Dunn, R.; Kehry, M.R.; Driver, J.P.; Haller, M.; Wasserfall, C.; Atkinson, M.A. Loss of intra-islet CD20 expression may complicate efficacy of B-cell-directed type 1 diabetes therapies. Diabetes 2011, 60, 2914–2921. [Google Scholar] [CrossRef] [PubMed]
- Pollinger, B.; Krishnamoorthy, G.; Berer, K.; Lassmann, H.; Bosl, M.R.; Dunn, R.; Domingues, H.S.; Holz, A.; Kurschus, F.C.; Wekerle, H. Spontaneous relapsing-remitting eae in the SJL/J mouse: Mog-reactive transgenic T cells recruit endogenous mog-specific B cells. J. Exp. Med. 2009, 206, 1303–1316. [Google Scholar] [CrossRef] [PubMed]
- Flach, A.C.; Litke, T.; Strauss, J.; Haberl, M.; Gomez, C.C.; Reindl, M.; Saiz, A.; Fehling, H.J.; Wienands, J.; Odoardi, F.; et al. Autoantibody-boosted t-cell reactivation in the target organ triggers manifestation of autoimmune CNS disease. Proc. Natl. Acad. Sci. USA 2016, 113, 3323–3328. [Google Scholar] [CrossRef] [PubMed]
- Lino, A.C.; Dorner, T.; Bar-Or, A.; Fillatreau, S. Cytokine-producing B cells: A translational view on their roles in human and mouse autoimmune diseases. Immunol. Rev. 2016, 269, 130–144. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.; Fillatreau, S. Antibody-independent functions of B cells: A focus on cytokines. Nat. Rev. Immunol. 2015, 15, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Noorchashm, H.; Lieu, Y.K.; Noorchashm, N.; Rostami, S.Y.; Greeley, S.A.; Schlachterman, A.; Song, H.K.; Noto, L.E.; Jevnikar, A.M.; Barker, C.F.; et al. I-ag7-mediated antigen presentation by B lymphocytes is critical in overcoming a checkpoint in T cell tolerance to islet beta cells of nonobese diabetic mice. J. Immunol. 1999, 163, 743–750. [Google Scholar] [PubMed]
- O’Neill, S.K.; Shlomchik, M.J.; Glant, T.T.; Cao, Y.; Doodes, P.D.; Finnegan, A. Antigen-specific B cells are required as apcs and autoantibody-producing cells for induction of severe autoimmune arthritis. J. Immunol. 2005, 174, 3781–3788. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, S.K.; Cao, Y.; Hamel, K.M.; Doodes, P.D.; Hutas, G.; Finnegan, A. Expression of CD80/86 on B cells is essential for autoreactive T cell activation and the development of arthritis. J. Immunol. 2007, 179, 5109–5116. [Google Scholar] [CrossRef] [PubMed]
- Marino, E.; Tan, B.; Binge, L.; Mackay, C.R.; Grey, S.T. B-cell cross-presentation of autologous antigen precipitates diabetes. Diabetes 2012, 61, 2893–2905. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.K.; Maresz, K.; Shriver, L.P.; Tan, Y.; Dittel, B.N. B cell regulation of CD4+CD25+ T regulatory cells and IL-10 via B7 is essential for recovery from experimental autoimmune encephalomyelitis. J. Immunol. 2007, 178, 3447–3456. [Google Scholar] [CrossRef] [PubMed]
- Lund, F.E.; Randall, T.D. Effector and regulatory B cells: Modulators of CD4+ T cell immunity. Nat. Rev. Immunol. 2010, 10, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Basu, S.; Williams, C.B.; Salzman, N.H.; Dittel, B.N. A novel IL-10-independent regulatory role for B cells in suppressing autoimmunity by maintenance of regulatory T cells via GITR ligand. J. Immunol. 2012, 188, 3188–3198. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, T.; Horikawa, M.; Iwata, Y.; Tedder, T.F. Regulatory B cells (B10 cells) and regulatory T cells have independent roles in controlling experimental autoimmune encephalomyelitis initiation and late-phase immunopathogenesis. J. Immunol. 2010, 185, 2240–2252. [Google Scholar] [CrossRef] [PubMed]
- Ireland, S.J.; Blazek, M.; Harp, C.T.; Greenberg, B.; Frohman, E.M.; Davis, L.S.; Monson, N.L. Antibody-independent B cell effector functions in relapsing remitting multiple sclerosis: Clues to increased inflammatory and reduced regulatory B cell capacity. Autoimmunity 2012, 45, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Pierson, E.R.; Stromnes, I.M.; Goverman, J.M. B cells promote induction of experimental autoimmune encephalomyelitis by facilitating reactivation of T cells in the central nervous system. J. Immunol. 2014, 192, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.K.; Ray, A.; Basu, S.; Karp, C.L.; Dittel, B.N. Pathogenic and regulatory roles for B cells in experimental autoimmune encephalomyelitis. Autoimmunity 2012, 45, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Rezk, A.; Miyazaki, Y.; Hilgenberg, E.; Touil, H.; Shen, P.; Moore, C.S.; Michel, L.; Althekair, F.; Rajasekharan, S.; et al. Proinflammatory gm-csf-producing B cells in multiple sclerosis and B cell depletion therapy. Sci. Transl. Med. 2015, 7, 310ra166. [Google Scholar] [CrossRef] [PubMed]
- Lykken, J.M.; Candando, K.M.; Tedder, T.F. Regulatory B10 cell development and function. Int. Immunol. 2015, 27, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Miyagaki, T.; Fujimoto, M.; Sato, S. Regulatory B cells in human inflammatory and autoimmune diseases: From mouse models to clinical research. Int. Immunol. 2015, 27, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Silveira, P.A.; Johnson, E.; Chapman, H.D.; Bui, T.; Tisch, R.M.; Serreze, D.V. The preferential ability of B lymphocytes to act as diabetogenic APC in NOD mice depends on expression of self-antigen-specific immunoglobulin receptors. Eur. J. Immunol. 2002, 32, 3657–3666. [Google Scholar] [CrossRef]
- Yu, S.; Maiti, P.K.; Dyson, M.; Jain, R.; Braley-Mullen, H. B cell-deficient NOD.H-2h4 mice have CD4+CD25+ T regulatory cells that inhibit the development of spontaneous autoimmune thyroiditis. J. Exp. Med. 2006, 203, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Chan, O.T.; Hannum, L.G.; Haberman, A.M.; Madaio, M.P.; Shlomchik, M.J. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J. Exp. Med. 1999, 189, 1639–1648. [Google Scholar] [CrossRef] [PubMed]
- Giles, J.R.; Kashgarian, M.; Koni, P.A.; Shlomchik, M.J. B cell-specific mhc class II deletion reveals multiple nonredundant roles for B cell antigen presentation in murine lupus. J. Immunol. 2015, 195, 2571–2579. [Google Scholar] [CrossRef] [PubMed]
- Molnarfi, N.; Schulze-Topphoff, U.; Weber, M.S.; Patarroyo, J.C.; Prod’homme, T.; Varrin-Doyer, M.; Shetty, A.; Linington, C.; Slavin, A.J.; Hidalgo, J.; et al. MHC class II-dependent B cell APC function is required for induction of cns autoimmunity independent of myelin-specific antibodies. J. Exp. Med. 2013, 210, 2921–2937. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.A.; Kendall, P.L.; Thomas, J.W. Autoantigen-specific B-cell depletion overcomes failed immune tolerance in type 1 diabetes. Diabetes 2012, 61, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Leeth, C.M.; Racine, J.; Chapman, H.D.; Arpa, B.; Carrillo, J.; Carrascal, J.; Wang, Q.; Ratiu, J.; Egia-Mendikute, L.; Rosell-Mases, E.; et al. B-lymphocytes expressing an immunoglobulin specificity recognizing the pancreatic ß-cell autoantigen peripherin are potent contributors to type 1 diabetes development in NOD mice. Diabetes 2016, 65, 1977–1987. [Google Scholar] [CrossRef] [PubMed]
- Marino, E.; Villanueva, J.; Walters, S.; Liuwantara, D.; Mackay, F.; Grey, S.T. Cd4(+)CD25(+) T-cells control autoimmunity in the absence of B-cells. Diabetes 2009, 58, 1568–1577. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Ellis, J.S.; Dunn, R.; Kehry, M.R.; Braley-Mullen, H. Transient depletion of b cells in young mice results in activation of regulatory T cells that inhibit development of autoimmune disease in adults. Int. Immunol. 2012, 24, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Hamel, K.M.; Cao, Y.; Ashaye, S.; Wang, Y.; Dunn, R.; Kehry, M.R.; Glant, T.T.; Finnegan, A. B cell depletion enhances T regulatory cell activity essential in the suppression of arthritis. J. Immunol. 2011, 187, 4900–4906. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Sung, S.S.; Fu, S.M.; Ju, S.T. Regulation of multi-organ inflammation in the regulatory T cell-deficient scurfy mice. J. Biomed. Sci. 2009, 16, 20. [Google Scholar] [CrossRef] [PubMed]
- Aschermann, S.; Lehmann, C.H.; Mihai, S.; Schett, G.; Dudziak, D.; Nimmerjahn, F. B cells are critical for autoimmune pathology in scurfy mice. Proc. Natl. Acad. Sci. USA 2013, 110, 19042–19047. [Google Scholar] [CrossRef] [PubMed]
- Alard, P.; Thompson, C.; Agersborg, S.S.; Thatte, J.; Setiady, Y.; Samy, E.; Tung, K.S. Endogenous oocyte antigens are required for rapid induction and progression of autoimmune ovarian disease following day-3 thymectomy. J. Immunol. 2001, 166, 4363–4369. [Google Scholar] [CrossRef] [PubMed]
- Horie, I.; Abiru, N.; Sakamoto, H.; Iwakura, Y.; Nagayama, Y. Induction of autoimmune thyroiditis by depletion of CD4+CD25+ regulatory T cells in thyroiditis-resistant IL-17, but not interferon-gamma receptor, knockout nonobese diabetic-h2h4 mice. Endocrinology 2011, 152, 4448–4454. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, M.; Nagayama, Y.; Ichikawa, T.; Yu, L.; Eisenbarth, G.S.; Abiru, N. The effect of regulatory T-cell depletion on the spectrum of organ-specific autoimmune diseases in nonobese diabetic mice at different ages. Autoimmunity 2011, 44, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Nagayama, Y.; Horie, I.; Saitoh, O.; Nakahara, M.; Abiru, N. CD4+CD25+ naturally occurring regulatory T cells and not lymphopenia play a role in the pathogenesis of iodide-induced autoimmune thyroiditis in NOD-h2h4 mice. J. Autoimmun. 2007, 29, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Medling, B.; Yagita, H.; Braley-Mullen, H. Characteristics of inflammatory cells in spontaneous autoimmune thyroiditis of NOD.H-2h4 mice. J. Autoimmun. 2001, 16, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Karnell, J.L.; Mahmoud, T.I.; Herbst, R.; Ettinger, R. Discerning the kinetics of autoimmune manifestations in a model of Sjogren’s syndrome. Mol. Immunol. 2014, 62, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.Y.; Rodriguez-Pinto, D.; Du, W.; Ahuja, A.; Henegariu, O.; Wong, F.S.; Shlomchik, M.J.; Wen, L. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J. Clin. Investig. 2007, 117, 3857–3867. [Google Scholar] [CrossRef] [PubMed]
- Feldman, A.G.; Tucker, R.M.; Fenner, E.K.; Pelanda, R.; Mack, C.L. B cell deficient mice are protected from biliary obstruction in the rotavirus-induced mouse model of biliary atresia. PLoS ONE 2013, 8, e73644. [Google Scholar] [CrossRef] [PubMed]
- Olalekan, S.A.; Cao, Y.; Hamel, K.M.; Finnegan, A. B cells expressing ifn-gamma suppress treg-cell differentiation and promote autoimmune experimental arthritis. Eur. J. Immunol. 2015, 45, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Hoehlig, K.; Shen, P.; Lampropoulou, V.; Roch, T.; Malissen, B.; O’Connor, R.; Ries, S.; Hilgenberg, E.; Anderton, S.M.; Fillatreau, S. Activation of CD4(+) Foxp3(+) regulatory T cells proceeds normally in the absence of B cells during eae. Eur. J. Immunol. 2012, 42, 1164–1173. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Jensen, P.E. Cutting edge: Primary B lymphocytes preferentially expand allogeneic Foxp3+ CD4 T cells. J. Immunol. 2007, 179, 2046–2050. [Google Scholar] [CrossRef] [PubMed]
- Walters, S.N.; Webster, K.E.; Daley, S.; Grey, S.T. A role for intrathymic B cells in the generation of natural regulatory T cells. J. Immunol. 2014, 193, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.T.; Yang, W.; Wang, Y.H.; Ma, H.D.; Tang, W.; Yang, J.B.; Li, L.; Ansari, A.A.; Lian, Z.X. Thymic B cells promote thymus-derived regulatory T cell development and proliferation. J. Autoimmun. 2015, 61, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.B.; Flach, C.F.; Czerkinsky, C.; Holmgren, J. B lymphocytes promote expansion of regulatory T cells in oral tolerance: Powerful induction by antigen coupled to cholera toxin B subunit. J. Immunol. 2008, 181, 8278–8287. [Google Scholar] [CrossRef] [PubMed]
- Shao, T.Y.; Hsu, L.H.; Chien, C.H.; Chiang, B.L. Novel Foxp3(-) IL-10(-) regulatory T-cells induced by B-cells alleviate intestinal inflammation in vivo. Sci. Rep. 2016, 6, 32415. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.H.; Li, K.P.; Chu, K.H.; Chiang, B.L. A B-1A cell subset induces Foxp3− T cells with regulatory activity through an IL-10-independent pathway. Cell. Mol. Immunol. 2015, 12, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.S.; Prod’homme, T.; Patarroyo, J.C.; Molnarfi, N.; Karnezis, T.; Lehmann-Horn, K.; Danilenko, D.M.; Eastham-Anderson, J.; Slavin, A.J.; Linington, C.; et al. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann. Neurol. 2010, 68, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Peng, J.; Tai, N.; Hu, C.; Zhou, Z.; Wong, F.S.; Wen, L. The dual effects of b cell depletion on antigen-specific T cells in BDC2.5NOD mice. J. Immunol. 2012, 188, 4747–4758. [Google Scholar] [CrossRef] [PubMed]
- Tadmor, T.; Zhang, Y.; Cho, H.M.; Podack, E.R.; Rosenblatt, J.D. The absence of b lymphocytes reduces the number and function of T-regulatory cells and enhances the anti-tumor response in a murine tumor model. Cancer Immunol. Immunother. 2011, 60, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Visperas, A.; Vignali, D.A.A. Are regulatory T cells defective in type 1 diabetes and can we fix them? J. Immunol. 2016, 197, 3762–3770. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.S.; Braley-Mullen, H. Regulatory t cells in B-cell-deficient and wild-type mice differ functionally and in expression of cell surface markers. Immunology 2015, 144, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Fillatreau, S. Pathogenic functions of b cells in autoimmune diseases: IFN-gamma production joins the criminal gang. Eur. J. Immunol. 2015, 45, 966–970. [Google Scholar] [CrossRef] [PubMed]
- Barr, T.A.; Shen, P.; Brown, S.; Lampropoulou, V.; Roch, T.; Lawrie, S.; Fan, B.; O’Connor, R.A.; Anderton, S.M.; Bar-Or, A.; et al. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J. Exp. Med. 2012, 209, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Ehrenstein, M.R.; Evans, J.G.; Singh, A.; Moore, S.; Warnes, G.; Isenberg, D.A.; Mauri, C. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J. Exp. Med. 2004, 200, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Olson, T.S.; Bamias, G.; Naganuma, M.; Rivera-Nieves, J.; Burcin, T.L.; Ross, W.; Morris, M.A.; Pizarro, T.T.; Ernst, P.B.; Cominelli, F.; et al. Expanded B cell population blocks regulatory T cells and exacerbates ileitis in a murine model of crohn disease. J. Clin. Investig. 2004, 114, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.B.; Liao, G.; Faubion, W.A.; Abadia-Molina, A.C.; Cozzo, C.; Laroux, F.S.; Caton, A.; Terhorst, C. Cutting edge: The natural ligand for glucocorticoid-induced TNF receptor-related protein abrogates regulatory T cell suppression. J. Immunol. 2004, 172, 5823–5827. [Google Scholar] [CrossRef] [PubMed]
- Ephrem, A.; Epstein, A.L.; Stephens, G.L.; Thornton, A.M.; Glass, D.; Shevach, E.M. Modulation of treg cells/T effector function by GITR signaling is context-dependent. Eur. J. Immunol. 2013, 43, 2421–2429. [Google Scholar] [CrossRef] [PubMed]
- Nowakowska, D.J.; Kissler, S. Ptpn22 modifies regulatory T cell homeostasis via GITR upregulation. J. Immunol. 2016, 196, 2145–2152. [Google Scholar] [CrossRef] [PubMed]
- Ronchetti, S.; Ricci, E.; Petrillo, M.G.; Cari, L.; Migliorati, G.; Nocentini, G.; Riccardi, C. Glucocorticoid-induced tumour necrosis factor receptor-related protein: A key marker of functional regulatory T cells. J. Immunol. Res. 2015, 2015, 171520. [Google Scholar] [CrossRef] [PubMed]
- Egwuagu, C.E.; Yu, C.R. Interleukin 35-producing B cells (i35-breg): A new mediator of regulatory B-cell functions in CNS autoimmune diseases. Crit. Rev. Immunol. 2015, 35, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Rosser, E.C.; Oleinika, K.; Tonon, S.; Doyle, R.; Bosma, A.; Carter, N.A.; Harris, K.A.; Jones, S.A.; Klein, N.; Mauri, C. Regulatory B cells are induced by gut microbiota-driven interleukin-1beta and interleukin-6 production. Nat. Med. 2014, 20, 1334–1339. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.X.; Yu, C.R.; Dambuza, I.M.; Mahdi, R.M.; Dolinska, M.B.; Sergeev, Y.V.; Wingfield, P.T.; Kim, S.H.; Egwuagu, C.E. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat. Med. 2014, 20, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Wang, L.; Dittel, B.N. IL-10-independent regulatory B-cell subsets and mechanisms of action. Int. Immunol. 2015, 27, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.; Roch, T.; Lampropoulou, V.; O’Connor, R.A.; Stervbo, U.; Hilgenberg, E.; Ries, S.; Dang, V.D.; Jaimes, Y.; Daridon, C.; et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 2014, 507, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Sun, L.; Fan, X.; Wang, Z.; Cheng, Y.; Zhu, J.; Jin, T. Role of regulatory B cells in neuroimmunologic disorders. J. Neurosci. Res. 2016, 94, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Mauri, C.; Menon, M. The expanding family of regulatory B cells. Int. Immunol. 2015, 27, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Ramsdell, F.; Ziegler, S.F. Foxp3 and scurfy: How it all began. Nat. Rev. Immunol. 2014, 14, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Kohm, A.P.; McMahon, J.S.; Podojil, J.R.; Begolka, W.S.; DeGutes, M.; Kasprowicz, D.J.; Ziegler, S.F.; Miller, S.D. Cutting edge: Anti-CD25 monoclonal antibody injection results in the functional inactivation, not depletion, of CD4+CD25+ T regulatory cells. J. Immunol. 2006, 176, 3301–3305. [Google Scholar] [CrossRef] [PubMed]
- Harakal, J.; Rival, C.; Qiao, H.; Tung, K.S. Regulatory T cells control TH2-dominant murine autoimmune gastritis. J. Immunol. 2016, 197, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Teh, C.E.; Gray, D.H. Can you rely on treg cells on the rebound? Eur. J. Immunol. 2014, 44, 3504–3507. [Google Scholar] [CrossRef] [PubMed]
- Nystrom, S.N.; Bourges, D.; Garry, S.; Ross, E.M.; van Driel, I.R.; Gleeson, P.A. Transient treg-cell depletion in adult mice results in persistent self-reactive CD4(+) T-cell responses. Eur. J. Immunol. 2014, 44, 3621–3631. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.W.; Ngiow, S.F.; von Scheidt, B.; McLaughlin, N.; Sparwasser, T.; Smyth, M.J. Conditional regulatory T-cell depletion releases adaptive immunity preventing carcinogenesis and suppressing established tumor growth. Cancer Res. 2010, 70, 7800–7809. [Google Scholar] [CrossRef] [PubMed]
- Klages, K.; Mayer, C.T.; Lahl, K.; Loddenkemper, C.; Teng, M.W.; Ngiow, S.F.; Smyth, M.J.; Hamann, A.; Huehn, J.; Sparwasser, T. Selective depletion of Foxp3+ regulatory T cells improves effective therapeutic vaccination against established melanoma. Cancer Res. 2010, 70, 7788–7799. [Google Scholar] [CrossRef] [PubMed]
- Dietze, K.K.; Zelinskyy, G.; Gibbert, K.; Schimmer, S.; Francois, S.; Myers, L.; Sparwasser, T.; Hasenkrug, K.J.; Dittmer, U. Transient depletion of regulatory T cells in transgenic mice reactivates virus-specific CD8+ T cells and reduces chronic retroviral set points. Proc. Natl. Acad. Sci. USA 2011, 108, 2420–2425. [Google Scholar] [CrossRef] [PubMed]
- Bos, P.D.; Plitas, G.; Rudra, D.; Lee, S.Y.; Rudensky, A.Y. Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. J. Exp. Med. 2013, 210, 2435–2466. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.P.; Piconese, S. Regulatory-T-cell inhibition versus depletion: The right choice in cancer immunotherapy. Nat. Rev. Cancer 2007, 7, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Rech, A.J.; Mick, R.; Martin, S.; Recio, A.; Aqui, N.A.; Powell, D.J., Jr.; Colligon, T.A.; Trosko, J.A.; Leinbach, L.I.; Pletcher, C.H.; et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci. Transl. Med. 2012, 4, 134ra62. [Google Scholar] [CrossRef] [PubMed]
- Duhen, T.; Duhen, R.; Lanzavecchia, A.; Sallusto, F.; Campbell, D.J. Functionally distinct subsets of human Foxp3+ treg cells that phenotypically mirror effector TH cells. Blood 2012, 119, 4430–4440. [Google Scholar] [CrossRef] [PubMed]
- Hori, S. Lineage stability and phenotypic plasticity of Foxp3+ regulatory T cells. Immunol. Rev. 2014, 259, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Koenecke, C.; Lee, C.-W.; Thamm, K.; Föhse, L.; Schafferus, M.; Mittrücker, H.-W.; Floess, S.; Huehn, J.; Ganser, A.; Förster, R.; et al. IFN-γ production by allogeneic Foxp3+ regulatory T cells is essential for preventing experimental graft-versus-host disease. J. Immunol. 2012, 189, 2890–2896. [Google Scholar] [CrossRef] [PubMed]
- Leonardo, S.M.; De Santis, J.L.; Malherbe, L.P.; Gauld, S.B. Cutting edge: In the absence of regulatory T cells, a unique th cell population expands and leads to a loss of B cell anergy. J. Immunol. 2012, 188, 5223–5226. [Google Scholar] [CrossRef] [PubMed]
- Leonardo, S.M.; De Santis, J.L.; Gehrand, A.; Malherbe, L.P.; Gauld, S.B. Expansion of follicular helper T cells in the absence of treg cells: Implications for loss of B-cell anergy. Eur. J. Immunol. 2012, 42, 2597–2607. [Google Scholar] [CrossRef] [PubMed]
- Leonardo, S.M.; Josephson, J.A.; Hartog, N.L.; Gauld, S.B. Altered B cell development and anergy in the absence of Foxp3. J. Immunol. 2010, 185, 2147–2156. [Google Scholar] [CrossRef] [PubMed]
- Riewaldt, J.; Düber, S.; Boernert, M.; Dembinski, M.; Weiss, S.; Garbe, A.; Kretschmer, K. Severe developmental B lymphopoietic defects in Foxp3-deficient mice are refractory to adoptive regulatory T cell therapy. Front. Immunol. 2012, 3, 141. [Google Scholar] [CrossRef] [PubMed]
- Mercadante, E.R.; Lorenz, U.M. Breaking free of control: How conventional T cells overcome regulatory T cell suppression. Front. Immunol. 2016, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Sanz, I.; Anolik, J.H.; Looney, R.J. B cell depletion therapy in autoimmune diseases. Front. Biosci. 2007, 12, 2546–2567. [Google Scholar] [CrossRef] [PubMed]
- Uchida, J.; Lee, Y.; Hasegawa, M.; Liang, Y.; Bradney, A.; Oliver, J.A.; Bowen, K.; Steeber, D.A.; Haas, K.M.; Poe, J.C.; et al. Mouse CD20 expression and function. Int. Immunol. 2004, 16, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Wong, F.S.; Wen, L. Translational mini-review series on B cell-directed therapies: B cell-directed therapy for autoimmune diseases. Clin. Exp. Immunol. 2009, 157, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Faurschou, M.; Jayne, D.R. Anti-B cell antibody therapies for inflammatory rheumatic diseases. Annu. Rev. Med. 2014, 65, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, J.W.; Jayne, D.R. B-cell depletion in the treatment of lupus nephritis. Nat. Rev. Nephrol. 2012, 8, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Braley-Mullen, H. Follicular B cells in thyroids of mice with spontaneous autoimmune thyroiditis contribute to disease pathogenesis and are targets of anti-CD20 antibody therapy. J. Immunol. 2014, 192, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, A.; Shupe, J.; Dunn, R.; Kashgarian, M.; Kehry, M.R.; Shlomchik, M.J. Depletion of B cells in murine lupus: Efficacy and resistance. J. Immunol. 2007, 179, 3351–3361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.H.; Skupsky, J.; Scott, D.W. Effect of B-cell depletion using anti-CD20 therapy on inhibitory antibody formation to human FVIII in hemophilia a mice. Blood 2011, 117, 2223–2226. [Google Scholar] [CrossRef] [PubMed]
- Stolp, J.; Marino, E.; Batten, M.; Sierro, F.; Cox, S.L.; Grey, S.T.; Silveira, P.A. Intrinsic molecular factors cause aberrant expansion of the splenic marginal zone B cell population in nonobese diabetic mice. J. Immunol. 2013, 191, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Marino, E.; Batten, M.; Groom, J.; Walters, S.; Liuwantara, D.; Mackay, F.; Grey, S.T. Marginal-zone B-cells of nonobese diabetic mice expand with diabetes onset, invade the pancreatic lymph nodes, and present autoantigen to diabetogenic T-cells. Diabetes 2008, 57, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Gao, C.; Suresh, L.; Xian, Z.; Song, N.; Chaves, L.D.; Yu, M.; Ambrus, J.L., Jr. Central role for marginal zone B cells in an animal model of Sjogren’s syndrome. Clin. Immunol. 2016, 168, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Niu, H.; Zheng, Y.Y.; Morel, L. Autoreactive marginal zone B cells enter the follicles and interact with CD4+ T cells in lupus-prone mice. BMC Immunol. 2011, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.; Cariappa, A.; Moran, S.T. Marginal zone B cells. Annu. Rev. Immunol. 2005, 23, 161–196. [Google Scholar] [CrossRef] [PubMed]
- Bodogai, M.; Lee Chang, C.; Wejksza, K.; Lai, J.; Merino, M.; Wersto, R.P.; Gress, R.E.; Chan, A.C.; Hesdorffer, C.; Biragyn, A. Anti-CD20 antibody promotes cancer escape via enrichment of tumor-evoked regulatory B cells expressing low levels of CD20 and CD137L. Cancer Res. 2013, 73, 2127–2138. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Ireland, S.J.; Davis, L.S.; Kong, X.; Stowe, A.M.; Wang, Y.; White, W.I.; Herbst, R.; Monson, N.L. Autoreactive CD19+CD20- plasma cells contribute to disease severity of experimental autoimmune encephalomyelitis. J. Immunol. 2016, 196, 1541–1549. [Google Scholar] [CrossRef] [PubMed]
- Gong, Q.; Ou, Q.; Ye, S.; Lee, W.P.; Cornelius, J.; Diehl, L.; Lin, W.Y.; Hu, Z.; Lu, Y.; Chen, Y.; et al. Importance of cellular microenvironment and circulatory dynamics in B cell immunotherapy. J. Immunol. 2005, 174, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Leandro, M.J. B-cell subpopulations in humans and their differential susceptibility to depletion with anti-CD20 monoclonal antibodies. Arthritis Res. Ther. 2013, 15, S3. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.A.; Rakocevic, G.; Leung, C.S.; Quast, I.; Lukacisin, M.; Goebels, N.; Munz, C.; Wardemann, H.; Dalakas, M.; Lunemann, J.D. Rituximab induces sustained reduction of pathogenic B cells in patients with peripheral nervous system autoimmunity. J. Clin. Investig. 2012, 122, 1393–1402. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Seshasayee, D.; Lee, W.P.; Caplazi, P.; McVay, S.; Suto, E.; Nguyen, A.; Lin, Z.; Sun, Y.; DeForge, L.; et al. Dual B cell immunotherapy is superior to individual anti-CD20 depletion or baff blockade in murine models of spontaneous or accelerated lupus. Arthritis Rheumatol. 2015, 67, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Roll, P.; Tony, H.P. B-cell-targeted therapy in the treatment of autoimmune diseases. Z. Rheumatol. 2009, 68, 255–259. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ellis, J.S.; Braley-Mullen, H. Mechanisms by Which B Cells and Regulatory T Cells Influence Development of Murine Organ-Specific Autoimmune Diseases. J. Clin. Med. 2017, 6, 13. https://doi.org/10.3390/jcm6020013
Ellis JS, Braley-Mullen H. Mechanisms by Which B Cells and Regulatory T Cells Influence Development of Murine Organ-Specific Autoimmune Diseases. Journal of Clinical Medicine. 2017; 6(2):13. https://doi.org/10.3390/jcm6020013
Chicago/Turabian StyleEllis, Jason S., and Helen Braley-Mullen. 2017. "Mechanisms by Which B Cells and Regulatory T Cells Influence Development of Murine Organ-Specific Autoimmune Diseases" Journal of Clinical Medicine 6, no. 2: 13. https://doi.org/10.3390/jcm6020013
APA StyleEllis, J. S., & Braley-Mullen, H. (2017). Mechanisms by Which B Cells and Regulatory T Cells Influence Development of Murine Organ-Specific Autoimmune Diseases. Journal of Clinical Medicine, 6(2), 13. https://doi.org/10.3390/jcm6020013