Discovery of Potential Plant-Derived Peptide Deformylase (PDF) Inhibitors for Multidrug-Resistant Bacteria Using Computational Studies

Abstract
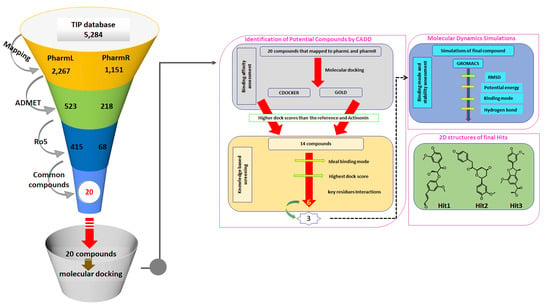
:
1. Introduction
2. Experimental Section
2.1. Ligand-Based Approach
2.1.1. Dataset Construction and Its Composition
2.1.2. Generation of the Pharmacophore Model
2.2. Generation of the Receptor-Based Pharmacophore Model
2.3. Validation of the Pharmacophore Models
2.3.1. Ligand-Based Pharmacophore Model Validation
2.3.2. Receptor-Based Pharmacophore Model Validation
2.3.3. Decoy Set Method of Validation
2.4. Virtual Screening of the TIP Database
2.5. Drug-Like Assessment
2.6. Molecular Docking Studies
2.7. Molecular Dynamics Simulation Studies
2.8. Novelty Assessment of the Compounds
3. Results
3.1. Generation of the Pharmacophore Model
3.1.1. Ligand-Based Pharmacophore Generation
3.1.2. Generation of Structure-Based Pharmacophore Generation
3.2. Validation of the Pharmacophore Models
3.2.1 Validation of PharmL
Fischer’s Randomization Method
Test Set Method
3.2.2. Validation of PharmR
Receiver Operating Characteristic (ROC) Plot Analysis
Decoy Set Method of Validation for PharmL and PharmR
3.3. Virtual Screening of Taiwan Indigenous Plants (TIP) Database
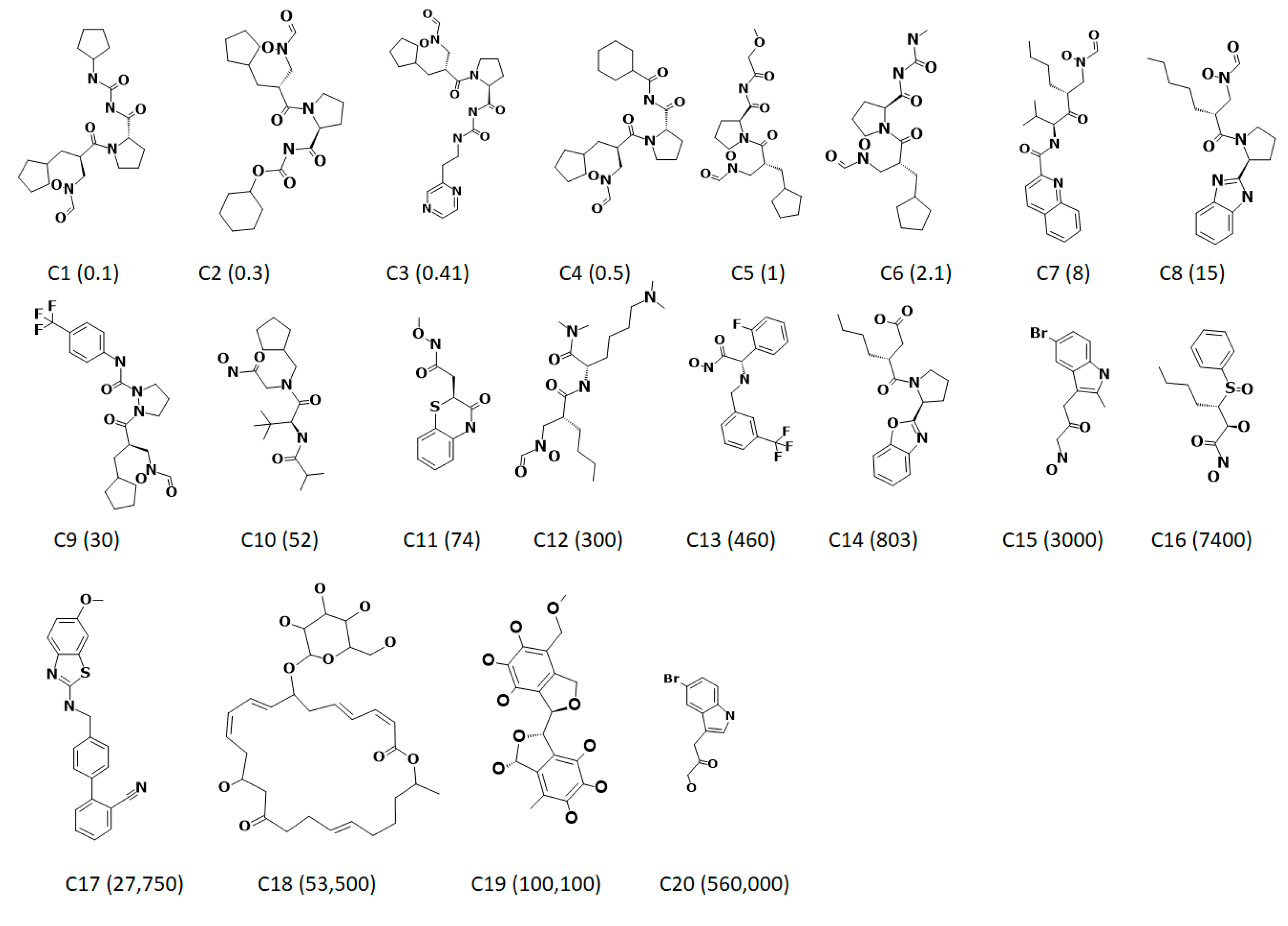
3.4. Molecular Docking-Based Screening
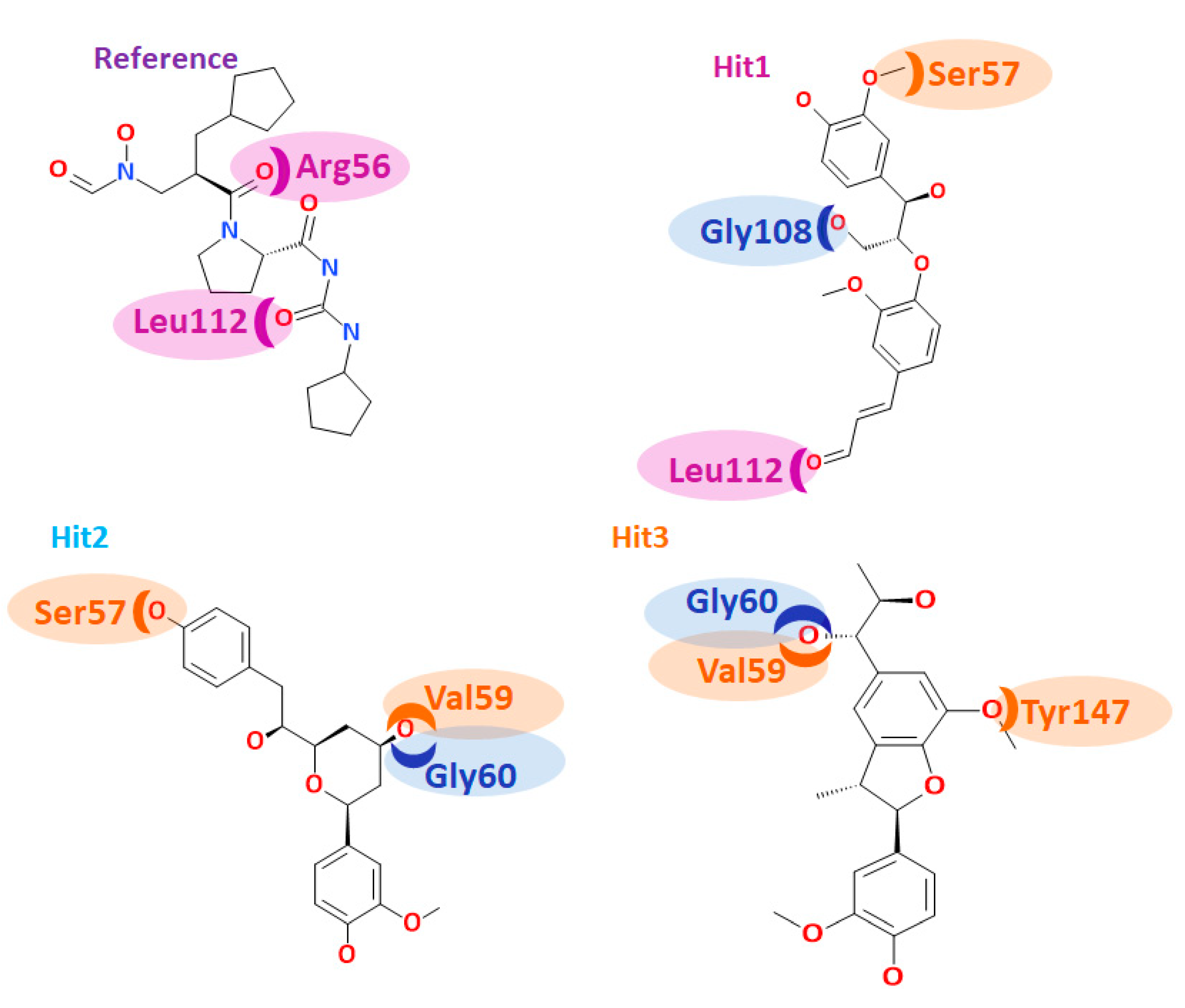
3.5. Molecular Dynamics Simulations
3.6. Probing the Novelty of the Hits
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lee, S.J.; Lee, S.J.; Lee, S.K.; Yoon, H.J.; Lee, H.H.; Kim, K.K.; Lee, B.J.; Lee, B., II; Suh, S.W. Structures of staphylococcus aureus peptide deformylase in complex with two classes of new inhibitors. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Ananthula, R.S.; Ravikumar, M.; Mahmood, S.K.; Kumar, M.N.S.P. Insights from ligand and structure based methods in virtual screening of selective Ni-peptide deformylase inhibitors. J. Mol. Model. 2012, 18, 693–708. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Cheng, Y.; Cui, W.; Zhang, F.; Zhang, H.; Du, Y.; Ji, M. prediction of the binding modes between macrolactin N and peptide deformylase from staphylococcus aureus by molecular docking and molecular dynamics simulations. Med. Chem. Res. 2013, 22, 2889–2901. [Google Scholar] [CrossRef]
- Zaman, S.B.; Hussain, M.A.; Nye, R.; Mehta, V.; Mamun, K.T.; Hossain, N. A review on antibiotic resistance: Alarm bells are ringing. Cureus 2017. [Google Scholar] [CrossRef] [PubMed]
- Spellberg, B.; Blaser, M.; Guidos, R.J.; Boucher, H.W.; Bradley, J.S.; Eisenstein, B.I.; Gerding, D.; Lynfield, R.; Reller, L.B.; Rex, J.; et al. Combating antimicrobial resistance: Policy recommendations to save lives. Clin. Infect. Dis. 2011, 52 (Suppl. 5). [Google Scholar] [CrossRef]
- Van Duin, D.; Paterson, D.L. Multidrug-resistant bacteria in the community: Trends and lessons learned. Infect. Dis. Clin. N. Am. 2016, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Tanwar, J.; Das, S.; Fatima, Z.; Hameed, S. Multidrug resistance: An emerging crisis. Interdiscip. Perspect. Infect. Dis. 2014. [Google Scholar] [CrossRef] [PubMed]
- Hooper, D.C. Mechanisms of action and resistance of older and newer fluoroquinolones. Clin. Infect. Dis. 2000, 31 (Suppl. 2), 24–28. [Google Scholar] [CrossRef]
- Weisblum, B. Erythromycin resistance by ribosome modification. Antimicrob. Agents Chemother. 1995, 577–585. [Google Scholar] [CrossRef]
- Poehlsgaard, J.; Douthwaite, S. The bacterial ribosome as a target for antibiotics. Nat. Rev. Microbiol. 2005, 3, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Wright, G.D. Bacterial resistance to aminoglycoside antibiotics. Trends Microbiol. 1997, 234–240. [Google Scholar] [CrossRef]
- Shaw, K.J.; Rather, P.N.; Hare, R.S.; Miller, G.H. Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol. Rev. 1993, 57, 138–163. [Google Scholar] [CrossRef] [PubMed]
- Benveniste, R.; Davies, J. Aminoglycoside antibiotic-inactivating enzymes in actinomycetes similar to those present in clinical isolates of antibiotic-resistant bacteria. Proc. Natl. Acad. Sci. USA 1973, 70, 2276–2280. [Google Scholar] [CrossRef] [PubMed]
- Datta, N.; Kontomichalou, P. Penicillinase synthesis controlled by infectious R factors in Enterobacteriaceae. Nature 1965, 208, 239–241. [Google Scholar] [CrossRef] [PubMed]
- Queenan, A.M.; Bush, K. Carbapenemases: The versatile β-lactamases. Clin. Microbiol. Rev. 2007, 440–458. [Google Scholar] [CrossRef] [PubMed]
- Robicsek, A.; Strahilevitz, J.; Jacoby, G.A.; Macielag, M.; Abbanat, D.; Chi, H.P.; Bush, K.; Hooper, D.C. Fluoroquinolone-modifying enzyme: A new adaptation of a common aminoglycoside acetyltransferase. Nat. Med. 2006, 12, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Spratt, B. Resistance to antibiotics mediated by target alterations. Science 1994, 264, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Connell, S.R.; Tracz, D.M.; Nierhaus, K.H.; Taylor, D.E. Ribosomal protection proteins and their mechanism of tetracycline resistance. Antimicrob. Agents Chemother. 2003, 3675–3681. [Google Scholar] [CrossRef]
- Achouak, W.; Heulin, T.; Pagès, J.M. Multiple facets of bacterial porins. FEMS Microbiol. Lett. 2001, 1–7. [Google Scholar] [CrossRef]
- Tamura, N.; Konishi, S.; Yamaguchi, A. Mechanisms of drug/H+ antiport: Complete cysteine-scanning mutagenesis and the protein engineering approach. Curr. Opin. Chem. Biol. 2003, 570–579. [Google Scholar] [CrossRef]
- Robicsek, A.; Jacoby, G.A.; Hooper, D.C. The worldwide emergence of plasmid-mediated quinolone resistance. Lancet Infect. Dis. 2006, 629–640. [Google Scholar] [CrossRef]
- Loeffler, J.; Stevens, D.A. Antifungal drug resistance. Clin. Infect. Dis. 2003, 36, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Khalilzadeh, S.; Boloorsaz, M.R.; Safavi, A.; Farnia, P.; Velayati, A.A. Primary and acquired drug resistance in childhood tuberculosis. East. Mediterr. Heal. J. 2006, 12, 909–914. [Google Scholar]
- Marks, S.M.; Flood, J.; Seaworth, B.; Hirsch-Moverman, Y.; Armstrong, L.; Mase, S.; Salcedo, K.; Oh, P.; Graviss, E.A.; Colson, P.W.; et al. Treatment practices, outcomes, and costs of multidrug-resistant and extensively drug-resistant tuberculosis, united states, 2005–2007. Emerg. Infect. Dis. 2014, 20, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Hackbarth, C.; Ni, Z.J.; Wu, C.; Wang, W.; Jain, R.; He, Y.; Bracken, K.; Weidmann, B.; Patel, D.V.; et al. Peptide deformylase inhibitors as antibacterial agents: Identification of VRC3375, a proline-3-alkylsuccinyl hydroxamate derivative, by using an integrated combinatorial and medicinal chemistry approach. Antimicrob. Agents Chemother. 2004. [Google Scholar] [CrossRef]
- Lv, F.; Chen, C.; Tang, Y.; Wei, J.; Zhu, T.; Hu, W. New peptide deformylase inhibitors design, synthesis and pharmacokinetic assessment. Bioorganic Med. Chem. Lett. 2016, 26, 3714–3718. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, F.; Genilloud, O. Antimicrobials: New and Old Molecules in the Fight against Multi-Resistant Bacteria; Springer International Publishing: Cham, Switzerland, 2014. [Google Scholar]
- Gordon, J.J.; Kelly, B.K.; Miller, G.A. Actinonin: An antibiotic substance produced by an actinomycete. Nature 1962, 195, 701–702. [Google Scholar] [CrossRef] [PubMed]
- Fieulaine, S.; Alves De Sousa, R.; Maigre, L.; Hamiche, K.; Alimi, M.; Bolla, J.M.; Taleb, A.; Denis, A.; Pagès, J.M.; Artaud, I.; et al. A unique peptide deformylase platform to rationally design and challenge novel active compounds. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Antczak, C.; Shum, D.; Escobar, S.; Bassit, B.; Kim, E.; Seshan, V.E.; Wu, N.; Yang, G.; Ouerfelli, O.; Li, Y.M.; et al. High-throughput identification of inhibitors of human mitochondrial peptide deformylase. J. Biomol. Screen. 2007, 12, 521–535. [Google Scholar] [CrossRef]
- Lee, M.D.; Antczak, C.; Li, Y.; Sirotnak, F.M.; Bornmann, W.G.; Scheinberg, D.A. A new human peptide deformylase inhibitable by actinonin. Biochem. Biophys. Res. Commun. 2003, 312, 309–315. [Google Scholar] [CrossRef]
- Lee, M.D.; She, Y.; Soskis, M.J.; Borella, C.P.; Gardner, J.R.; Hayes, P.A.; Dy, B.M.; Heaney, M.L.; Philips, M.R.; Bornmann, W.G.; et al. Human mitochondrial peptide deformylase, a new anticancer target of actinonin-based antibiotics. J. Clin. Investig. 2004, 114, 1107–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grujić, M.; Renko, M. aminopeptidase inhibitors bestatin and actinonin inhibit cell proliferation of myeloma cells predominantly by intracellular interactions. Cancer Lett. 2002, 182, 113–119. [Google Scholar] [CrossRef]
- Apfel, C.; Banner, D.W.; Bur, D.; Dietz, M.; Hirata, T.; Hubschwerlen, C.; Locher, H.; Page, M.G.; Pirson, W.; Rossé, G.; et al. Hydroxamic acid derivatives as potent peptide deformylase inhibitors and antibacterial agents. J. Med. Chem. 2000, 43, 2324–2331. [Google Scholar] [CrossRef] [PubMed]
- Goemaere, E.; Melet, A.; Larue, V.; Lieutaud, A.; Alves de sousa, R.; Chevalier, J.; Yimga-djapa, L.; Giglione, C.; Huguet, F.; Alimi, M.; et al. New peptide deformylase inhibitors and cooperative interaction: a combination to improve antibacterial activity. J. Antimicrob. Chemother. 2012, 67, 1392–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margolis, P.; Hackbarth, C.; Lopez, S.; Maniar, M.; Wang, W.; Yuan, Z.; White, R.; Trias, J. Resistance of streptococcus pneumoniae to deformylase inhibitors is due to mutations in defB. Antimicrob. Agents Chemother. 2001, 45, 2432–2435. [Google Scholar] [CrossRef]
- Zorzet, A.; Andersen, J.M.; Nilsson, A.I.; Møller, N.F.; Andersson, D.I. Compensatory mutations in AgrC partly restore fitness in vitro to peptide deformylase inhibitor-resistant staphylococcus aureus. J. Antimicrob. Chemother. 2012, 67, 1835–1842. [Google Scholar] [CrossRef] [PubMed]
- Guilloteau, J.P.; Mathieu, M.; Giglione, C.; Blanc, V.; Dupuy, A.; Chevrier, M.; Gil, P.; Famechon, A.; Meinnel, T.; Mikol, V. The crystal structures of four peptide deformylases bound to the antibiotic actinonin reveal two distinct types: A platform for the structure-based design of antibacterial agents. J. Mol. Biol. 2002, 320, 951–962. [Google Scholar] [CrossRef]
- Liang, L.; Zhou, Q.; Hao, Z.; Wang, F.; Zhu, Y.; Lin, Q.; Gao, J. The discovery of antibacterial natural compound based on peptide deformylase. Comb. Chem. High Throughput Screen. 2018, 21, 292–297. [Google Scholar] [CrossRef]
- Robien, M.A. An improved crystal form of plasmodium falciparum peptide deformylase. Protein Sci. 2004, 13, 1155–1163. [Google Scholar] [CrossRef]
- Nguyen, K.T.; Hu, X.; Colton, C.; Chakrabarti, R.; Zhu, M.X.; Pei, D. Characterization of a human peptide deformylase: implications for antibacterial drug design. Biochemistry 2003, 42, 9952–9958. [Google Scholar] [CrossRef]
- Boularot, A.; Giglione, C.; Petit, S.; Duroc, Y.; De Sousa, R.A.; Larue, V.; Cresteil, T.; Dardel, F.; Artaud, I.; Meinnel, T. Discovery and refinement of a new structural class of potent peptide deformylase inhibitors. J. Med. Chem. 2007, 50, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Petit, S.; Duroc, Y.; Larue, V.; Giglione, C.; Léon, C.; Soulama, C.; Denis, A.; Dardel, F.; Meinnel, T.; Artaud, I. Structure-activity relationship analysis of the peptide deformylase inhibitor 5-bromo-1h-indole-3-acetohydroxamic acid. ChemMedChem 2009, 4, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Petrovska, B. Historical review of medicinal plants’ usage. Pharmacogn. Rev. 2012, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Osbourn, A.E. Preformed antimicrobial compounds and plant defense against fungal attack. Plant Cell. 1996, 8, 1821–1831. [Google Scholar] [CrossRef] [PubMed]
- Tagboto, S.; Townson, S. Antiparasitic properties of medicinal plants and other naturally occurring products. Adv. Parasitol. 2001, 199–295. [Google Scholar] [CrossRef]
- Nash, R.J.; Kato, A.; Yu, C.-Y.; Fleet, G.W. Iminosugars as therapeutic agents: recent advances and promising trends. Future Med. Chem. 2011, 3, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Arruda, A.L.A.; Vieira, C.J.B.; Sousa, D.G.; Oliveira, R.F.; Castilho, R.O. Jacaranda Cuspidifolia Mart. (Bignoniaceae) as an antibacterial agent. J. Med. Food 2011, 14, 1604–1608. [Google Scholar] [CrossRef]
- Ahmad, I.; Aqil, F.; Owais, M. Modern Phytomedicine: Turning Medicinal Plants into Drugs; John Wiley & Sons: Hoboken, NJ, USA, 2006. [Google Scholar] [CrossRef]
- Savoia, D. Plant-derived antimicrobial compounds: alternatives to antibiotics. Future Microbiol. 2012, 7, 979–990. [Google Scholar] [CrossRef]
- Cowan, M.M. Plant products as antimicrobial agents. Clin. Microbiol. Rev. 1999, 12, 564–582. [Google Scholar] [CrossRef]
- Subramani, R.; Narayanasamy, M.; Feussner, K.D. Plant-derived antimicrobials to fight against multi-drug-resistant human pathogens. 3 Biotech 2017. [Google Scholar] [CrossRef]
- Rampogu, S.; Baek, A.; Gajula, R.G.; Zeb, A.; Bavi, R.S.; Kumar, R.; Kim, Y.; Kwon, Y.J.; Lee, K.W. Ginger (Zingiber officinale) phytochemicals-gingerenone—A and shogaol inhibit SaHPPK: Molecular docking, molecular dynamics simulations and in vitro approaches. Ann. Clin. Microbiol. Antimicrob. 2018, 17, 16. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, P.; Purkayastha, S. Phytochemical screening and antimicrobial activity of some medicinal plants against multi-drug resistant bacteria from clinical isolates. Indian J. Pharm Sci. 2012, 74, 443–450. [Google Scholar] [CrossRef]
- Al-Saleem, M.S.; Awaad, A.S.; Alothman, M.R.; Alqasoumi, S.I. Phytochemical standardization and biological activities of certain desert plants growing in Saudi Arabia. Saudi Pharm. J. 2018, 26, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Borges, A.; Abreu, A.C.; Dias, C.; Saavedra, M.J.; Borges, F.; Simoes, M. New perspectives on the use of phytochemicals as an emergent strategy to control bacterial infections including biofilms. Molecules 2016, 21, 877. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Prabhakaran, V.S.; Kim, K. sun. The multi-faceted potential of plant-derived metabolites as antimicrobial agents against multidrug-resistant pathogens. Microbial Pathogenesis. 2018, 209–214. [Google Scholar] [CrossRef]
- Chew, Y.L.; Mahadi, A.M.; Wong, K.M.; Goh, J.K. Anti-methicillin-resistance Staphylococcus aureus (MRSA) Compounds from Bauhinia kockiana Korth. and their mechanism of antibacterial activity. BMC Complement. Altern. Med. 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Khalil, N.; Ashour, M.; Fikry, S.; Singab, A.N.; Salama, O. Chemical composition and antimicrobial activity of the essential oils of selected apiaceous fruits. Futur. J. Pharm. Sci. 2018. [Google Scholar] [CrossRef]
- Upadhyay, A.; Upadhyaya, I.; Kollanoor-Johny, A.; Venkitanarayanan, K. Combating pathogenic microorganisms using plant-derived antimicrobials: A minireview of the mechanistic basis. Biomed. Res. Int. 2014. [Google Scholar] [CrossRef]
- Tung, C.W.; Lin, Y.C.; Chang, H.S.; Wang, C.C.; Chen, I.S.; Jheng, J.L.; Li, J.H. TIPdb-3D: The three-dimensional structure database of phytochemicals from Taiwan indigenous plants. Database 2014, 2014. [Google Scholar] [CrossRef]
- Lin, Y.C.; Wang, C.C.; Chen, I.S.; Jheng, J.L.; Li, J.H.; Tung, C.W. TIPdb: A database of anticancer, antiplatelet, and antituberculosis phytochemicals from indigenous plants in Taiwan. Sci. World J. 2013, 2013. [Google Scholar] [CrossRef]
- Tung, C.-W. Public databases of plant natural products for computational drug discovery. Curr. Comput. Aided-Drug Des. 2015, 10, 191–196. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, Y.; Zhou, T.; Zheng, J.; Li, S.; Li, H.B. Dietary natural products for prevention and treatment of liver cancer. Nutrients 2016, 8, 156. [Google Scholar] [CrossRef] [PubMed]
- Aung, T.N.; Qu, Z.; Kortschak, R.D.; Adelson, D.L. Understanding the effectiveness of natural compound mixtures in cancer through their molecular mode of action. Int. J. Mol. Sci. 2017, 18, 656. [Google Scholar] [CrossRef] [PubMed]
- Rampogu, S.; Son, M.; Park, C.; Kim, H.-H.; Suh, J.-K.; Lee, K. Sulfonanilide derivatives in identifying novel aromatase inhibitors by applying docking, virtual screening, and MD simulations studies. Biomed. Res. Int. 2017, 2017, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M. polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089. [Google Scholar] [CrossRef]
- Rampogu, S.; Baek, A.; Zeb, A.; Lee, K.W. Exploration for novel inhibitors showing back-to-front approach against VEGFR-2 kinase domain (4AG8) employing molecular docking mechanism and molecular dynamics simulations. BMC Cancer 2018, 18, 264. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hypo Number | Total Cost | Cost Difference a | RMSD | Correlation | Features b | Maximum Fit |
|---|---|---|---|---|---|---|
| Hypo1 | 127.67 | 113.10 | 1.77 | 0.90 | 2HBA, HBD, HyP | 13.23 |
| Hypo2 | 131.722 | 109.06 | 2.06 | 0.86 | 2HBA, HyP, HyP, HyP | 13.34 |
| Hypo3 | 132.165 | 108.62 | 1.94 | 0.88 | 2HBA, HBD, HyP | 12.71 |
| Hypo4 | 133.398 | 107.38 | 2.00 | 0.87 | 2HBA, HBD, HyP | 12.27 |
| Hypo5 | 133.808 | 106.97 | 2.14 | 0.85 | 2HBA, HyP, HyP, HyP | 12.08 |
| Hypo6 | 133.895 | 106.89 | 1.87 | 0.89 | 2HBA, HBD, HyP | 13.88 |
| Hypo7 | 135.009 | 105.77 | 2.09 | 0.86 | 2HBA, HBD, HyP | 11.53 |
| Hypo8 | 135.104 | 105.68 | 2.17 | 0.85 | 2HBD, HyP, HyP, HyP | 12.29 |
| Hypo9 | 135.444 | 105.34 | 1.97 | 0.88 | 2HBA, HBD, HyP | 13.30 |
| Hypo10 | 135.564 | 105.22 | 2.01 | 0.87 | 2HBA, HBD, HyP | 12.91 |
| Name | Fit | IC50 (nmol/L) | RMSE a | Activity Scale | ||
|---|---|---|---|---|---|---|
| Experimental | Predicted | Experimental | Predicted | |||
| C1 | 13.03 | 0.1 | 0.55 | 5.5 | +++ | +++ |
| C2 | 12.29 | 0.3 | 3 | 10 | +++ | +++ |
| C3 | 12.99 | 0.41 | 0.61 | 1.5 | +++ | +++ |
| C4 | 12.98 | 0.5 | 0.62 | 1.2 | +++ | +++ |
| C5 | 12.72 | 1 | 1.1 | 1.1 | +++ | +++ |
| C6 | 12.54 | 2.1 | 1.7 | −1.2 | +++ | +++ |
| C7 | 12.72 | 8 | 1.1 | −7 | +++ | +++ |
| C8 | 11.44 | 15 | 22 | 1.4 | +++ | +++ |
| C9 | 11.16 | 30 | 41 | 1.4 | +++ | +++ |
| C10 | 10.24 | 52 | 350 | 6.6 | +++ | +++ |
| C11 | 9.5 | 74 | 190 | 6 | +++ | +++ |
| C12 | 9.46 | 300 | 2100 | 6.9 | +++ | ++ |
| C13 | 9.66 | 430 | 130 | 3 | +++ | +++ |
| C14 | 9.32 | 800 | 2800 | 3.5 | ++ | ++ |
| C15 | 9.61 | 3000 | 1400 | −2.1 | ++ | ++ |
| C16 | 9.97 | 7400 | 630 | −12 | ++ | ++ |
| C17 | 8.07 | 28,000 | 51,000 | 1.8 | + | + |
| C18 | 9.65 | 54,000 | 1300 | −40 | + | ++ |
| C19 | 8.79 | 100,000 | 9600 | −10 | + | ++ |
| C20 | 8.8 | 560,000 | 9400 | −6.0 | + | ++ |
| Pharmacophore | Number of Features | Feature Set | Selectivity Score |
|---|---|---|---|
| Pharmacophore_1 | 6 | HBA, HBD, HBD, HyP, HyP, HyP | 11.498 |
| Pharmacophore_2 | 6 | HBA, HBD, HBD, HyP, HyP, HyP | 11.498 |
| Pharmacophore_3 | 6 | HBA, HBD, HBD, HyP, HyP, HyP | 11.498 |
| Pharmacophore_4 | 6 | HBA, HBD, HBD, HyP, HyP, HyP | 11.498 |
| Pharmacophore_5 | 6 | HBA, HBD, HBD, HyP, HyP, HyP | 11.498 |
| Pharmacophore_6 | 6 | HBA, HBD, HBD, HyP, HyP, HyP | 11.498 |
| Pharmacophore_7 | 6 | HBA, HBD, HBD, HyP, HyP, HyP | 11.498 |
| Pharmacophore_8 | 6 | HBA, HBD, HBD, HyP, HyP, HyP | 11.498 |
| Pharmacophore_9 | 6 | HBA, HBD, HBD, HyP, HyP, HyP | 11.498 |
| Pharmacophore_10 | 6 | HBA, HBD, HBD, HyP, HyP, HyP | 11.498 |
| Model Number | RMSD |
|---|---|
| 1 | 1.41 |
| 2 | 1.63 |
| 3 | 1.87 |
| 4 | 2.03 |
| 5 | 2.11 |
| 6 | 2.14 |
| 7 | 2.47 |
| 8 | 2.92 |
| 9 | 2.94 |
| 10 | 2.99 |
| Name | Fit | IC50 (nmol/L) | RMSE a | Activity Scale | ||
|---|---|---|---|---|---|---|
| Experimental | Predicted | Experimental | Predicted | |||
| C1 | 13.21 | 0.19 | 0.45 | 2.3 | +++ | +++ |
| C2 | 13.21 | 0.19 | 0.45 | 2.3 | +++ | +++ |
| C3 | 12.9 | 0.22 | 0.92 | 4.2 | +++ | +++ |
| C4 | 13.07 | 0.31 | 0.61 | 2 | +++ | +++ |
| C5 | 12.15 | 3 | 5.1 | 1.7 | +++ | +++ |
| C6 | 13.07 | 4.4 | 0.62 | −7.1 | +++ | +++ |
| C7 | 11.54 | 7 | 21 | 3 | +++ | +++ |
| C8 | 11.81 | 10 | 11 | 1.1 | +++ | +++ |
| C9 | 10.95 | 16 | 81 | 5.1 | +++ | +++ |
| C10 | 11.64 | 20 | 17 | −1.2 | +++ | +++ |
| C11 | 11.54 | 40 | 21 | −1.9 | +++ | +++ |
| C12 | 10.14 | 64 | 520 | 8.1 | +++ | +++ |
| C13 | 10.81 | 100 | 98 | 1.1 | +++ | +++ |
| C14 | 10.75 | 120 | 130 | 1.1 | ++ | ++ |
| C15 | 10.02 | 170 | 690 | 4.1 | ++ | ++ |
| C16 | 9.7 | 180 | 1400 | 8.2 | ++ | ++ |
| C17 | 10.11 | 290 | 560 | 1.9 | ++ | ++ |
| C18 | 9.43 | 330 | 2700 | 8.1 | ++ | ++ |
| C19 | 10.01 | 590 | 700 | 1.2 | ++ | ++ |
| C20 | 9.7 | 1000 | 1400 | 1.4 | ++ | ++ |
| C21 | 9.86 | 1400 | 990 | −1.4 | ++ | ++ |
| C22 | 9.88 | 2200 | 950 | −2.3 | ++ | ++ |
| C23 | 8.89 | 4100 | 9200 | 2.2 | ++ | ++ |
| C24 | 8.92 | 7500 | 8600 | 1.1 | ++ | ++ |
| C25 | 8.39 | 21,000 | 29,000 | 1.4 | + | + |
| C26 | 8.36 | 34,000 | 32,000 | −1.1 | + | + |
| C27 | 9.36 | 61,000 | 31,000 | 2.0 | + | + |
| C28 | 9.48 | 80,000 | 24,000 | 3.4 | + | + |
| C29 | 9.57 | 100,000 | 1900 | −52 | + | ++ |
| C30 | 8.95 | 200,000 | 8100 | −25 | + | ++ |
| C31 | 7.18 | 380,000 | 480,000 | 1.3 | + | + |
| Parameters | PharmL | PharmR |
|---|---|---|
| Total number of molecules in database (D) | 1000 | 1000 |
| Total number of actives in database (A) | 20 | 20 |
| Total number of hit molecules (Ht) | 25 | 24 |
| Total number of active molecules (Ha) | 19 | 20 |
| % Yield of active ((Ha/Ht) × 100) | 76.0 | 83.3 |
| % Ratio of actives ((Ha/A) × 100) | 95 | 100 |
| Enrichment factor (EF) | 38.0 | 41.5 |
| False negatives (A-Ha) | 1 | 0 |
| False positives (Ht–Ha) | 5 | 4 |
| Goodness of fit score (GF) | 0.79 | 0.83 |
| Name | Hydrogen Bond (<3 Å) | Alkyl/π- alkyl | Van der Waals Interactions |
|---|---|---|---|
| Ref | Arg56: HH21-O5 (2.7) Leu112: HN-O24 (2.9) Asn117: HD22-O5 (2.8) Asn117: OD1-H35 (2.1) | Val59, Val151, His159 | Ser57, Gly58, Gly60, Leu105, Gly108, Glu109, CDS111, Tyr147, Glu155, Glu185 |
| Hit1 | Ser57: HG-O17 (2.1) Gln65: HE22-O23 (1.8) Gly108: O-H49 (2.4) Leu112: HN-O23 (2.6) | - | Gly58, Val59, Leu61, Leu105, Thr107, Glu109, CSD111, Ile150, Val151, His154, Glu185 His186 |
| Hit2 | Ser57: HG-O16 (2.7) Val59: HN1-O23 (1.8) Gly60: O-H44 (1.7) | Val151 | Arg56, Gly58, Gln65, Leu61, Leu105, Gly108, Glu109, CDS111, Leu112, Arg124, Tyr147, Ile150, Glu155, Glu185 |
| Hit3 | Val59: HN-O22 (2.9) Gly60: O-H50 (1.8) Gly110: O-H46 (1.8) Tyr147: HH-O1 (2.1) | Val59 | Leu41, Arg56, Ser57, Gly58, Leu61, Gln65, Pro78, Ile77, Glu109, Gly110, CSD111, Leu112, Ile150, His154, His158, Glu185 |
| Compound Name | -CDOCKER Energy | -CDOCKER Interaction Energy | GoldScore | ChemScore |
|---|---|---|---|---|
| Reference1 | 4.00 | 46.42 | 42.86 | –19.68 |
| Actinonin | 29.12 | 45.94 | 41.05 | –18.56 |
| Hit1 | 30.66 | 51.11 | 51.29 | –26.55 |
| Hit2 | 30.76 | 50.13 | 48.55 | –27.00 |
| Hit3 | 21.78 | 47.32 | 55.33 | –28.97 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rampogu, S.; Zeb, A.; Baek, A.; Park, C.; Son, M.; Lee, K.W. Discovery of Potential Plant-Derived Peptide Deformylase (PDF) Inhibitors for Multidrug-Resistant Bacteria Using Computational Studies. J. Clin. Med. 2018, 7, 563. https://doi.org/10.3390/jcm7120563
Rampogu S, Zeb A, Baek A, Park C, Son M, Lee KW. Discovery of Potential Plant-Derived Peptide Deformylase (PDF) Inhibitors for Multidrug-Resistant Bacteria Using Computational Studies. Journal of Clinical Medicine. 2018; 7(12):563. https://doi.org/10.3390/jcm7120563
Chicago/Turabian StyleRampogu, Shailima, Amir Zeb, Ayoung Baek, Chanin Park, Minky Son, and Keun Woo Lee. 2018. "Discovery of Potential Plant-Derived Peptide Deformylase (PDF) Inhibitors for Multidrug-Resistant Bacteria Using Computational Studies" Journal of Clinical Medicine 7, no. 12: 563. https://doi.org/10.3390/jcm7120563