Endothelial Cell Aging: How miRNAs Contribute?



{kind=link}
Abstract
:1. Introduction
2. Endothelial Cells: The Importance of this Silent Guardian of the Body
3. Endothelial Senescence
4. Endothelial miRNAs
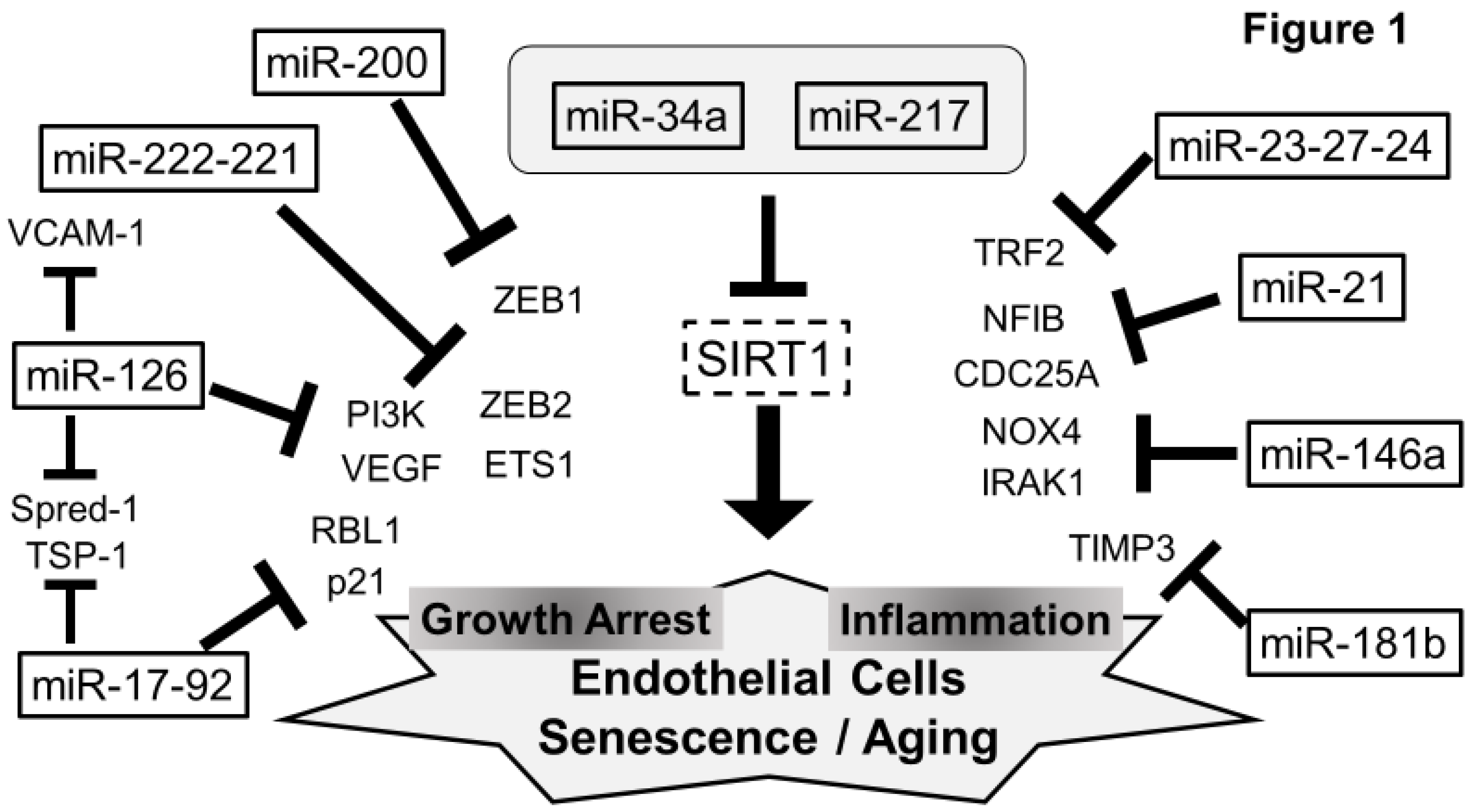
5. Senescent miRNAs in ECs (Figure 1)
5.1. SIRT1 and miRNAs—miR-217, miR-34a, and miR-21
5.2. miR-126
5.3. miR-17-92 Cluster
5.4. miR-23-27-24 Cluster
5.5. miR-222-221 Cluster
5.6. miR-200 Family
5.7. miR-146a
5.8. miR-181b
6. Conclusion
Conflicts of Interest
References
- Bulterijs, S.; Hull, R.S.; Bjork, V.C.; Roy, A.G. It is time to classify biological aging as a disease. Front. Genet. 2015, 6, 205. [Google Scholar] [CrossRef] [PubMed]
- Gems, D. The aging-disease false dichotomy: Understanding senescence as pathology. Front. Genet. 2015, 6, 212. [Google Scholar] [CrossRef] [PubMed]
- Niccoli, T.; Partridge, L. Ageing as a risk factor for disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef] [PubMed]
- Regina, C.; Panatta, E.; Candi, E.; Melino, G.; Amelio, I.; Balistreri, C.R.; Annicchiarico-Petruzzelli, M.; Di Daniele, N.; Ruvolo, G. Vascular ageing and endothelial cell senescence: Molecular mechanisms of physiology and diseases. Mech. Ageing Dev. 2016, 159, 14–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Liu, H.; Ha, Y.; Tilton, R.G.; Zhang, W. Oxidative stress induces endothelial cell senescence via downregulation of Sirt6. Biomed. Res. Int. 2014, 2, 902842. [Google Scholar] [CrossRef] [PubMed]
- Ota, H.; Eto, M.; Kano, M.R.; Ogawa, S.; Iijima, K.; Akishita, M.; Ouchiet, Y. Cilostazol inhibits oxidative stress-induced premature senescence via upregulation of Sirt1 in human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1634–1639. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Xu, J.; Qu, W.; Peng, X.; Xin, P.; Yang, X.; Ying, C.; Sun, X.; Hao, L. Resveratrol reduces vascular cell senescence through attenuation of oxidative stress by SIRT1/NADPH oxidase-dependent mechanisms. J. Nutr. Biochem. 2012, 23, 1410–1416. [Google Scholar] [CrossRef] [PubMed]
- El Assar, M.; Angulo, J.; Rodriguez-Manas, L. Oxidative stress and vascular inflammation in aging. Free Radic. Biol. Med. 2013, 65, 380–401. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Boerma, M.; Zhou, D. Ionizing Radiation-Induced Endothelial Cell Senescence and Cardiovascular Diseases. Radiat. Res. 2016, 186, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Bloom, S.I.; Donato, A.J. The Role of Senescence, Telomere Dysfunction and Shelterin in Vascular Aging. Microcirculation 2018. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29924435 (accessed on 12 June 2018). [CrossRef] [PubMed]
- Donato, A.J.; Morgan, R.G.; Walker, A.E.; Lesniewski, L.A. Cellular and molecular biology of aging endothelial cells. J. Mol. Cell Cardiol. 2015, 89, 122–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Reif, M.M.; Craige, S.M.; Kant, S.; Keaney, J.F., Jr. Endothelial AMPK activation induces mitochondrial biogenesis and stress adaptation via eNOS-dependent mTORC1 signaling. Nitric. Oxide. 2016, 55–56, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Berezikov, E. Evolution of microRNA diversity and regulation in animals. Nat. Rev. Genet. 2011, 12, 846–860. [Google Scholar] [CrossRef] [PubMed]
- Dangwal, S.; Thum, T. MicroRNA therapeutics in cardiovascular disease models. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, G.; Latronico, M.V.; Cavarretta, E. MicroRNAs in cardiovascular diseases: current knowledge and the road ahead. J. Am. Coll. Cardiol. 2014, 63, 2177–2187. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Jazbutyte, V.; Fiedler, J.; Kneitz, S.; Galuppo, P.; Just, A.; Holzmann, A.; Bauersachs, J.; Thum, T. MicroRNA-22 increases senescence and activates cardiac fibroblasts in the aging heart. Age 2013, 5, 747–762. [Google Scholar] [CrossRef] [PubMed]
- Voghel, G.; Thorin-Trescases, N.; Farhat, N.; Nguyen, A.; Villeneuve, L.; Mamarbachi, A.M.; Fortier, A.; Perrault, L.P.; Carrier, M.; Thorin, E. Cellular senescence in endothelial cells from atherosclerotic patients is accelerated by oxidative stress associated with cardiovascular risk factors. Mech. Ageing Dev. 2007, 128, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Menghini, R.; Casagrande, V.; Cardellini, M.; Martelli, E.; Terrinoni, A.; Amati, F.; Vasa-Nicotera, M.; Ippoliti, A.; Novelli, G.; Melino, G. MicroRNA 217 modulates endothelial cell senescence via silent information regulator 1. Circulation 2009, 120, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Yagi, S.; Yamakuchi, M. MicroRNA-34a regulation of endothelial senescence. Biochem. Biophys Res. Commun. 2010, 398, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.S.; Sivachandran, N.; Lau, A.; Boudreau, E.; Zhao, J.L.; Baltimore, D.; Delgado-Olguin, P.; Cybulsky, M.I.; Fish, J.E. MicroRNA-146 represses endothelial activation by inhibiting pro-inflammatory pathways. EMBO Mol. Med. 2013, 5, 1017–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augustin, H.G.; Kozian, D.H.; Johnson, R.C. Differentiation of endothelial cells: Analysis of the constitutive and activated endothelial cell phenotypes. Bioessays 1994, 16, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, E.A. Cell biology of endothelial cells. Hum. Pathol. 1987, 18, 234–239. [Google Scholar] [CrossRef]
- Zhou, G.; Hamik, A.; Nayak, L.; Tian, H.; Shi, H.; Lu, Y.; Sharma, N.; Liao, X.; Hale, A.; Boerboom, L. Endothelial Kruppel-like factor 4 protects against atherothrombosis in mice. J. Clin. Invest. 2012, 122, 4727–4731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial cell control of thrombosis. BMC Cardiovasc. Disord. 2015, 15, 130. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M. Endothelial mitochondria and heart disease. Cardiovasc. Res. 2010, 88, 58–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawler, P.R.; Lawler, J. Molecular basis for the regulation of angiogenesis by thrombospondin-1 and-2. Cold Spring Harb. Perspect. Med. 2012, 2, a006627. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalizing tumor microenvironment to treat cancer: Bench to bedside to biomarkers. J. Clin. Oncol. 2013, 31, 2205–2218. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Campisi, J. The biology of replicative senescence. Eur. J. Cancer 1997, 33, 703–709. [Google Scholar] [CrossRef]
- Kong, Y.; Trabucco, S.E.; Zhang, H. Oxidative stress, mitochondrial dysfunction and the mitochondria theory of aging. Interdiscip. Top. Gerontol. 2014, 39, 86–107. [Google Scholar] [PubMed]
- Barrientos, A. Complementary roles of mitochondrial respiration and ROS signaling on cellular aging and longevity. Aging (Albany NY) 2012, 4, 578–579. [Google Scholar] [CrossRef] [PubMed]
- Aan, G.J.; Hairi, H.A.; Makpol, S.; Rahman, M.A.; Karsani, S.A. Differences in protein changes between stress-induced premature senescence and replicative senescence states. Electrophoresis 2013, 34, 2209–2217. [Google Scholar] [CrossRef] [PubMed]
- Seals, D.R.; Jablonski, K.L.; Donato, A.J. Aging and vascular endothelial function in humans. Clin. Sci. 2011, 120, 357–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loscalzo, J. Nitric oxide insufficiency, platelet activation, and arterial thrombosis. Circ. Res. 2001, 88, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.; Qadri, S.M.; Liu, L. Inhibition of nitric oxide synthesis enhances leukocyte rolling and adhesion in human microvasculature. J. Inflamm. 2012, 9, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsikas, D.; Haufe, S.; Stichtenoth, D.O.; Jordan, J. Nitric oxide and hypertension. J. Hypertens. 2012, 30, 625–626. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.; Donnelly, T.; Lyons, D. Impaired endothelial nitric oxide bioavailability: A common link between aging, hypertension, and atherogenesis? J. Am. Geriatr. Soc. 2009, 57, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chang, E.; Cherry, A.M.; Bangs, C.D.; Oei, Y.; Bodnar, A.; Bronstein, A.; Chiu, C.-P.; Scott Herron, G. Human endothelial cell life extension by telomerase expression. J. Biol. Chem. 1999, 274, 26141–26148. [Google Scholar] [CrossRef] [PubMed]
- Vasa, M.; Breitschopf, K.; Zeiher, A.M.; Dimmeler, S. Nitric oxide activates telomerase and delays endothelial cell senescence. Circ. Res. 2000, 87, 540–542. [Google Scholar] [CrossRef] [PubMed]
- Rosso, A.; Balsamo, A.; Gambino, R.; Dentelli, P.; Falcioni, R.; Cassader, M.; Pegoraro, L.; Pagano, G.; Brizzi, M.F. p53 Mediates the accelerated onset of senescence of endothelial progenitor cells in diabetes. J. Biol. Chem. 2006, 281, 4339–4347. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, A.; Zheng, Q.; Adam, A.; Vincent, P.; Pumiglia, K. Activation of endothelial ras signaling bypasses senescence and causes abnormal vascular morphogenesis. Cancer Res. 2010, 70, 3803–3812. [Google Scholar] [CrossRef] [PubMed]
- Foreman, K.E.; Tang, J. Molecular mechanisms of replicative senescence in endothelial cells. Exp. Gerontol. 2003, 38, 1251–1257. [Google Scholar] [CrossRef] [PubMed]
- Shelton, D.N.; Chang, E.; Whittier, P.S.; Choi, D.; Funk, W.D. Microarray analysis of replicative senescence. Curr. Biol. 1999, 9, 939–945. [Google Scholar] [CrossRef]
- Tang, J.; Gordon, G.M.; Nickoloff, B.J.; Foreman, K.E. The helix-loop-helix protein id-1 delays onset of replicative senescence in human endothelial cells. Lab. Investig. 2002, 82, 1073–1079. [Google Scholar]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P.; Chen, C.Z. Micromanagers of gene expression: The potentially widespread influence of metazoan microRNAs. Nat. Rev. Genet. 2004, 5, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Ketting, R.F.; Fischer, S.E.; Bernstein, E.; Sijen, T.; Hannon, G.J.; Plasterk, R.H. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. Elegans. Genes Dev. 2001, 15, 2654–2659. [Google Scholar] [CrossRef] [PubMed]
- Suarez, Y.; Fernandez-Hernando, C.; Pober, J.S.; Sessa, W.C. Dicer dependent microRNAs regulate gene expression and functions in human endothelial cells. Circ. Res. 2007, 100, 1164–1173. [Google Scholar] [CrossRef] [PubMed]
- Suarez, Y.; Fernandez-Hernando, C.; Yu, J.; Gerber, S.A.; Harrison, K.D.; Pober, J.S.; Luisa Iruela-Arispe, M.; Merkenschlager, M.; Sessa, W.C. Dicer-dependent endothelial microRNAs are necessary for postnatal angiogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 14082–14087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuehbacher, A.; Urbich, C.; Zeiher, A.M.; Dimmeler, S. Role of Dicer and Drosha for endothelial microRNA expression and angiogenesis. Circ. Res. 2007, 101, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of tissue-specific microRNAs from mouse. Curr. Biol. 2002, 12, 735–739. [Google Scholar] [CrossRef]
- Fehlmann, T.; Ludwig, N.; Backes, C.; Meese, E.; Keller, A. Distribution of microRNA biomarker candidates in solid tissues and body fluids. RNA Biol. 2016, 13, 1084–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ. Res. 2007, 100, 174–190. [Google Scholar]
- Bhasin, M.; Yuan, L.; Keskin, D.B.; Out, H.H.; Libermann, T.A.; Oettgen, P. Bioinformatic identification and characterization of human endothelial cell-restricted genes. BMC Genom. 2010, 11, 342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCall, M.N.; Kent, O.A.; Yu, J.; Fox-Talbot, K.; Zaiman, A.L.; Halushka, M.K. MicroRNA profiling of diverse endothelial cell types. BMC Med. Genom. 2011, 4, 78. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.; Roberts, H.; Burton, J.; Pan, J.; States, V.; Rai, S.N.; Galandiuk, S. Assay reproducibility in clinical studies of plasma miRNA. PLoS ONE 2015, 10, e0121948. [Google Scholar] [CrossRef] [PubMed]
- Eriksen, A.H.; Andersen, R.F.; Pallisgaard, N.; Sorensen, F.B.; Jakobsen, A.; Hansen, T.F. MicroRNA Expression Profiling to Identify and Validate Reference Genes for the Relative Quantification of microRNA in Rectal Cancer. PLoS ONE 2016, 11, e0150593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fichtlscherer, S.; Zeiher, A.M.; Dimmeler, S. Circulating microRNAs: biomarkers or mediators of cardiovascular diseases? Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, S.; Weber, J.; Baxter, D.; Galas, D.J. Export of microRNAs and microRNA-protective protein by mammalian cells. Nucleic Acids Res. 2010, 38, 7248–7259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Noren Hooten, N.; Fitzpatrick, M.; Wood, W.H.; De, S.; Ejiogu, N.; Zhang, Y.; Mattison, J.A.; Becker, K.G.; Zonderman, A.B.; Evans, M.K. Age-related changes in microRNA levels in serum. Aging (Albany NY) 2013, 5, 725–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.K.; Austriaco, N.R., Jr.; Zhang, J.; Guarente, L. Mutation in the silencing gene SIR4 can delay aging in S. cerevisiae. Cell 1995, 80, 485–496. [Google Scholar] [CrossRef]
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999, 13, 2570–2580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L.; Picard, F. Calorie restriction—The SIR2 connection. Cell 2005, 120, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K. SIRT1: Regulation of longevity via autophagy. Cell Signal. 2009, 21, 1356–1360. [Google Scholar] [CrossRef] [PubMed]
- Nisoli, E.; Tonello, C.; Cardile, A.; Cozzi, V.; Bracale, R.; Tedesco, L.; Falcone, S.; Valerio, A.; Cantoni, O.; Clementi, E.; et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 2005, 310, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Mattagajasingh, I.; Ki, C.S.; Naqvi, A.; Yamamori, T.; Hoffman, T.A.; Jung, S.B.; DeRicco, J.; Kasuno, K.; Irani, K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2007, 104, 14855–14860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potente, M.; Ghaeni, L.; Baldessari, D.; Mostoslavsky, R.; Rossig, L.; Dequiedt, F.; Haendeler, J.; Mione, M.; Dejana, E.; Alt, F.W.; et al. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev. 2007, 21, 2644–2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gracia-Sancho, J.; Villarreal G, Jr.; Zhang, Y.; Garcia-Cardena, G. Activation of SIRT1 by resveratrol induces KLF2 expression conferring an endothelial vasoprotective phenotype. Cardiovasc. Res. 2010, 85, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welch, C.; Chen, Y.; Stallings, R.L. MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene 2007, 26, 5017–5022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bommer, G.T.; Gerin, I.; Feng, Y.; Kaczorowski, A.J.; Kuick, R.; Love, R.E. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr. Biol. 2007, 17, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.C.; Wentzel, E.A.; Kent, O.A.; Ramachandran, K.; Mullendore, M.; Lee, K.H. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell 2007, 26, 745–752. [Google Scholar] [CrossRef] [PubMed]
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y. A microRNA component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarasov, V.; Jung, P.; Verdoodt, B.; Lodygin, D.; Epanchintsev, A.; Menssen, A. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle 2007, 6, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Fang, D. The Roles of SIRT1 in Cancer. Genes Cancer 2013, 4, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Cheng, H.W.; Qiu, Y.; Dupee, D.; Noonan, M.; Lin, Y.D. MicroRNA-34a Plays a Key Role in Cardiac Repair and Regeneration Following Myocardial Infarction. Circ. Res. 2015, 117, 450–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Wang, K.; Li, P.F. MicroRNA-34 Family and Its Role in Cardiovascular Disease. Crit. Rev. Eukaryot. Gene Expr. 2015, 25, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Iekushi, K.; Lechner, S.; Seeger, T.; Fischer, A.; Heydt, S. MicroRNA-34a regulates cardiac ageing and function. Nature 2013, 495, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Li, J.; Chen, A.F. MicroRNA-34a induces endothelial progenitor cell senescence and impedes its angiogenesis via suppressing silent information regulator 1. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E110–E116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamakuchi, M.; Lowenstein, C.J. MiR-34, SIRT1 and p53: the feedback loop. Cell Cycle 2009, 8, 712–715. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Tillman, D.M.; Wang, M.Y.; Margolius, H.S.; Chao, L. Identification of a new tissue-kallikrein-binding protein. Biochem. J. 1986, 239, 325–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, J.; Shen, B.; Gao, L.; Xia, C.F.; Bledsoe, G.; Chao, L. Tissue kallikrein in cardiovascular, cerebrovascular and renal diseases and skin wound healing. Biol. Chem. 2010, 391, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Bledsoe, G.; Chao, L. Protective Role of Kallistatin in Vascular and Organ Injury. Hypertension 2016, 68, 533–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Li, P.; Gao, L.; Zhang, J.; Yang, Z.; Bledsoe, G. Kallistatin reduces vascular senescence and aging by regulating microRNA-34a-SIRT1 pathway. Aging Cell 2017, 16, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Kumarswamy, R.; Volkmann, I.; Jazbutyte, V.; Dangwal, S.; Park, D.H.; Thum, T. Transforming growth factor-beta-induced endothelial-to-mesenchymal transition is partly mediated by microRNA-21. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.A.; Yamakuchi, M.; Ferlito, M.; Mendell, J.T.; Lowenstein, C.J. MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc. Natl. Acad. Sci. USA 2008, 105, 1516–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, T.A.; Yamakuchi, M.; Kondo, M.; Oettgen, P.; Lowenstein, C.J. Ets-1 and Ets-2 regulate the expression of microRNA-126 in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1990–1997. [Google Scholar] [CrossRef] [PubMed]
- Fish, J.E.; Santoro, M.M.; Morton, S.U.; Yu, S.; Yeh, R.F.; Wythe, J.D. miR-126 regulates angiogenic signaling and vascular integrity. Dev. Cell 2008, 15, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Aurora, A.B.; Johnson, B.A.; Qi, X.; McAnally, J.; Hill, J.A. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev. Cell 2008, 15, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Kuhnert, F.; Mancuso, M.R.; Hampton, J.; Stankunas, K.; Asano, T.; Chen, C.Z. Attribution of vascular phenotypes of the murine Egfl7 locus to the microRNA miR-126. Development 2008, 135, 3989–3993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicoli, S.; Standley, C.; Walker, P.; Hurlstone, A.; Fogarty, K.E.; Lawson, N.D. MicroRNA-mediated integration of haemodynamics and Vegf signalling during angiogenesis. Nature 2010, 464, 1196–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schober, A.; Nazari-Jahantigh, M.; Wei, Y.; Bidzhekov, K.; Gremse, F.; Grommes, J. MicroRNA-126-5p promotes endothelial proliferation and limits atherosclerosis by suppressing Dlk1. Nat. Med. 2014, 20, 368–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zampetaki, A.; Kiechl, S.; Drozdov, I.; Willeit, P.; Mayr, U.; Prokopi, M. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ. Res. 2010, 107, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Y.; Zhao, X.; Liu, Y.Z.; Meng, Z.; Wang, D.; Yang, F. Plasma MicroRNA-126-5p is Associated with the Complexity and Severity of Coronary Artery Disease in Patients with Stable Angina Pectoris. Cell Physiol. Biochem. 2016, 39, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, F.; Bonafe, M.; Spazzafumo, L.; Gobbi, M.; Prattichizzo, F.; Recchioni, R. Age-and glycemia-related miR-126-3p levels in plasma and endothelial cells. Aging (Albany NY) 2014, 6, 771–787. [Google Scholar] [CrossRef] [PubMed]
- Kuhnert, F.; Kuo, C.J. miR-17-92 angiogenesis micromanagement. Blood 2010, 115, 4631–4633. [Google Scholar] [CrossRef] [PubMed]
- Doebele, C.; Bonauer, A.; Fischer, A.; Scholz, A.; Reiss, Y.; Urbich, C. Members of the microRNA-17-92 cluster exhibit a cell-intrinsic antiangiogenic function in endothelial cells. Blood 2010, 115, 4944–4950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, R.; Wang, R.; Guo, L.; Zhang, W.; Lu, Y. MiR-17-3p inhibits angiogenesis by downregulating flk-1 in the cell growth signal pathway. J. Vasc. Res. 2013, 50, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Olive, V.; Jiang, I.; He, L. mir-17-92, a cluster of miRNAs in the midst of the cancer network. Int. J. Biochem. Cell Biol. 2010, 42, 1348–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Almen, G.C.; Verhesen, W.; van Leeuwen, R.E.; van de Vrie, M.; Eurlings, C.; Schellings, M.W. MicroRNA-18 and microRNA-19 regulate CTGF and TSP-1 expression in age-related heart failure. Aging Cell 2011, 10, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Lai, M.; Chen, M.; Xie, C.; Liao, R.; Kang, Y.J. The miR-17-92 cluster of microRNAs confers tumorigenicity by inhibiting oncogene-induced senescence. Cancer Res. 2010, 70, 8547–8557. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Herbert, B.S.; Rajashekhar, G.; Ingram, D.A.; Yoder, M.C.; Clauss, M. Premature senescence of highly proliferative endothelial progenitor cells is induced by tumor necrosis factor-alpha via the p38 mitogen-activated protein kinase pathway. FASEB J. 2009, 23, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.F.; Jamal, J.; Tong, K.L.; Khor, E.S.; Yeap, C.E.; Jong, H.L. Deregulation of has-miR-20b expression in TNF-alpha-induced premature senescence of human pulmonary microvascular endothelial cells. Microvasc. Res. 2017, 114, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Bang, C.; Fiedler, J.; Thum, T. Cardiovascular importance of the microRNA-23/27/24 family. Microcirculation 2012, 19, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Wang, J.; Pan, Q.; Yu, Y.; Zhang, Y.; Wan, Y. Characterization of function and regulation of miR-24-1 and miR-31. Biochem. Biophys. Res. Commun. 2009, 380, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, J.; Jazbutyte, V.; Kirchmaier, B.C.; Gupta, S.K.; Lorenzen, J.; Hartmann, D. MicroRNA-24 regulates vascularity after myocardial infarction. Circulation 2011, 124, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhao, Y.; Lu, Y.; Ritchie, W.; Grau, G.; Vadas, M. The Poly-cistronic miR-23-27-24 Complexes Target Endothelial Cell Junctions: Differential Functional and Molecular Effects of miR-23a and miR-23b. Mol. Ther. Nucleic. Acids. 2016, 5, e354. [Google Scholar] [CrossRef] [PubMed]
- Dellago, H.; Preschitz-Kammerhofer, B.; Terlecki-Zaniewicz, L.; Schreiner, C.; Fortschegger, K.; Chang, M.W. High levels of oncomiR-21 contribute to the senescence-induced growth arrest in normal human cells and its knock-down increases the replicative lifespan. Aging Cell 2013, 12, 446–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, M.; Nasu, T.; Takahashi, Y.; Osaki, T.; Hitomi, S.; Morino, Y. Expression of miR-23a induces telomere shortening and is associated with poor clinical outcomes in patients with coronary artery disease. Clin. Sci. 2017, 131, 2007–2017. [Google Scholar] [CrossRef] [PubMed]
- Lal, A.; Kim, H.H.; Abdelmohsen, K.; Kuwano, Y.; Pullmann, R., Jr.; Srikantan, S. p16(INK4a) translation suppressed by miR-24. PLoS ONE 2008, 3, e1864. [Google Scholar] [CrossRef] [PubMed]
- Urbich, C.; Kuehbacher, A.; Dimmeler, S. Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc. Res. 2008, 79, 581–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Cheng, Y.; Yang, J.; Xu, L.; Zhang, C. Cell-specific effects of miR-221/222 in vessels: Molecular mechanism and therapeutic application. J. Mol. Cell. Cardiol. 2012, 52, 245–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Y.; Wei, Z.; Ding, H.; Wang, Q.; Zhou, Z.; Zheng, S. MicroRNA-19b/221/222 induces endothelial cell dysfunction via suppression of PGC-1alpha in the progression of atherosclerosis. Atherosclerosis 2015, 241, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.W.; Sung, H.C.; Lin, S.R.; Wu, C.W.; Lee, C.W.; Lee, I.T. Resveratrol attenuates ICAM-1 expression and monocyte adhesiveness to TNF-alpha-treated endothelial cells: evidence for an anti-inflammatory cascade mediated by the miR-221/222/AMPK/p38/NF-kappaB pathway. Sci. Rep. 2017, 7, 44689. [Google Scholar] [CrossRef] [PubMed]
- Nicoli, S.; Knyphausen, C.P.; Zhu, L.J.; Lakshmanan, A.; Lawson, N.D. miR-221 is required for endothelial tip cell behaviors during vascular development. Dev. Cell 2012, 22, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Banda, M.; Speyer, C.L.; Smith, J.S.; Rabson, A.B.; Gorski, D.H. Regulation of the expression and activity of the antiangiogenic homeobox gene GAX/MEOX2 by ZEB2 and microRNA-221. Mol. Cell. Biol. 2010, 30, 3902–3913. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Qiu, X.F.; Yu, W.; Zhang, Q.P.; Chen, Q.; Zhang, C.Y. MicroRNA-200a is up-regulated in aged rats with erectile dysfunction and could attenuate endothelial function via SIRT1 inhibition. Asian. J. Androl. 2016, 18, 74–79. [Google Scholar] [PubMed]
- Magenta, A.; Cencioni, C.; Fasanaro, P.; Zaccagnini, G.; Greco, S.; Sarra-Ferraris, G. miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ. 2011, 18, 1628–1639. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Veronese, A.; Pichiorri, F.; Lee, T.J.; Jeon, Y.J.; Volinia, S. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J. Exp. Med. 2011, 208, 875–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, S.; Wang, H.; Jia, C.; Zhu, S.; Chu, X.; Ma, Q. MicroRNA-146a Induces Lineage-Negative Bone Marrow Cell Apoptosis and Senescence by Targeting Polo-Like Kinase 2 Expression. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; Haupt, Y. Mutant p53 subverts PLK2 function in a novel, reinforced loop of corruption. Cell Cycle 2012, 11, 217–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strebhardt, K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 643–660. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, F.; Lazzarini, R.; Recchioni, R.; Marcheselli, F.; Rippo, M.R.; Di Nuzzo, S. MiR-146a as marker of senescence-associated pro-inflammatory status in cells involved in vascular remodelling. Age 2013, 35, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, F.; Lazzarini, R.; Babini, L.; Prattichizzo, F.; Rippo, M.R.; Tiano, L. Anti-inflammatory effect of ubiquinol-10 on young and senescent endothelial cells via miR-146a modulation. Free Radic. Biol. Med. 2013, 63, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Komuro, I. Vascular cell senescence: Contribution to atherosclerosis. Circ. Res. 2007, 100, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U. Oxidative stress in vascular disease: Causes, defense mechanisms and potential therapies. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Vasa-Nicotera, M.; Chen, H.; Tucci, P.; Yang, A.L.; Saintigny, G.; Menghini, R. miR-146a is modulated in human endothelial cell with aging. Atherosclerosis 2011, 217, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Hori, D.; Dunkerly-Eyring, B.; Nomura, Y.; Biswas, D.; Steppan, J.; Henao-Mejia, J. miR-181b regulates vascular stiffness age dependently in part by regulating TGF-beta signaling. PLoS ONE 2017, 12, e0174108. [Google Scholar] [CrossRef] [PubMed]
- Heath, J.M.; Fernandez Esmerats, J.; Khambouneheuang, L.; Kumar, S.; Simmons, R.; Jo, H. Mechanosensitive microRNA-181b Regulates Aortic Valve Endothelial Matrix Degradation by Targeting TIMP3. Cardiovasc. Eng. Technol. 2018, 9, 141–150. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamakuchi, M.; Hashiguchi, T. Endothelial Cell Aging: How miRNAs Contribute? J. Clin. Med. 2018, 7, 170. https://doi.org/10.3390/jcm7070170
Yamakuchi M, Hashiguchi T. Endothelial Cell Aging: How miRNAs Contribute? Journal of Clinical Medicine. 2018; 7(7):170. https://doi.org/10.3390/jcm7070170
Chicago/Turabian StyleYamakuchi, Munekazu, and Teruto Hashiguchi. 2018. "Endothelial Cell Aging: How miRNAs Contribute?" Journal of Clinical Medicine 7, no. 7: 170. https://doi.org/10.3390/jcm7070170
APA StyleYamakuchi, M., & Hashiguchi, T. (2018). Endothelial Cell Aging: How miRNAs Contribute? Journal of Clinical Medicine, 7(7), 170. https://doi.org/10.3390/jcm7070170