Consecutive Prostate Cancer Specimens Revealed Increased Aldo–Keto Reductase Family 1 Member C3 Expression with Progression to Castration-Resistant Prostate Cancer

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Prostate Tissue Samples
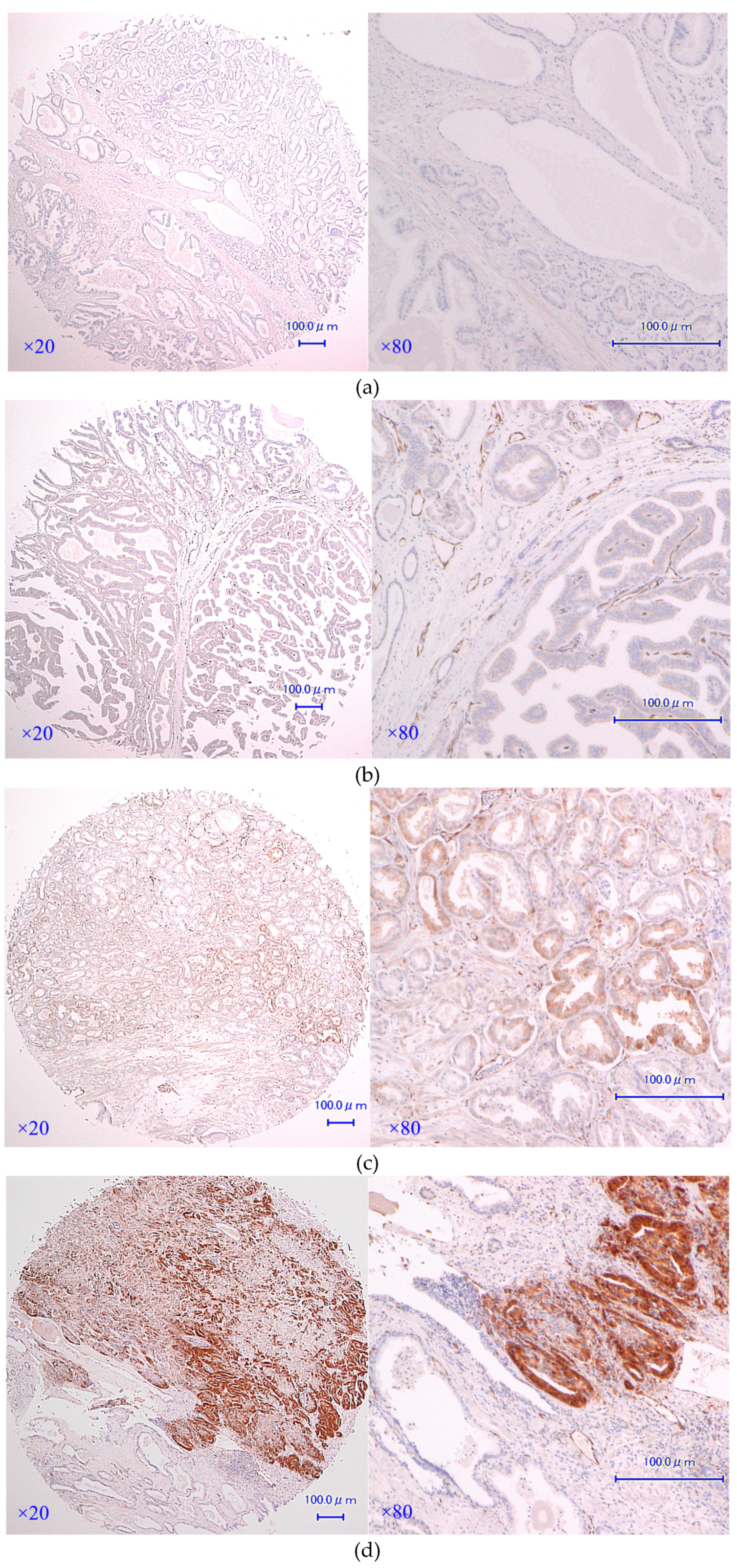
2.2. Immunohistochemistry
2.3. Statistical Analysis
3. Results
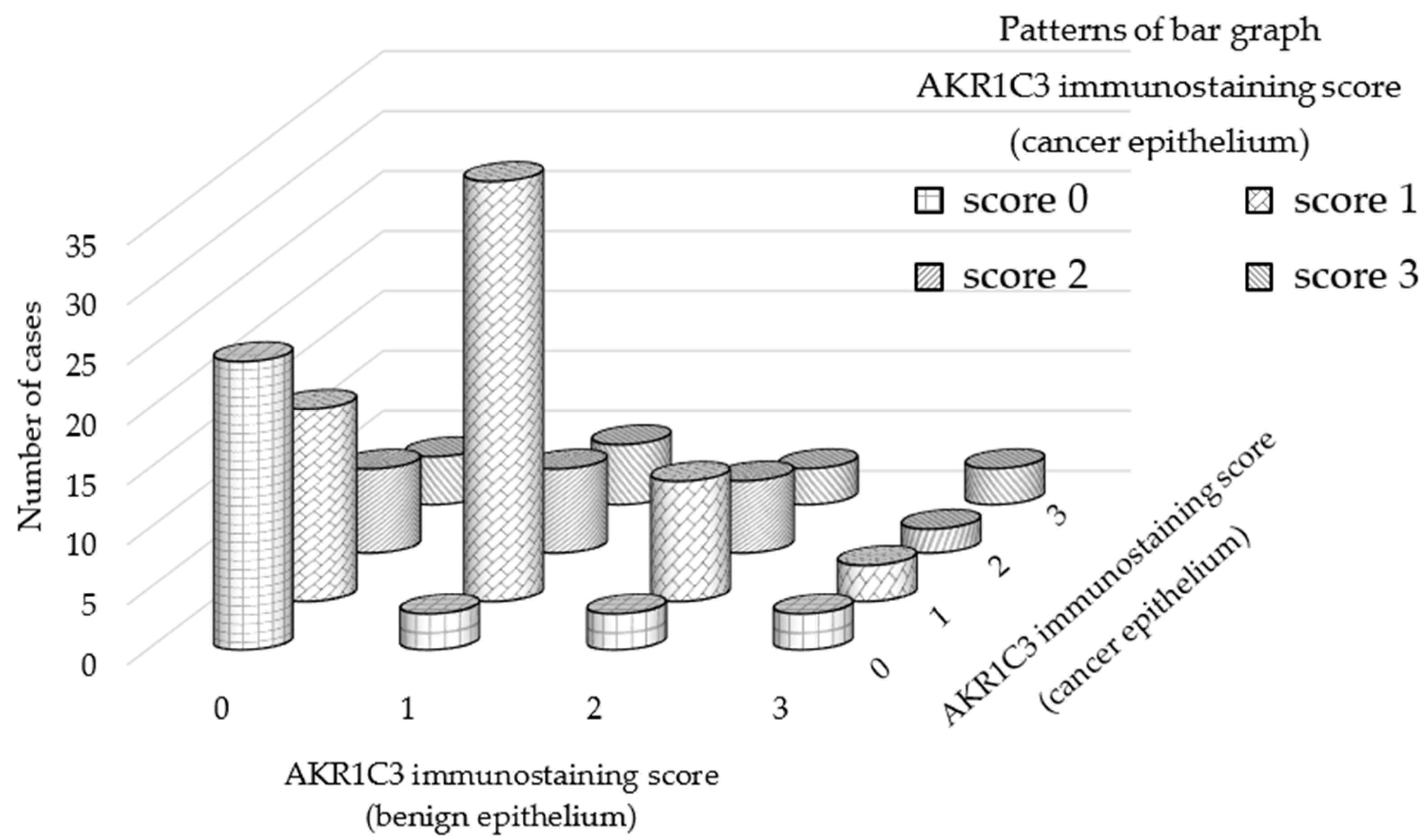
3.1. AKR1C3 Immunostaining of Cancer Epithelium Is Significantly Stronger than That of Benign Epithelia in Patients with Localized Hormone-Naïve Prostate Cancer
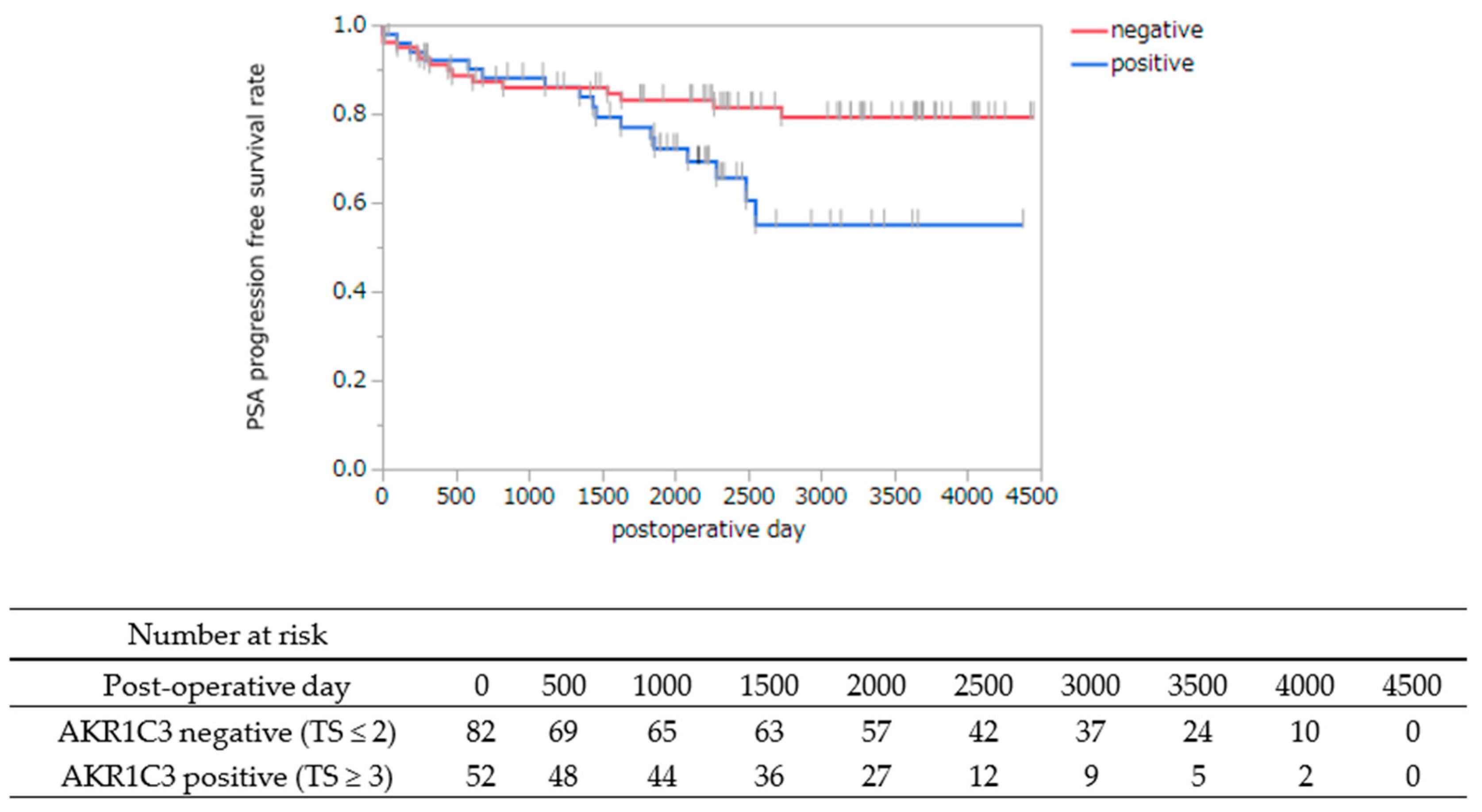
3.2. AKR1C3 Immunostaining of Cancer Cells Is Statistically Associated with PSA Progression-Free Survival after Radical Prostatectomy
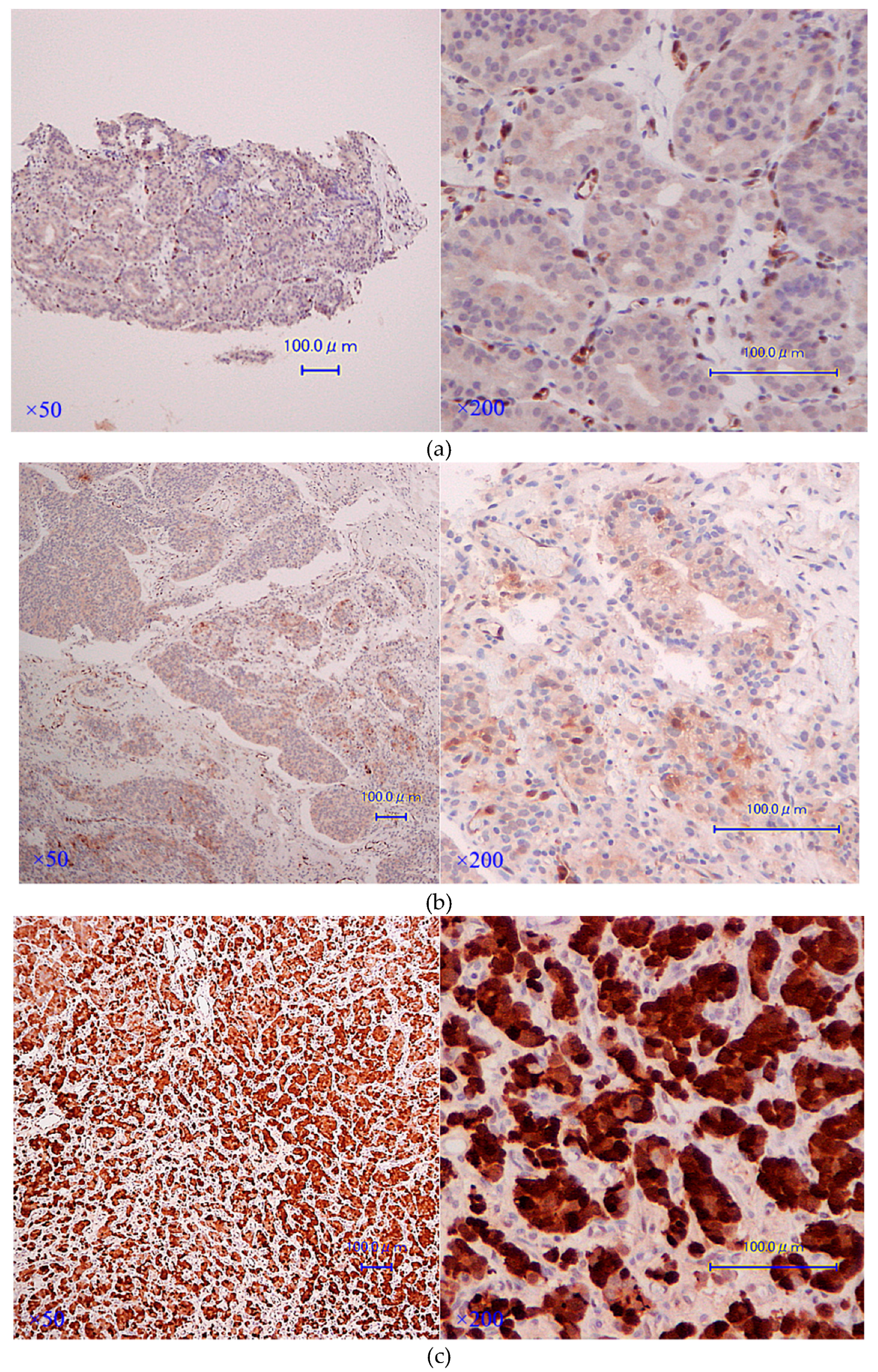
3.3. AKR1C3 Immunostaining Is Significantly Stronger in CRPC Tissues rather than in Hormone-Naïve Ones in the Same Cases
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AKR1C3 | Aldo-Keto Reductase Family 1 Member C3 |
| PCa | prostate cancer |
| CRPC | castration-resistant prostate cancer |
| HNPC | hormone-naïve prostate cancer |
| ADT | androgen deprivation therapy |
| T | testosterone |
| DHT | 5α-dihydrotestosterone |
| AR | androgen receptor |
| DHEA | dehydroepiandrosterone |
| ER | estrogen receptor |
| PG | prostaglandin |
| TMA | tissue-microarray |
| RP | radical prostatectomy |
| TUR-P | transurethral resection of the prostate |
| PFS | progression-free survival |
| TS | total score |
| IS | intensity score |
| PS | proportion score |
| GS | Gleason score |
| CAB | combined androgen blockade |
| DOC | docetaxel |
| Abi | abiraterone |
| Enz | enzalutamide |
| LHRH | luteinizing hormone-releasing hormone |
| ARATs | androgen receptor-axis-targeted agents |
| GABAAR | gamma-aminobutyric acid type A receptor |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Cancer Statistics in Japan-2017. Available online: https://ganjoho.jp/en/professional/statistics/brochure/2017_en.html (accessed on 1 April 2019).
- Huggins, C.; Hodges, C.V. Studies on Prostatic Cancer. I. The Effect of Castration, of Estrogen and of Androgen Injection on Serum Phosphatases in Metastatic Carcinoma of the Prostate. Cancer Res. 1941, 1, 293–297. [Google Scholar]
- Page, S.T.; Lin, D.W.; Mostaghel, E.A.; Hess, D.L.; True, L.D.; Amory, J.K.; Nelson, P.S.; Matsumoto, A.M.; Bremner, W.J. Persistent intraprostatic androgen concentrations after medical castration in healthy men. J. Clin. Endocrinol. MeTable 2006, 91, 3850–3856. [Google Scholar] [CrossRef]
- Nishiyama, T.; Hashimoto, Y.; Takahashi, K. The influence of androgen deprivation therapy on dihydrotestosterone levels in the prostatic tissue of patients with prostate cancer. Clin. Cancer Res. 2004, 10, 7121–7126. [Google Scholar] [CrossRef]
- Dai, C.; Heemers, H.; Sharifi, N. Androgen Signaling in Prostate Cancer. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Locke, J.A.; Guns, E.S.; Lubik, A.A.; Adomat, H.H.; Hendy, S.C.; Wood, C.A.; Ettinger, S.L.; Gleave, M.E.; Nelson, C.C. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008, 68, 6407–6415. [Google Scholar] [CrossRef]
- Hofland, J.; van Weerden, W.M.; Dits, N.F.; Steenbergen, J.; van Leenders, G.J.; Jenster, G.; Schroder, F.H.; de Jong, F.H. Evidence of limited contributions for intratumoral steroidogenesis in prostate cancer. Cancer Res. 2010, 70, 1256–1264. [Google Scholar] [CrossRef]
- Penning, T.M. Mechanisms of drug resistance that target the androgen axis in castration resistant prostate cancer (CRPC). J. Steroid Biochem. Mol. Biol. 2015, 153, 105–113. [Google Scholar] [CrossRef]
- Byrns, M.C.; Jin, Y.; Penning, T.M. Inhibitors of type 5 17beta-hydroxysteroid dehydrogenase (AKR1C3): Overview and structural insights. J. Steroid Biochem. Mol. Biol. 2011, 125, 95–104. [Google Scholar] [CrossRef]
- Ricke, W.A.; McPherson, S.J.; Bianco, J.J.; Cunha, G.R.; Wang, Y.; Risbridger, G.P. Prostatic hormonal carcinogenesis is mediated by in situ estrogen production and estrogen receptor alpha signaling. FASEB J. 2008, 22, 1512–1520. [Google Scholar] [CrossRef]
- Di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di Donato, M.; Di Santi, A.; Cernera, G.; Rossi, V.; Abbondanza, C.; Moncharmont, B.; Sinisi, A.A.; et al. Prostate cancer stem cells: The role of androgen and estrogen receptors. Oncotarget 2016, 7, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di Donato, M.; Castoria, G. Estrogens and Their Receptors in Prostate Cancer: Therapeutic Implications. Front. Oncol. 2018, 8, 2. [Google Scholar] [CrossRef]
- Komoto, J.; Yamada, T.; Watanabe, K.; Takusagawa, F. Crystal structure of human prostaglandin F synthase (AKR1C3). Biochemistry 2004, 43, 2188–2198. [Google Scholar] [CrossRef]
- Sun, S.Q.; Gu, X.; Gao, X.S.; Li, Y.; Yu, H.; Xiong, W.; Yu, H.; Wang, W.; Li, Y.; Teng, Y.; et al. Overexpression of AKR1C3 significantly enhances human prostate cancer cells resistance to radiation. Oncotarget 2016, 7, 48050–48058. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Zhao, L.; Zhang, H.; Liu, X.; Zhao, L.; Zhao, X.; Li, Y.; Li, J. AKR1C3 overexpression may serve as a promising biomarker for prostate cancer progression. Diagn. Pathol. 2014, 9, 42. [Google Scholar] [CrossRef]
- Hamid, A.R.; Pfeiffer, M.J.; Verhaegh, G.W.; Schaafsma, E.; Brandt, A.; Sweep, F.C.; Sedelaar, J.P.; Schalken, J.A. Aldo-keto reductase family 1 member C3 (AKR1C3) is a biomarker and therapeutic target for castration-resistant prostate cancer. Mol. Med. 2013, 18, 1449–1455. [Google Scholar] [CrossRef]
- Jernberg, E.; Thysell, E.; Bovinder Ylitalo, E.; Rudolfsson, S.; Crnalic, S.; Widmark, A.; Bergh, A.; Wikstrom, P. Characterization of prostate cancer bone metastases according to expression levels of steroidogenic enzymes and androgen receptor splice variants. PLoS ONE 2013, 8, e77407. [Google Scholar] [CrossRef]
- Wang, B.; Gu, Y.; Hui, K.; Huang, J.; Xu, S.; Wu, S.; Li, L.; Fan, J.; Wang, X.; Hsieh, J.T.; et al. AKR1C3, a crucial androgenic enzyme in prostate cancer, promotes epithelial-mesenchymal transition and metastasis through activating ERK signaling. Urol. Oncol. 2018, 36, 472.e11–472.e20. [Google Scholar] [CrossRef]
- Barnard, M.; Quanson, J.L.; Mostaghel, E.; Pretorius, E.; Snoep, J.L.; Storbeck, K.H. 11-Oxygenated androgen precursors are the preferred substrates for aldo-keto reductase 1C3 (AKR1C3): Implications for castration resistant prostate cancer. J. Steroid Biochem. Mol. Biol. 2018, 183, 192–201. [Google Scholar] [CrossRef]
- Fan, L.; Peng, G.; Hussain, A.; Fazli, L.; Guns, E.; Gleave, M.; Qi, J. The Steroidogenic Enzyme AKR1C3 Regulates Stability of the Ubiquitin Ligase Siah2 in Prostate Cancer Cells. J. Biol. Chem. 2015, 290, 20865–20879. [Google Scholar] [CrossRef]
- Stanbrough, M.; Bubley, G.J.; Ross, K.; Golub, T.R.; Rubin, M.A.; Penning, T.M.; Febbo, P.G.; Balk, S.P. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006, 66, 2815–2825. [Google Scholar] [CrossRef]
- Uegaki, M.; Kita, Y.; Shirakawa, R.; Teramoto, Y.; Kamiyama, Y.; Saito, R.; Yoshikawa, T.; Sakamoto, H.; Goto, T.; Akamatsu, S.; et al. Downregulation of RalGTPase-activating protein promotes invasion of prostatic epithelial cells and progression from intraepithelial neoplasia to cancer during prostate carcinogenesis. Carcinogenesis 2019, in press. [Google Scholar]
- Lin, H.K.; Steckelbroeck, S.; Fung, K.M.; Jones, A.N.; Penning, T.M. Characterization of a monoclonal antibody for human aldo-keto reductase AKR1C3 (type 2 3alpha-hydroxysteroid dehydrogenase/type 5 17beta-hydroxysteroid dehydrogenase); immunohistochemical detection in breast and prostate. Steroids 2004, 69, 795–801. [Google Scholar] [CrossRef]
- Liu, C.; Lou, W.; Zhu, Y.; Yang, J.C.; Nadiminty, N.; Gaikwad, N.W.; Evans, C.P.; Gao, A.C. Intracrine Androgens and AKR1C3 Activation Confer Resistance to Enzalutamide in Prostate Cancer. Cancer Res. 2015, 75, 1413–1422. [Google Scholar] [CrossRef]
- Liu, C.; Armstrong, C.M.; Lou, W.; Lombard, A.; Evans, C.P.; Gao, A.C. Inhibition of AKR1C3 Activation Overcomes Resistance to Abiraterone in Advanced Prostate Cancer. Mol. Cancer Ther. 2017, 16, 35–44. [Google Scholar] [CrossRef]
- Fung, K.M.; Samara, E.N.; Wong, C.; Metwalli, A.; Krlin, R.; Bane, B.; Liu, C.Z.; Yang, J.T.; Pitha, J.V.; Culkin, D.J.; et al. Increased expression of type 2 3alpha-hydroxysteroid dehydrogenase/type 5 17beta-hydroxysteroid dehydrogenase (AKR1C3) and its relationship with androgen receptor in prostate carcinoma. Endocr. Relat. Cancer 2006, 13, 169–180. [Google Scholar] [CrossRef]
- Nelson, C.P.; Dunn, R.L.; Wei, J.T.; Rubin, M.A.; Montie, J.E.; Sanda, M.G. Contemporary preoperative parameters predict cancer-free survival after radical prostatectomy: A tool to facilitate treatment decisions. Urol. Oncol. 2003, 21, 213–218. [Google Scholar] [CrossRef]
- Penning, T.M. Aldo-Keto Reductase Regulation by the Nrf2 System: Implications for Stress Response, Chemotherapy Drug Resistance, and Carcinogenesis. Chem. Res. Toxicol. 2017, 30, 162–176. [Google Scholar] [CrossRef]
- Xia, D.; Lai, D.V.; Wu, W.; Webb, Z.D.; Yang, Q.; Zhao, L.; Yu, Z.; Thorpe, J.E.; Disch, B.C.; Ihnat, M.A.; et al. Transition from androgenic to neurosteroidal action of 5alpha-androstane-3alpha, 17beta-diol through the type A gamma-aminobutyric acid receptor in prostate cancer progression. J. Steroid Biochem. Mol. Biol. 2018, 178, 89–98. [Google Scholar] [CrossRef]
- Powell, K.; Semaan, L.; Conley-LaComb, M.K.; Asangani, I.; Wu, Y.M.; Ginsburg, K.B.; Williams, J.; Squire, J.A.; Maddipati, K.R.; Cher, M.L.; et al. ERG/AKR1C3/AR Constitutes a Feed-Forward Loop for AR Signaling in Prostate Cancer Cells. Clin. Cancer Res. 2015, 21, 2569–2579. [Google Scholar] [CrossRef] [Green Version]
- Verma, K.; Gupta, N.; Zang, T.; Wangtrakluldee, P.; Srivastava, S.K.; Penning, T.M.; Trippier, P.C. AKR1C3 Inhibitor KV-37 Exhibits Antineoplastic Effects and Potentiates Enzalutamide in Combination Therapy in Prostate Adenocarcinoma Cells. Mol. Cancer Ther. 2018, 17, 1833–1845. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n = 134 | n | p Value | |
|---|---|---|---|
| Age (mean ± SD) | 65.6 ± 6.31 | 0.6087 † | |
| PSA, ng/mL (median) | 7.25 (IQR 5.40–9.88) | 0.9429 † | |
| Pathological T stage | T2a | 7 | N.A. |
| T2b | 1 | ||
| T2c | 77 | ||
| T3a | 37 | ||
| T3b | 12 | ||
| Grade group (pathological) | 1 | 47 | 0.4119 †† |
| 2 | 38 | ||
| 3 | 37 | ||
| 4 | 9 | ||
| 5 | 3 | ||
| AKR1C3 Immunostaining | Score | n = 134 |
|---|---|---|
| AKR1C3 total score | 0 | 45 |
| 2 | 37 | |
| 3 | 11 | |
| 4 | 19 | |
| 5 | 14 | |
| 6 | 4 | |
| 7 | 4 | |
| AKR1C3 total score (median) | 2 (IQR 0–4) | |
| Variables | PSA PFS Rate | ||
|---|---|---|---|
| HR | 95% CI | p Value ††† | |
| PSA level before RP | 1.12 | 1.05–1.18 | 0.0003 |
| Grade group | 1.66 | 1.16–2.36 | 0.0053 |
| AKR1C3 (TS) | 2.19 | 1.07–4.55 | 0.032 |
| Case | Age at Diagnosis | Clinical Stage at Diagnosis | Gleason Score at Diagnosis | Excised CRPC Organ | Age at Excision | PSA at Excision | Days from Diagnosis to Castration | Treatment until Excision of CRPC Specimens | AKR1C3 Immunostaining Total Score | |
|---|---|---|---|---|---|---|---|---|---|---|
| at HNPC | at CRPC | |||||||||
| Case 1 | 57 | cT3bN1M0 | 3 + 3 | Prostate | 62 | 25 | 786 | CAB + DOC | 6 | 7 |
| Case 2 | 73 | cT3bN0M0 | 3 + 4 | Prostate | 84 | 5 | 2860 | CAB | 4 | 7 |
| Case 3 | 60 | cT3aN0M1c | 4 + 4 | Prostate | 65 | 392 | 603 | CAB + DOC | 4 | 4 |
| Case 4 | 79 | cT3aN0M0 | 4 + 4 | Penis | 82 | 16.1 | 1051 | CAB | 2 | 4 |
| Case 5 | 68 | cT3aN0M1c | 4 + 4 | Thoracic vertebra | 75 | 39.21 | 702 | CAB + DOC + Abi | 0 | 5 |
| Case 6 | 70 | cT3aN0M0 | 4 + 3 | Prostate | 78 | 408.2 | 2112 | CAB | 6 | 6 |
| Case 7 | 76 | cT4N1M1b | 4 + 5 | Prostate | 85 | 14.07 | 2320 | CAB + Enz | 4 | 7 |
| Case 8 | 78 | cT4N1M1b | 5 + 4 | Thoracic vertebra | 79 | 7.58 | 321 | CAB | 7 | 7 |
| Case 9 | 69 | cT4N0M0 | 4 + 5 | Prostate | 71 | 46.74 | 567 | CAB | 2 | 7 |
| Case 10 | 80 | cT3bN1M0 | 4 + 5 | Prostate | 82 | 6.21 | 544 | CAB | 1 | 1 |
| Case 11 | 69 | cT4N0M1b | 4 + 5 | Prostate | 70 | 127 | 318 | CAB + Enz | 6 | 5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyazaki, Y.; Teramoto, Y.; Shibuya, S.; Goto, T.; Okasho, K.; Mizuno, K.; Uegaki, M.; Yoshikawa, T.; Akamatsu, S.; Kobayashi, T.; et al. Consecutive Prostate Cancer Specimens Revealed Increased Aldo–Keto Reductase Family 1 Member C3 Expression with Progression to Castration-Resistant Prostate Cancer. J. Clin. Med. 2019, 8, 601. https://doi.org/10.3390/jcm8050601
Miyazaki Y, Teramoto Y, Shibuya S, Goto T, Okasho K, Mizuno K, Uegaki M, Yoshikawa T, Akamatsu S, Kobayashi T, et al. Consecutive Prostate Cancer Specimens Revealed Increased Aldo–Keto Reductase Family 1 Member C3 Expression with Progression to Castration-Resistant Prostate Cancer. Journal of Clinical Medicine. 2019; 8(5):601. https://doi.org/10.3390/jcm8050601
Chicago/Turabian StyleMiyazaki, Yu, Yuki Teramoto, Shinsuke Shibuya, Takayuki Goto, Kosuke Okasho, Kei Mizuno, Masayuki Uegaki, Takeshi Yoshikawa, Shusuke Akamatsu, Takashi Kobayashi, and et al. 2019. "Consecutive Prostate Cancer Specimens Revealed Increased Aldo–Keto Reductase Family 1 Member C3 Expression with Progression to Castration-Resistant Prostate Cancer" Journal of Clinical Medicine 8, no. 5: 601. https://doi.org/10.3390/jcm8050601