Clinical, Immunological, and Functional Characterization of Six Patients with Very High IgM Levels

 , ,
, ,
Abstract
:1. Introduction
2. Experimental Section
2.1. Patients
2.2. B-Cell Immunophenotyping and CD40-CD40L Expression
2.3. Study of CSR and SHM In Vitro
2.4. Whole Exome and Sanger Sequencing
2.5. Real-Time qRT-PCR of ITPKB Gene
3. Results
3.1. Clinical and Immunological Data of HIGM-Like Patients
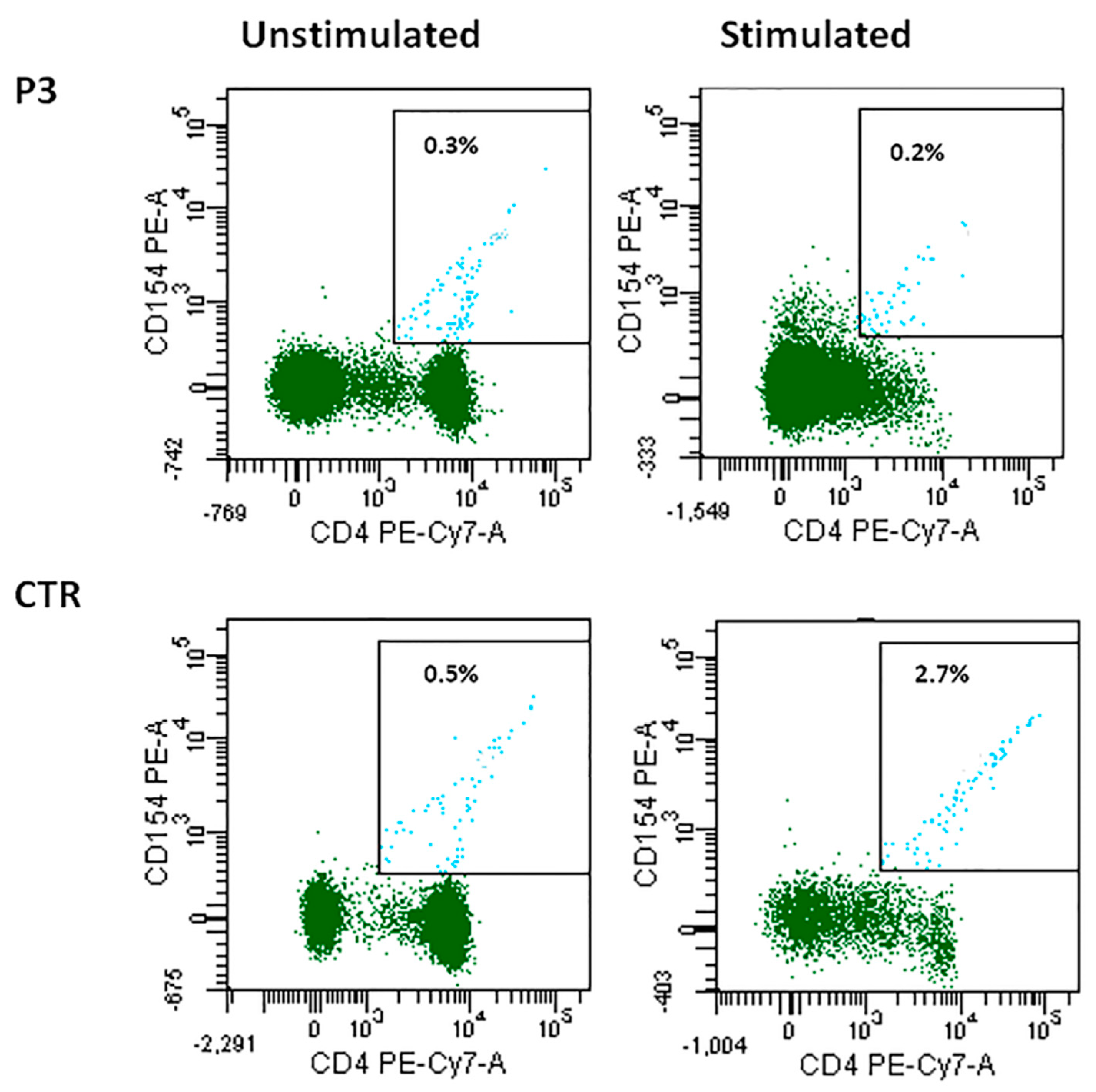
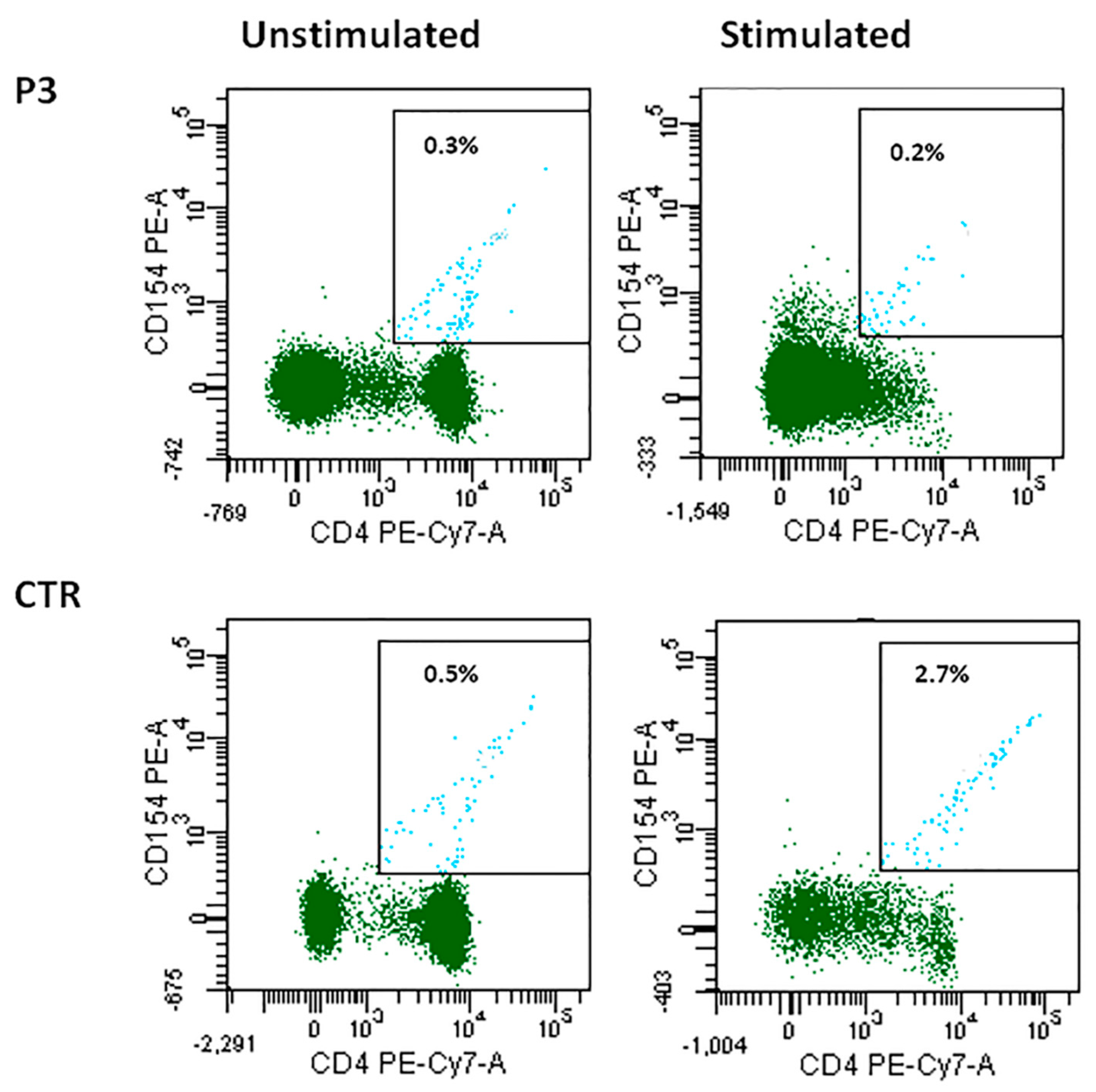
3.2. B-Lymphocyte Profiling and CD40-CD40L Expression
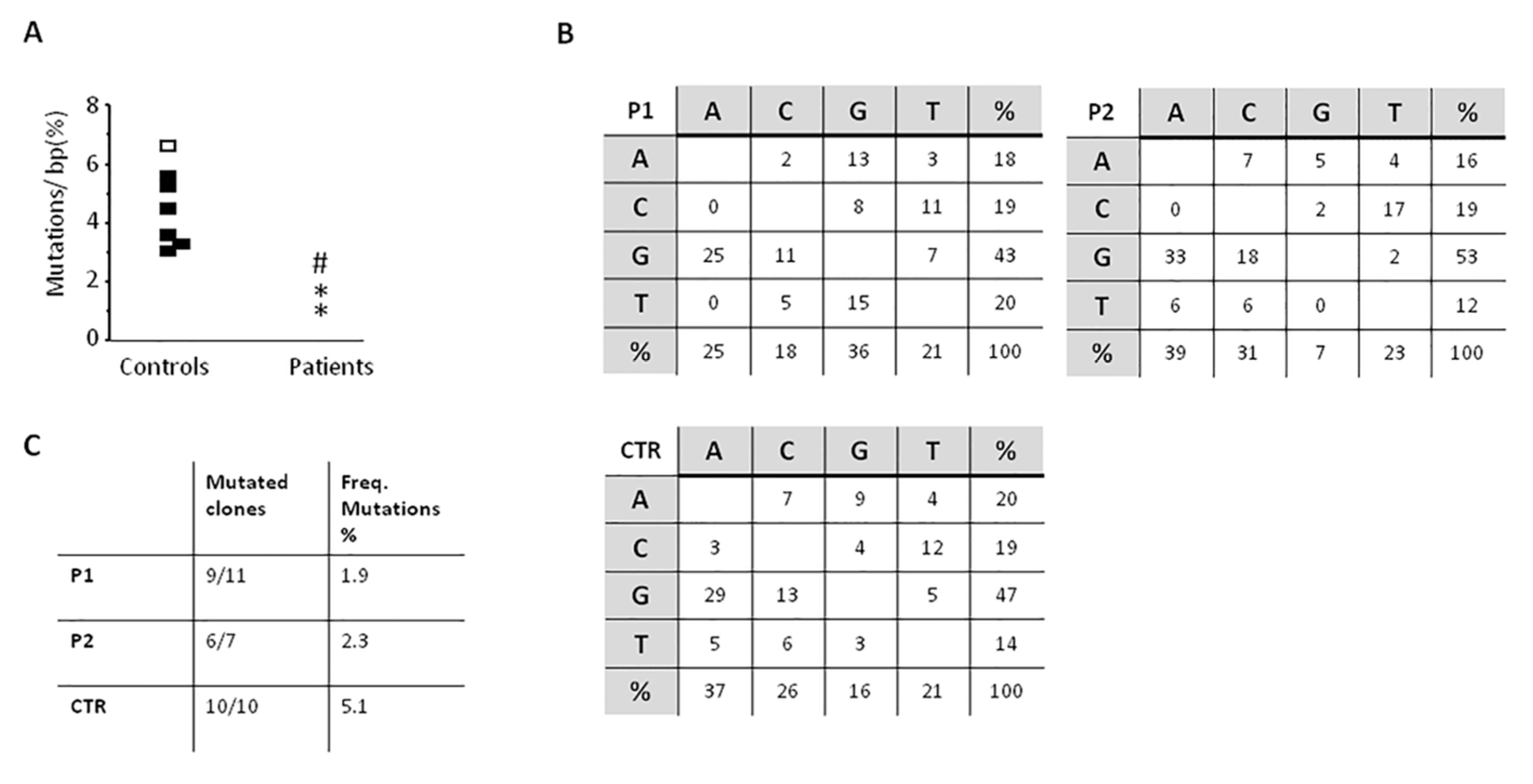
3.3. Reduced SHM with Normal In Vivo and In Vitro CSR
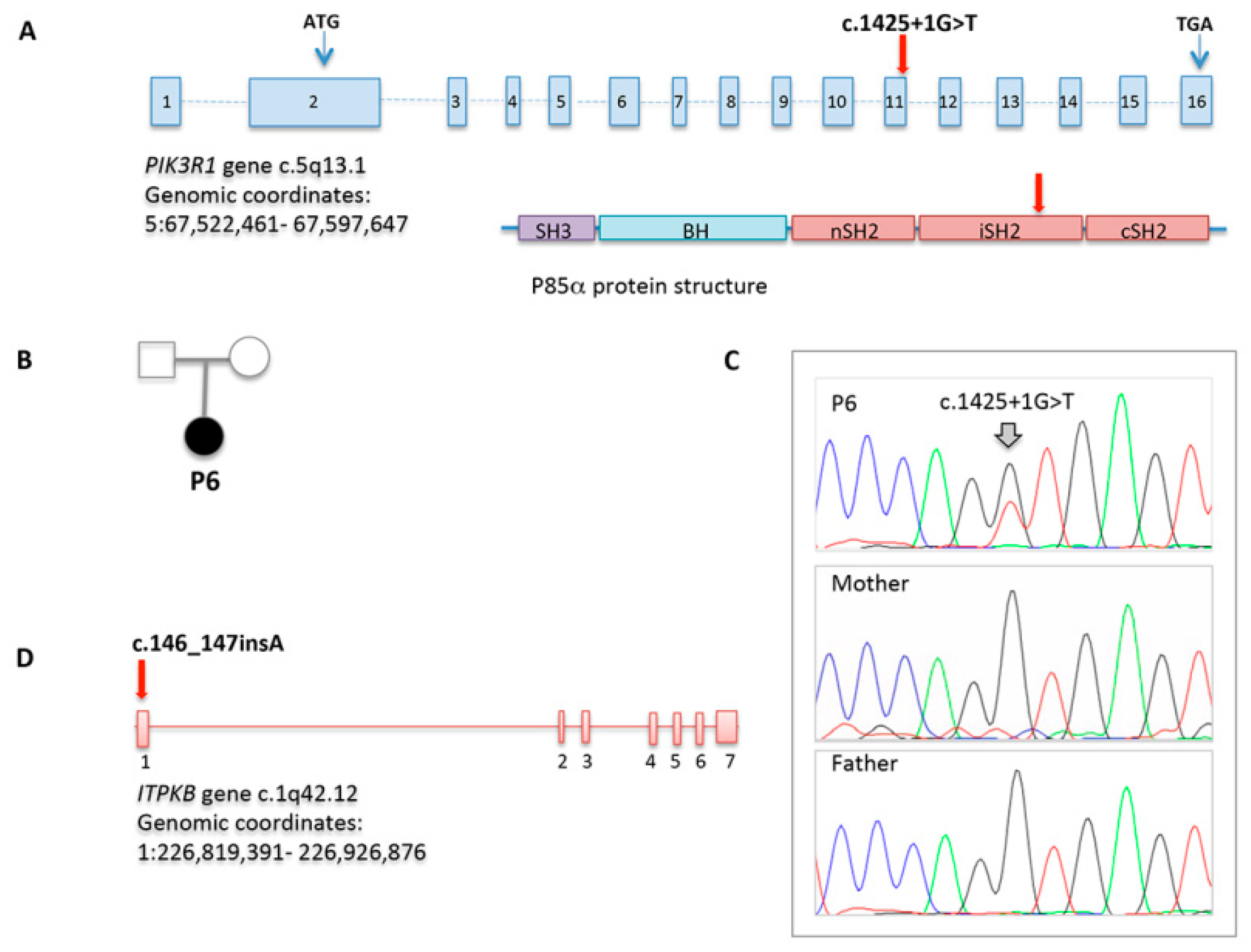
3.4. NGS
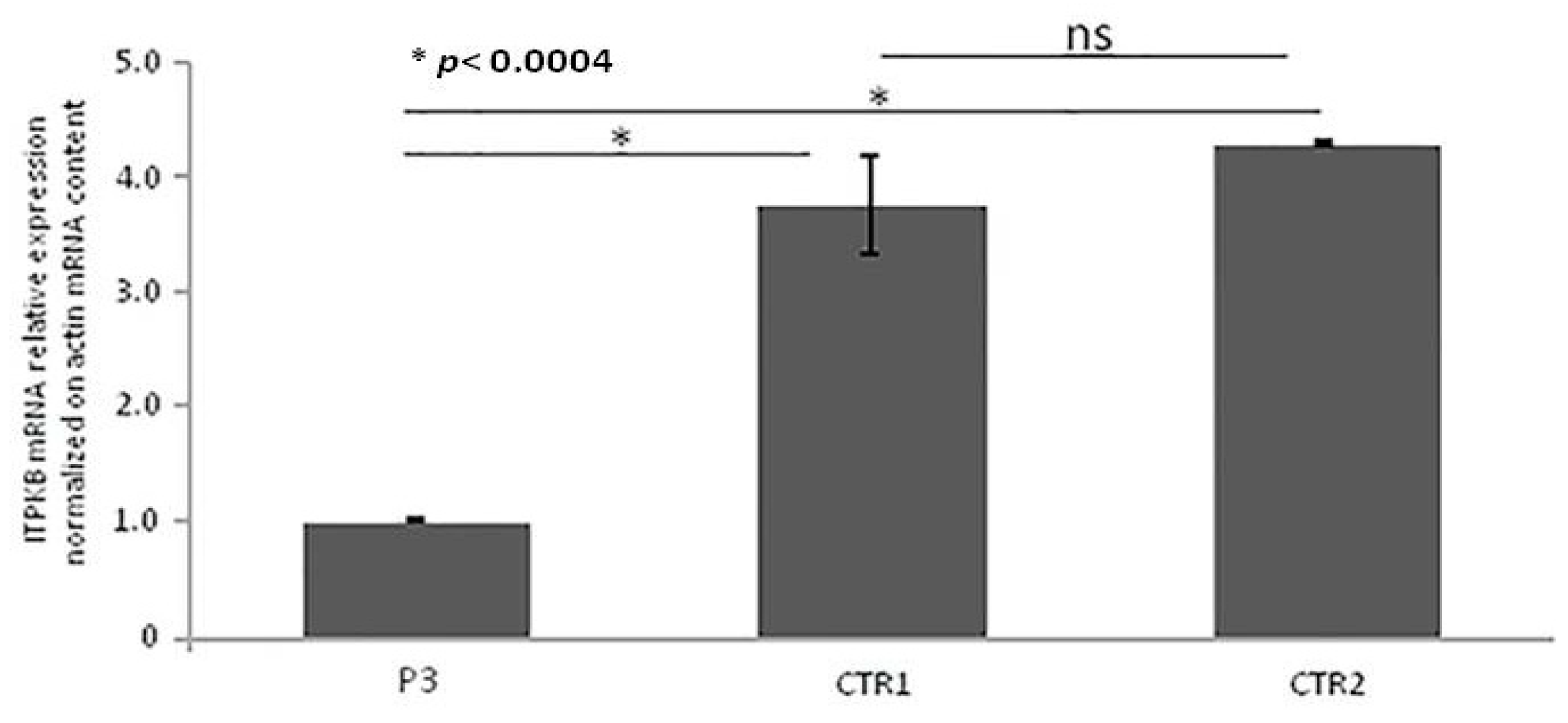
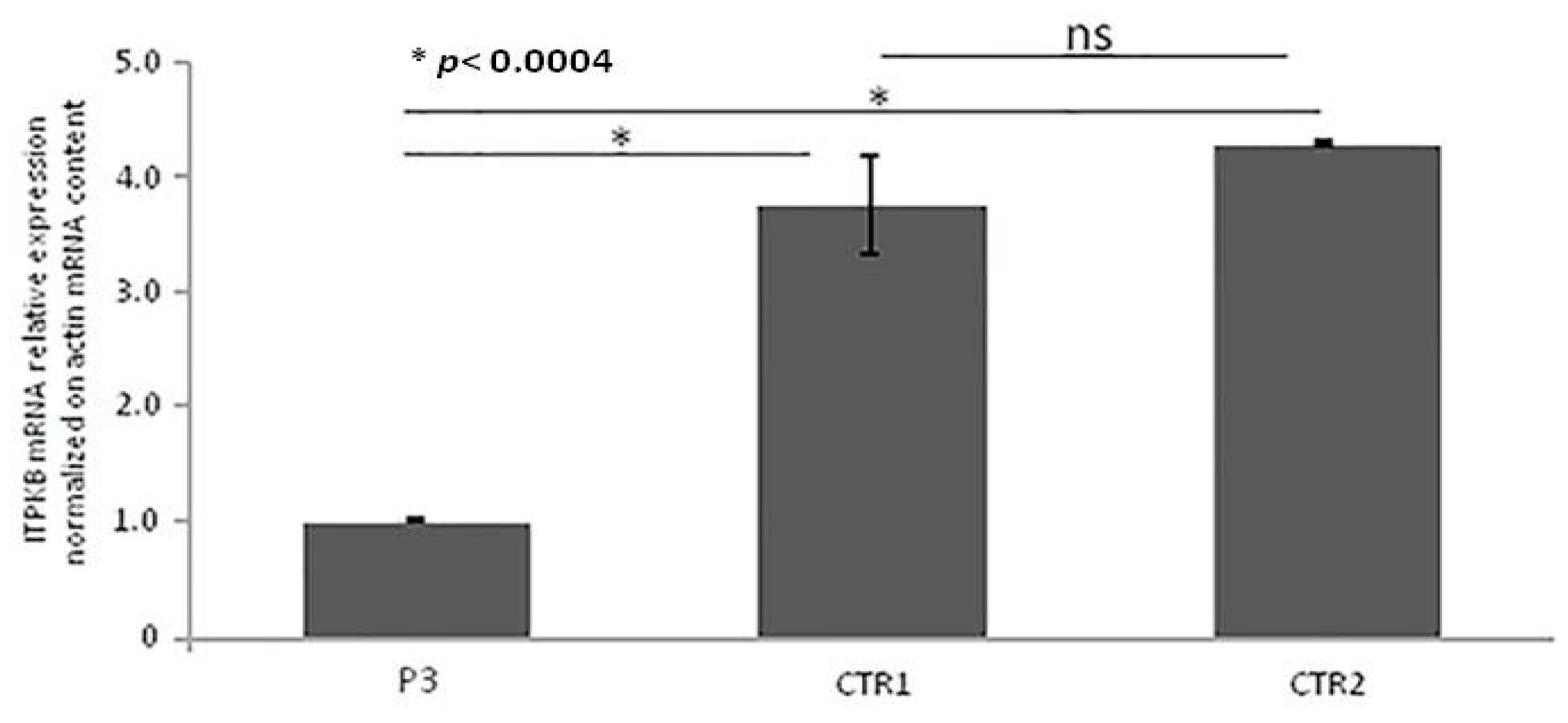
3.5. Real-Time qRT-PCR of ITPKB Gene from RNA
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AID | Activation-induced cytidine deaminase |
| APDS | Activated PI3 kinase delta syndrome |
| A-T | Ataxia telangiectasia |
| ATM | Ataxia telangiectasia mutated |
| CD40L | CD40 ligand |
| CMV | Cytomegalovirus |
| CSR | Class switch recombination |
| DOCK | Dedicator of cytokinesis |
| EBV | Epstein–Barr virus |
| HBV | Hepatitis B virus |
| HCV | Hepatitis C virus |
| HIGM | Hyper IgM |
| HSCT | Hematopoietic stem cell transplantation |
| IVIG | Intravenous immunoglobulin |
| LPD | Lymphoproliferative disorders |
| LRBA | Lipopolysaccharide-responsive beige-like anchor |
| MALT | Mucosa-associated lymphoid tissue |
| mAb | Monoclonal antibodies |
| NBS | Nijmegen breakage syndromes |
| NEMO | Nuclear factor κappa B essential modulator |
| NF-κB | Nuclear factor κappa B |
| NGS | Next generation sequencing |
| NHEJ | Non-homologous end joining |
| NH Lymphoma | Non-Hodgkin lymphoma |
| PBMCs | Peripheral blood mononuclear cells |
| PCR | Polymerase chain reaction |
| PIDs | Primary immunodeficiencies |
| PMS2 | Post-meiotic segregation 2 |
| RAG2 | Recombination-activating gene 2 |
| SHM | Somatic hypermutation |
| UNG | Uracil DNA glycosylase |
| VUS | Variant of unknown significance |
| WES | Whole exome sequencing |
| WHIM | Warts, hypogammaglobulinemia, infections, myelokathexis |
| XLP1 | X-linked lymphoproliferative syndrome type 1 |
References
- Davies, E.G.; Trasher, A.J. Update on the hyper immunoglobulin M syndromes. Br. J. Haematol. 2010, 149, 167–180. [Google Scholar] [CrossRef] [Green Version]
- Duarte-Rey, C.; Bogdanos, D.P.; Leung, P.S.; Anaya, J.M.; Gershwin, M.E. IgM predominance in autoimmune disease: Genetics and gender. Autoimmun. Rev. 2012, 11, A404–A412. [Google Scholar] [CrossRef]
- De la Morena, M.T. Clinical Phenotypes of Hyper-IgM Syndromes. J. Allergy Clin. Immunol. Pract. 2016, 4, 1023–1036. [Google Scholar] [CrossRef]
- Cox, M.C.; Di Napoli, A.; Scarpino, S.; Salerno, G.; Tatarelli, C.; Talerico, C.; Lombardi, M.; Monarca, B.; Amadori, S.; Ruco, L. Clinicopathologic characterization of diffuse-large-B-cell lymphoma with an associated serum monoclonal IgM component. PLoS ONE 2014, 9, e93903. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A. Waldenstrom macroglobulinemia: 2017 update on diagnosis, risk stratification, and management. Am. J. Hematol. 2017, 92, 209–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aruffo, A.; Farrington, M.; Hollenbaugh, D.; Li, X.; Milatovich, A.; Nonoyama, S.; Bajorath, J.; Grosmaire, L.S.; Stenkamp, R.; Neubauer, M.; et al. The CD40 ligand, gp39, is defective in activated T cells from patients with X-linked hyper-IgM syndrome. Cell 1993, 72, 291–300. [Google Scholar] [CrossRef]
- Ferrari, S.; Giliani, S.; Insalaco, A.; Al-Ghonaium, A.; Soresina, A.R.; Loubser, M.; Avanzini, M.A.; Marconi, M.; Badolato, R.; Ugazio, A.G.; et al. Mutations of CD40 gene cause an autosomal recessive form of immunodeficiency with hyper IgM. Proc. Natal. Acad. Sci. USA 2001, 98, 12614–12619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revy, P.; Muto, T.; Levy, Y.; Geissmann, F.; Plebani, A.; Sanal, O.; Catalan, N.; Forveille, M.; Dufourcq-Lagelouse, R.; Gennery, A.; et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell 2000, 102, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Qamar, N.; Fuleihan, R.L. The hyper IgM syndromes. Clin. Rev. Allergy Immunol. 2014, 46, 120–130. [Google Scholar] [CrossRef]
- Leven, E.A.; Maffucci, P.; Ochs, H.D.; Scholl, P.R.; Buckley, R.H.; Fuleihan, R.L.; Geha, R.S.; Cunningham, C.K.; Bonilla, F.A.; Conley, M.E.; et al. Hyper IgM Syndrome: A Report from the USIDNET Registry. J. Clin. Immunol. 2016, 36, 490–501. [Google Scholar] [CrossRef] [Green Version]
- Al Ismail, A.; Husain, A.; Kobayashi, M.; Honjo, T.; Begum, N.A. Depletion of recombination-specific cofactors by the C-terminal mutant of the activation-induced cytidine deaminase causes the dominant negative effect on class switch recombination. Int. Immunol. 2017, 29, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, R.; Fekrvand, S.; Shahkarami, S.; Azizi, G.; Moazzami, B.; Abolhassani, H.; Aghamohammadi, A. The hyper IgM syndromes: Epidemiology, pathogenesis, clinical manifestations, diagnosis and management. Clin. Immunol. 2019, 198, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Winkelstein, J.A.; Marino, M.C.; Ochs, H.; Fuleihan, R.; Scholl, P.R.; Geha, R.; Stiehm, E.R.; Conley, M.E. The X-linked hyper-IgM syndrome: Clinical and immunologic features of 79 patients. Medicine 2003, 82, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Lanzi, G.; Ferrari, S.; Vihinen, M.; Caraffi, S.; Kutukculer, N.; Schiaffonati, L.; Plebani, A.; Notarangelo, L.; Fra, A.M.; Giliani, S. Different molecular behavior of CD40 mutants causing hyper-IgM syndrome. Blood 2010, 116, 5867–5874. [Google Scholar] [CrossRef] [Green Version]
- Imai, K.; Morio, T.; Zhu, Y.; Jin, Y.; Itoh, S.; Kajiwara, M.; Yata, J.; Mizutani, S.; Ochs, H.D.; Nonoyama, S. Clinical course of patients with WASP gene mutations. Blood 2004, 103, 456–646. [Google Scholar] [CrossRef] [Green Version]
- Péron, S.; Metin, A.; Gardès, P.; Alyanakian, M.A.; Sheridan, E.; Kratz, C.P.; Fischer, A.; Durandy, A. Human PMS2 deficiency is associated with impaired immunoglobulin class switch recombination. J. Exp. Med. 2008, 205, 2465–2472. [Google Scholar] [CrossRef]
- Kracker, S.; Di Virgilio, M.; Schwartzentruber, J.; Cuenin, C.; Forveille, M.; Deau, M.C.; McBride, K.M.; Majewski, J.; Gazumyan, A.; Seneviratne, S.; et al. An inherited immunoglobulin class-switch recombination deficiency associated with a defect in the INO80 chromatin remodeling complex. J. Allergy Clin. Immunol. 2015, 135, 998–1007. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Ma, C.A.; Liu, S.; Brown, M.; Cohen, J.; Strober, W. Specific missense mutations in NEMO result in hyper-IgM syndrome with hypohydrotic ectodermal dysplasia. Nat. Immunol. 2001, 2, 223–228. [Google Scholar] [CrossRef]
- Chou, J.; Ohsumi, T.K.; Geha, R.S. Use of whole exome and genome sequencing in the identification of genetic causes of primary immunodeficiency. Curr. Opin. Allergy Clin. Immunol. 2012, 12, 623–628. [Google Scholar] [CrossRef]
- Shokri, S.; Nabavi, M.; Hirschmugl, T.; Aghamohammadi, A.; Arshi, S.; Bemanian, M.H.; Fallahpour, M.; Molatefi, R.; Rekabi, M.; Eslami, N.; et al. LPS-Responsive Beige-Like Anchor Gene Mutation Associated With Possible Bronchiolitis Obliterans Organizing Pneumonia Associated With Hypogammaglobulinemia and Normal IgM Phenotype and Low Number of B Cells. Acta Med. Iran. 2016, 54, 620–623. [Google Scholar]
- Aghamohammadi, A.; Imai, K.; Moazzami, K.; Abolhassani, H.; Tabatabaeiyan, M.; Parvaneh, N.; Nasiri Kalmarzi, R.; Nakagawa, N.; Oshima, K.; Ohara, O.; et al. Ataxia-telangiectasia in a patient presenting with hyper-immunoglobulin M syndrome. J. Investig. Allergol. Clin. Immunol. 2010, 20, 442–445. [Google Scholar] [PubMed]
- Bajin, I.Y.; Ayvaz, D.C.; Unal, S.; Ozgur, T.T.; Cetin, M.; Gumruk, F.; Tezcan, I.; de Villartay, J.P.; Sanal, O. Atypical combined immunodeficiency due to Artemis defect: A case presenting as hyperimmunoglobulin M syndrome and with LGLL. Mol. Immunol. 2013, 56, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, Z.; Mazinani, M.; Shakerian, L.; Nabavi, M.; Fazlollahi, M.R. DOCK2 Deficiency in a Patient with Hyper IgM Phenotype. J. Clin. Immunol. 2018, 38, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Durandy, A.; Schiff, C.; Bonnefoy, J.Y.; Forveille, M.; Rousset, F.; Mazzei, G.; Milili, M.A.F. Induction by anti-CD40 antibody or soluble CD40 ligand and cytokines of IgG, IgA and IgE production by B cells from patients with X-linked hyper IgM syndrome. Eur. J. Immunol. 1993, 23, 2294–2299. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.K.; Huang, C.; Stollar, B.D.; Schwartz, R.S. High-frequency representation of a single VH gene in the expressed human B cell repertoire. J. Exp. Med. 1993, 177, 1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, C.L.; Kuehn, H.S.; Zhao, F.; Niemela, J.E.; Deenick, E.K.; Palendira, U.; Avery, D.T.; Moens, L.; Cannons, J.L.; Biancalana, M.; et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat. Immunol. 2014, 15, 88–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crank, M.C.; Grossman, J.K.; Moir, S.; Pittaluga, S.; Buckner, C.M.; Kardava, L. Mutations in PIK3CD can cause hyper IgM syndrome (HIGM) associated with increased cancer susceptibility. J. Clin. Immunol. 2014, 34, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Deau, M.C.; Heurtier, L.; Frange, P.; Suarez, F.; Bole-Feysot, C.; Nitschke, P.; Cavazzana, M.; Picard, C.; Durandy, A.; Fischer, A.; et al. A human immunodeficiency caused by mutations in the PIK3R1 gene. J. Clin. Investig. 2014, 124, 3923–3928. [Google Scholar] [CrossRef] [Green Version]
- Lucas, C.L.; Zhang, Y.; Venida, A.; Wang, Y.; Hughes, J.; McElwee, J.; Butrick, M.; Matthews, H.; Price, S.; Biancalana, M.; et al. Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. J. Exp. Med. 2014, 211, 2537–2547. [Google Scholar] [CrossRef]
- Petrovski, S.; Parrott, R.E.; Roberts, J.L.; Huang, H.; Yang, J.; Gorentla, B. Dominant splice site mutations in PIK3R1 cause hyper IgM syndrome, lymphadenopathy and short stature. J. Clin. Immunol. 2016, 36, 426–471. [Google Scholar] [CrossRef] [Green Version]
- Saunders, C.J.; Miller, N.A.; Soden, S.E.; Dinwiddie, D.L.; Noll, A.; Alnadi, N.A.; Andraws, N.; Patterson, M.L.; Krivohlavek, L.A.; Fellis, J.; et al. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci. Transl. Med. 2012, 4, 154ra135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, V.; Dotta, L.; Giardino, G.; Cirillo, E.; Lougaris, V.; D’Assante, R.; Prandini, A.; Consolini, R.; Farrow, E.G.; Thiffault, I.; et al. Diagnostics of Primary Immunodeficiencies through Next-Generation Sequencing. Front. Immunol 2016, 7, 466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moschese, V.; Lintzman, J.; Callea, F.; Chini, L.; Devito, R.; Carsetti, R.; Di Cesare, S.; Geissmann, F.; Brousse, N.; Rossi, P.; et al. A novel form of non-X-linked HyperIgM associated with growth and pubertal disturbances and with lymphoma development. J. Pediatr. 2006, 148, 404–406. [Google Scholar] [CrossRef] [PubMed]
- Morbach, H.; Eichhorn, E.M.; Liese, J.G.; Girschick, H.J. Reference values for B cell subpopulations from infancy to adulthood. Clin. Exp. Immunol. 2010, 162, 271–279. [Google Scholar] [CrossRef]
- Aramburu, A.; Mortari, E.P.; Baban, A.; Jorda, E.; Cascioli, S.; Marcellini, V.; Scarsella, M.; Ceccarelli, S.; Corbelli, S.; Cantarutti, N.; et al. Human B-cell memory is shaped by age- and tissue-specific T-independent and GC-dependent events. Eur. J. Immunol. 2017, 47, 327–344. [Google Scholar] [CrossRef]
- Agematsu, K.; Futatani, T.; Hokibara, S.; Kobayashi, N.; Takamoto, M.; Tsukada, S.; Suzuki, H.; Koyasu, S.; Miyawaki, T.; Sugane, K.; et al. Absence of memory B cells in patients with common variable immunodeficiency. Clin. Immunol. 2002, 103, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.; Radigan, L.; Cunningham-Rundles, C. Immune competence and switched memory B cells in common variable immunodeficiency. Clin. Immunol. 2005, 116, 37–41. [Google Scholar] [CrossRef]
- Ma, C.S.; Pittaluga, S.; Avery, D.T.; Hare, N.J.; Maric, I.; Klion, A.D.; Nichols, K.E.; Tangye, S.G. Selective generation of functional somatically mutated IgM+CD27+, but not Ig isotype-switched, memory B cells in X-linked lymphoproliferative disease. J. Clin. Investig. 2006, 116, 322–333. [Google Scholar] [CrossRef]
- Gulino, A.V.; Moratto, D.; Sozzani, S.; Cavadini, P.; Otero, K.; Tassone, L.; Imberti, L.; Pirovano, S.; Notarangelo, L.D.; Soresina, R.; et al. Altered leukocyte response to CXCL12 in patients with warts hypogammaglobulinemia, infections, myelokathexis (WHIM) syndrome. Blood 2004, 104, 444–452. [Google Scholar] [CrossRef]
- Jabara, H.H.; McDonald, D.R.; Janssen, E.; Massaad, M.J.; Ramesh, N.; Borzutzky, A.; Rauter, I.; Benson, H.; Schneider, L.; Baxi, S.; et al. DOCK8 functions as an adaptor that links TLR-MyD88 signaling to B cell activation. Nat. Immunol. 2012, 13, 612–620. [Google Scholar] [CrossRef]
- Cunningham-Rundles, C. How I treat common variable immune deficiency. Blood 2010, 116, 7–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapel, H.; Lucas, M.; Lee, M.; Bjorkander, J.; Webster, D.; Grimbacher, B.; Fieschi, C.; Thon, V.; Abedi, M.R.; Hammarstrom, L. Common variable immunodeficiency disorders: Division into distinct clinical phenotypes. Blood 2008, 112, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan-Hammarstrom, Q.; Dai, S.; Zhao, Y.; van Dijk-Hard, I.F.; Gatti, R.A.; Borresen-Dale, A.L.; Hammarstrom, L. ATM is not required in somatic hypermutation of VH, but is involved in the introduction of mutations in the switch mu region. J. Immunol. 2003, 170, 3707–3716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kracker, S.; Bergmann, Y.; Demuth, I.; Frappart, P.O.; Hildebrand, G.; Christine, R.; Wang, Z.Q.; Sperling, K.; Digweed, M.; Radbruch, A. Nibrin functions in Ig class-switch recombination. Proc. Natal. Acad. Sci. USA 2005, 102, 1584–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, K.; Slupphaug, G.; Lee, W.I.; Revy, P.; Nonoyama, S.; Catalan, N.; Yel, L.; Forveille, M.; Kavli, B.; Krokan, H.E.; et al. Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat. Immunol. 2003, 4, 1023–1028. [Google Scholar] [CrossRef]
- Gardès, P.; Forveille, M.; Alyanakian, M.A.; Aucouturier, P.; Ilencikova, D.; Leroux, D.; Rahner, N.; Mazerolles, F.; Fischer, A.; Kracker, S.; et al. Human MSH6 deficiency is associated with impaired antibody maturation. J. Immunol. 2012, 188, 2023–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, A.G.; Yel, L.; Cao, J.N.; Agrawal, S.; Gupta, S. Common variable immunodeficiency associated with microdeletion of chromosome 1q42.1-q42.3 and inositol 1,4,5-trisphosphate kinase B (ITPKB) deficiency. Clin. Transl. Immunol. 2016, 5, e59. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.T.; Sandberg, M.; Huang, Y.H.; Young, M.; Sutton, S.; Sauer, K.; Cooke, M.P. Production of Ins(1,3,4,5)P4 mediated by the kinase Itpkb inhibits store-operated calcium channels and regulates B cell selection and activation. Nat. Immunol. 2007, 8, 514–521. [Google Scholar] [CrossRef]
- Pouillon, V.; Maréchal, Y.; Frippiat, C.; Erneux, C.; Schurmans, S. Inositol 1,4,5-trisphosphate 3-kinase B (Itpkb) controls survival, proliferation and cytokine production in mouse peripheral T cells. Adv. Biol. Regul. 2013, 53, 39–50. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P1 | P2 | P3 | P4 | P5 | P6† | |
|---|---|---|---|---|---|---|
| Age (y) | 16 | 49 | 45 | 8 | 15 | 15 |
| Sex | F | F | M | M | F | F |
| Clinical features | ||||||
| Infections | ||||||
| Bacterial | + | + | − | + | + | + |
| Opportunistic | − | − | − | − | − | − |
| Viral | + * | + ** | − | − | − | − |
| Lung disease | ||||||
| Bronchiectasis | + | − | − | − | − | − |
| Atelectasis | + | − | − | + | − | − |
| Interstitial lung disease | − | + | − | − | − | − |
| Lymphadenopathy | + | − | +/− | + | + | ++ |
| Autoimmunity | − | − | − | + | − | − |
| Inflammatory disease | − | + | − | − | − | − |
| Recurrent fever | − | + | − | − | − | − |
| Musculoskeletal involvement | + | + | − | − | − | − |
| Liver and/or spleen enlargement | + | − | + | + | − | − |
| Cutaneous manifestations | + | + | + | + | − | − |
| Cancer | NH- Lymphoma | MALT- Lymphoma | − | − | Hodgkin- Lymphoma | Diffuse large B-cell Lymphoma |
| Other | Mood disorder | Behavioral disorder | Growth and pubertal delay; bone defects; Arnold Chiari syndrome | Growth and pubertal delay |
| P1 | P2 | P3 | P4 | P5 | P6† | |||
|---|---|---|---|---|---|---|---|---|
| Age (y) | 10 Before LPD | 16 After LPD | 34 Before LPD | 49 After LPD | 45 | 8 | 15 Before LPD | 15 Before LPD |
| Immunological features | ||||||||
| IgG, mg/dL | 442↓ (IVIG) | 660 | 858 | 788 | 1500 | 294↓ (IVIG) | 565↓ (IVIG) | 140↓ (IVIG) |
| IgA, mg/dL | 19↓ | 8.3↓ | 282 | 332 | 227 | 33 | 5↓ | 5↓ |
| IgM, mg/dL | 753↑ | 416↑ | 911↑ | 1060↑ | 516↑ | 800↑ | 443↑ | 596↑ |
| Lymphocyte absolute counts/mL | 7860 | 6950 | NK | 3.300 | 1400 | 3090 | 2100 | 2260 |
| T-cell subsets | ||||||||
| CD3+ (% of lympho) (absolute value) | 67 (5266) | 72 (5004) | NK | 79 (2607) | 86 (1204) | 74 (2286) | 90 (1890) | 95 (2147) |
| CD4+ (% of lympho) | 23 (1807) | 31 (2154) | NK | 57 (1881) | 48 (672) | 40 (1236) | 26↓ (546) | 14↓ (300) |
| CD8+ (% of lympho) | 40 (3144) | 31 (2154) | NK | 21 (693) | 36 (504) | 30 (927) | 62↑ (1302) | 70↑ (1502) |
| CD56+ (% of lympho) | 2.7 (212) | 4 (278) | NK | 14 (462) | 6 (84) | 6 (185) | NK | NK |
| B-cell subsets | ||||||||
| CD19+ (% of lympho) | 28.5↑ (2240↑) | 25 (1737) | NK | 5↓ (165↓) | 2↓ (28↓) | 12 (370) | 4↓ (84↓) | 0↓ (0) |
| CD19+CD27+IgM+ (IgM memory, % of CD19+) | 12.3 (275) | 3↓ (52.0↓) | NK | 20 (33↓) | 5↓ (1.4↓) | 1.6↓ (5.9↓) | NK | NK |
| CD19+CD27+IgM- (switched memory, % of CD19+) | 0↓ (0) | 2.9↓ (50.3↓) | NK | 0↓ (0) | 10 (2.8↓) | 12.5 (46.2) | NK | NK |
| Genetic alteration | ITPKB c.146_147insA | PIK3R1 c.1425+1G>T | ||||||
| Inheritance | NK | de novo | ||||||
| P1 | P2 | P3 | P4 | |
|---|---|---|---|---|
| In vivo CSR | ||||
| IgG anti-HbsAg | − | ND | − | − |
| IgG anti-Measles | + | + | ND | ND |
| IgG anti-CMV | + | + | + | + |
| IgG anti-EBV | + | + | + | + |
| IgG anti-VZV | + | + | + | + |
| IgG anti-Rubella | − | ND | + | ND |
| IgG anti-Mumps | − | ND | + | |
| In vitro CSR | ||||
| IgE pg/mL (not stimulated) | 173 | 2860 | ND | ND |
| IgE pg/mL (stimulated) | 14658 | 6120 | ND | ND |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallo, V.; Cirillo, E.; Prencipe, R.; Lepore, A.; Del Vecchio, L.; Scalia, G.; Martinelli, V.; Di Matteo, G.; Saunders, C.; Durandy, A.; et al. Clinical, Immunological, and Functional Characterization of Six Patients with Very High IgM Levels. J. Clin. Med. 2020, 9, 818. https://doi.org/10.3390/jcm9030818
Gallo V, Cirillo E, Prencipe R, Lepore A, Del Vecchio L, Scalia G, Martinelli V, Di Matteo G, Saunders C, Durandy A, et al. Clinical, Immunological, and Functional Characterization of Six Patients with Very High IgM Levels. Journal of Clinical Medicine. 2020; 9(3):818. https://doi.org/10.3390/jcm9030818
Chicago/Turabian StyleGallo, Vera, Emilia Cirillo, Rosaria Prencipe, Alessio Lepore, Luigi Del Vecchio, Giulia Scalia, Vincenzo Martinelli, Gigliola Di Matteo, Carol Saunders, Anne Durandy, and et al. 2020. "Clinical, Immunological, and Functional Characterization of Six Patients with Very High IgM Levels" Journal of Clinical Medicine 9, no. 3: 818. https://doi.org/10.3390/jcm9030818