Hypertension, Thrombosis, Kidney Failure, and Diabetes: Is COVID-19 an Endothelial Disease? A Comprehensive Evaluation of Clinical and Basic Evidence

 ,
, 


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
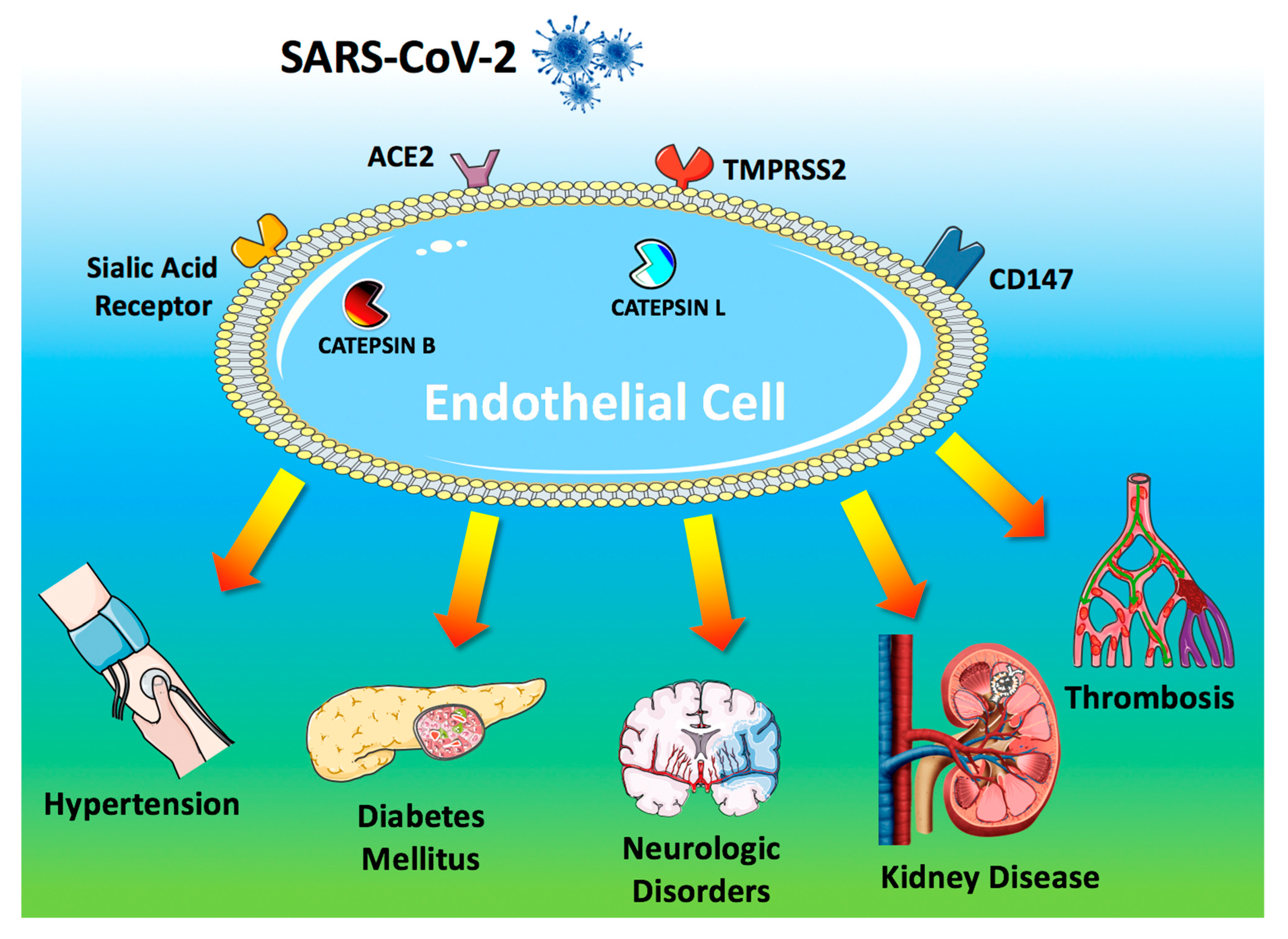
2. Pathogenesis of COVID-19
3. Hypertension and COVID-19
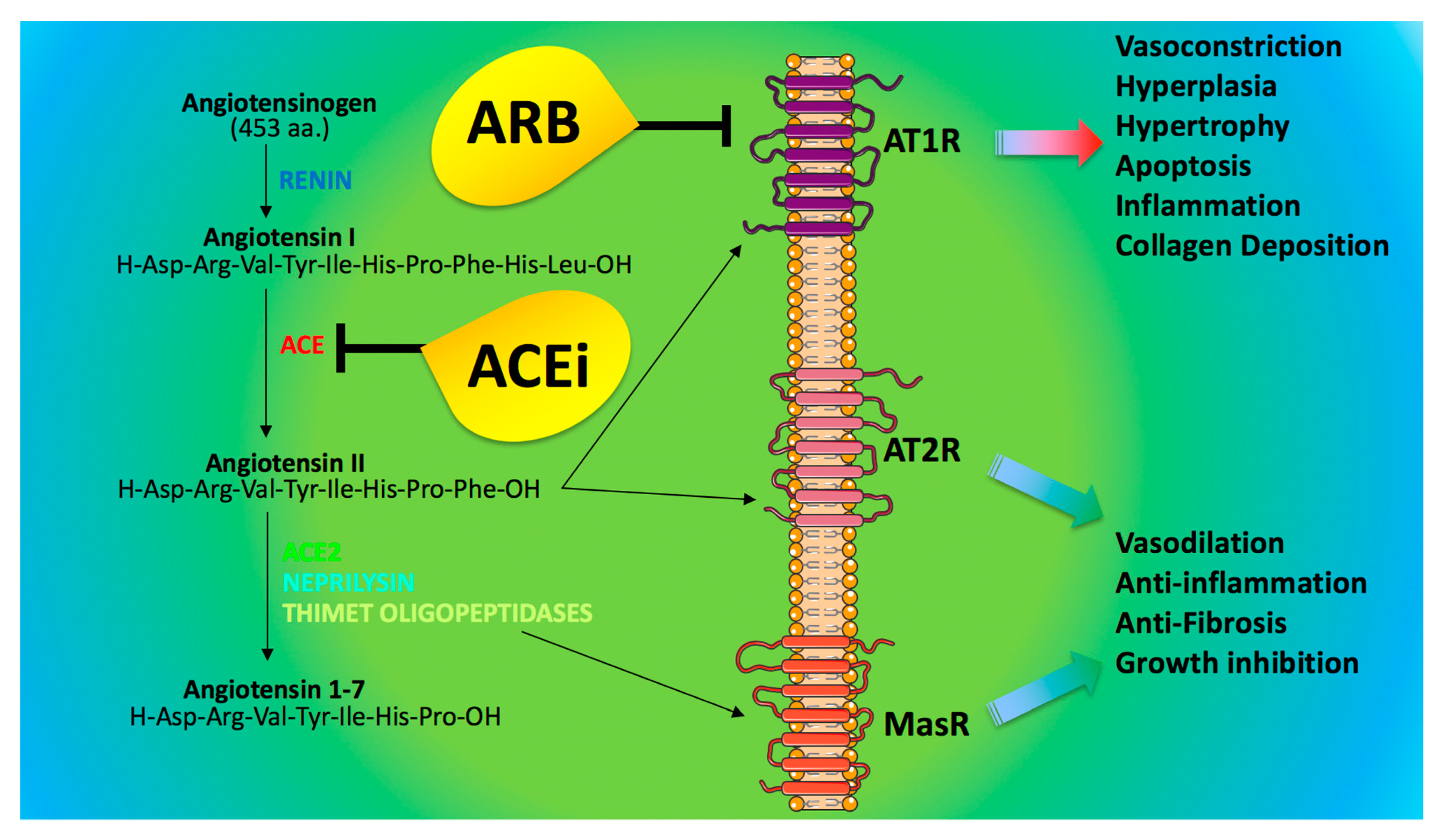
4. ACE2 and Anti-Hypertensive Drugs: What Do We Know?
5. Kidney Disease in COVID-19
6. Diabetes and COVID-19
7. Thromboembolism and COVID-19
8. Anticoagulation as a Key Therapy for COVID-19
Author Contributions
Funding
Conflicts of Interest
References
- Fauci, A.S.; Lane, H.C.; Redfield, R.R. Covid-19—Navigating the Uncharted. N. Engl. J. Med. 2020, 382, 1268–1269. [Google Scholar] [CrossRef] [PubMed]
- Paules, C.I.; Marston, H.D.; Fauci, A.S. Coronavirus Infections-More Than Just the Common Cold. JAMA 2020, 323, 707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, D.S.; Azhar, E.E.; Madani, T.A.; Ntoumi, F.; Kock, R.; Dar, O.; Ippolito, G.; McHugh, T.D.; Memish, Z.A.; Drosten, C.; et al. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health—The latest 2019 novel coronavirus outbreak in Wuhan, China. Int. J. Infect. Dis. 2020, 91, 264–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Lovren, F.; Pan, Y.; Quan, A.; Teoh, H.; Wang, G.; Shukla, P.C.; Levitt, K.S.; Oudit, G.Y.; Al-Omran, M.; Stewart, D.J.; et al. Angiotensin converting enzyme-2 confers endothelial protection and attenuates atherosclerosis. Am. J. Physiol. Circ. Physiol. 2008, 295, H1377–H1384. [Google Scholar] [CrossRef] [Green Version]
- Sluimer, J.C.; Gasc, J.M.; Hamming, I.; van Goor, H.; Michaud, A.; van den Akker, L.H.; Jutten, B.; Cleutjens, J.; Bijnens, A.P.; Corvol, P.; et al. Angiotensin-converting enzyme 2 (ACE2) expression and activity in human carotid atherosclerotic lesions. J. Pathol. 2008, 215, 273–279. [Google Scholar] [CrossRef]
- Schiffrin, E.L.; Flack, J.; Ito, S.; Muntner, P.; Webb, C. Hypertension and COVID-19. Am. J. Hypertens. 2020, 33, 33–373. [Google Scholar] [CrossRef]
- Richardson, S.; Hirsch, J.S.; Narasimhan, M.; Crawford, J.M.; McGinn, T.; Davidson, K.W. The Northwell COVID-19 Research Consortium. Presenting Characteristics, Comorbidities, and Outcomes among 5700 Patients Hospitalized With COVID-19 in the New York City Area. JAMA 2020. [Google Scholar] [CrossRef]
- Chen, T.; Wu, D.; Chen, H.; Yan, W.; Yang, D.; Chen, G.; Ma, K.; Xu, D.; Yu, H.; Wang, H.; et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: Retrospective study. BMJ 2020, 368, m1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, L.C.; Parodi, S.M.; Escobar, G.J.; Liu, V.X. Characteristics of Hospitalized Adults With COVID-19 in an Integrated Health Care System in California. JAMA 2020. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.J.; Liang, W.H.; Zhao, Y.; Liang, H.R.; Chen, Z.S.; Li, Y.M.; Liu, X.Q.; Chen, R.C.; Tang, C.L.; Wang, T.; et al. Comorbidity and its impact on 1590 patients with Covid-19 in China: A Nationwide Analysis. Eur. Respir. J. 2020, 2000547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Bikdeli, B.; Madhavan, M.V.; Jimenez, D.; Chuich, T.; Dreyfus, I.; Driggin, E.; Nigoghossian, C.; Ageno, W.; Madjid, M.; Guo, Y.; et al. Lip GYH. COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-up. J. Am. Coll. Cardiol. 2020. [Google Scholar] [CrossRef]
- Klok, F.A.; Kruip, M.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020. [Google Scholar] [CrossRef]
- Durvasula, R.; Wellington, T.; McNamara, E.; Watnick, S. COVID-19 and Kidney Failure in the Acute Care Setting: Our Experience From Seattle. Am. J. Kidney Dis. 2020. [Google Scholar] [CrossRef]
- Ronco, C.; Reis, T. Kidney involvement in COVID-19 and rationale for extracorporeal therapies. Nat. Rev. Nephrol. 2020, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Rotzinger, D.C.; Beigelman-Aubry, C.; von Garnier, C.; Qanadli, S.D. Pulmonary embolism in patients with COVID-19: Time to change the paradigm of computed tomography. Thromb. Res. 2020, 190, 58–59. [Google Scholar] [CrossRef]
- Poissy, J.; Goutay, J.; Caplan, M.; Parmentier, E.; Duburcq, T.; Lassalle, F.; Jeanpierre, E.; Rauch, A.; Labreuche, J.; Susen, S. Pulmonary Embolism in COVID-19 Patients: Awareness of an Increased Prevalence. Circulation 2020. [Google Scholar] [CrossRef]
- Aggarwal, G.; Lippi, G.; Michael Henry, B. Cerebrovascular disease is associated with an increased disease severity in patients with Coronavirus Disease 2019 (COVID-19): A pooled analysis of published literature. Int. J. Stroke 2020, 1747493020921664. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Jin, H.; Wang, M.; Hu, Y.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D.; et al. Neurologic Manifestations of Hospitalized Patients With Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santulli, G.; Morelli, M.; Gambardella, J. Is Endothelial Dysfunction the Concealed Cornerstone of COVID-19? BMJ 2020, in press. [Google Scholar]
- Cooke, J.P. The endothelium: A new target for therapy. Vasc. Med. 2000, 5, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Endothelium as an organ system. Crit. Care Med. 2004, 32, S271–S279. [Google Scholar] [CrossRef] [PubMed]
- Inagami, T.; Naruse, M.; Hoover, R. Endothelium as an endocrine organ. Annu. Rev. Physiol. 1995, 57, 171–189. [Google Scholar] [CrossRef] [PubMed]
- Riphagen, S.; Gomez, R.; Gonzalez-Martinez, C.; Wilkinson, N.; Theocharis, P. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet 2020, in press. [Google Scholar] [CrossRef]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020. [Google Scholar] [CrossRef]
- Guzzi, P.H.; Mercatelli, D.; Ceraolo, C.; Giorgi, F.M. Master Regulator Analysis of the SARS-CoV-2/Human Interactome. J. Clin. Med. 2020, 9, 982. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Zhong, L.; Deng, J.; Peng, J.; Dan, H.; Zeng, X.; Li, T.; Chen, Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 2020, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.P.; Look, D.C.; Shi, L.; Hickey, M.; Pewe, L.; Netland, J.; Farzan, M.; Wohlford-Lenane, C.; Perlman, S.; McCray, P.B., Jr. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J. Virol. 2005, 79, 14614–14621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perico, L.; Benigni, A.; Remuzzi, G. Should COVID-19 Concern Nephrologists? Why and to What Extent? The Emerging Impasse of Angiotensin Blockade. Nephron 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.; Turner, A.T.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System. Circ. Res. 2020, 126. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, L.; Zhang, J.; Xiong, C.; Li, X. The SARS-CoV-2 receptor ACE2 expression of maternal-fetal interface and fetal organs by single-cell transcriptome study. PLoS ONE 2020, 15, e0230295. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, T.M.; Buchmeier, M.J. Coronavirus spike proteins in viral entry and pathogenesis. Virology 2001, 279, 371–374. [Google Scholar] [CrossRef] [Green Version]
- Matsuyama, S.; Nao, N.; Shirato, K.; Kawase, M.; Saito, S.; Takayama, I.; Nagata, N.; Sekizuka, T.; Katoh, H.; Kato, F.; et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc. Natl. Acad. Sci. USA 2020, 117, 7001–7003. [Google Scholar] [CrossRef] [Green Version]
- Sungnak, W.; Huang, N.; Becavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- Tortorici, M.A.; Walls, A.C.; Lang, Y.; Wang, C.; Li, Z.; Koerhuis, D.; Boons, G.-J.; Bosch, B.-J.; Rey, F.A.; De Groot, R.J.; et al. Structural basis for human coronavirus attachment to sialic acid receptors. Nat. Struct. Mol. Biol. 2019, 26, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Hulswit, R.; Lang, Y.; Bakkers, M.J.G.; Li, W.; Li, Z.; Schouten, A.; Ophorst, B.; Van Kuppeveld, F.J.M.; Boons, G.-J.; Bosch, B.-J.; et al. Human coronaviruses OC43 and HKU1 bind to 9-O-acetylated sialic acids via a conserved receptor-binding site in spike protein domain A. Proc. Natl. Acad. Sci. USA 2019, 116, 2681–2690. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Mi, L.; Xu, J.; Yu, J.; Wang, X.; Jiang, J.; Xing, J.; Shang, P.; Qian, A.; Li, Y.; et al. Function of HAb18G/CD147 in invasion of host cells by severe acute respiratory syndrome coronavirus. J. Infect. Dis. 2005, 191, 755–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Feng, X.; Zhou, Q.; Cheng, W.; Shang, C.; Han, P.; Lin, C.-H.; Chen, H.-S.V.; Quertermous, T.; Chang, C.-P. Pathological Ace2-to-Ace enzyme switch in the stressed heart is transcriptionally controlled by the endothelial Brg1-FoxM1 complex. Proc. Natl. Acad. Sci. USA 2016, 113, E5628–E5635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aimes, R.; Zijlstra, A.; Hooper, J.; Ogbourne, S.; Sit, M.-L.; Fuchs, S.; Gotley, D.; Quigley, J.P.; Antalis, T. Endothelial cell serine proteases expressed during vascular morphogenesis and angiogenesis. Thromb. Haemost. 2003, 89, 561–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.T.-N.; Lu, C.-Y.; Chi, Y.; Li, W.-L.; Chang, L.-Y.; Lai, M.-J.; Chen, J.-S.; Hsu, W.-M.; Huang, L.-M. Adaptation of influenza A (H7N9) virus in primary human airway epithelial cells. Sci. Rep. 2017, 7, 11300. [Google Scholar] [CrossRef]
- Vanarsdall, A.L.; Pritchard, S.R.; Wisner, T.W.; Liu, J.; Jardetzky, T.S.; Johnson, D.C. CD147 Promotes Entry of Pentamer-Expressing Human Cytomegalovirus into Epithelial and Endothelial Cells. mBio 2018, 9, e00781-18. [Google Scholar] [CrossRef] [Green Version]
- Im, E.; Venkatakrishnan, A.; Kazlauskas, A. Cathepsin B regulates the intrinsic angiogenic threshold of endothelial cells. Mol. Biol. Cell. 2005, 16, 3488–3500. [Google Scholar] [CrossRef] [Green Version]
- Platt, M.O.; Shockey, W.A. Endothelial cells and cathepsins: Biochemical and biomechanical regulation. Biochimie 2016, 122, 314–323. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Zhong, H.; Wu, J.; Chen, R.-F.; Yang, H.; Al-Abed, Y.; Li, Y.; Li, X.; Jiang, W.; Montenegro, M.F.; et al. Cathepsin L promotes Vascular Intimal Hyperplasia after Arterial Injury. Mol. Med. 2017, 23, 92–100. [Google Scholar] [CrossRef]
- Rivellese, F.; Prediletto, E. ACE2 at the centre of COVID-19 from paucisymptomatic infections to severe pneumonia. Autoimmun. Rev. 2020, 102536. [Google Scholar] [CrossRef]
- Touyz, R.M.; Li, H.; Delles, C. ACE2 the Janus-faced protein—From cardiovascular protection to severe acute respiratory syndrome-coronavirus and COVID-19. Clin. Sci. (Lond.) 2020, 134, 747–750. [Google Scholar] [CrossRef] [Green Version]
- Leng, Z.; Zhu, R.; Hou, W.; Fengchun, Z.; Yangyang, Z.; Luchan, D.; Shan, G.; Meng, F.; Du, D.; Wang, S.; et al. Transplantation of ACE2(-) Mesenchymal Stem Cells Improves the Outcome of Patients with COVID-19 Pneumonia. Aging Dis. 2020, 11, 216–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brake, S.; Barnsley, K.; Lu, W.; McAlinden, K.; Eapen, M.S.; Sohal, S.S. Smoking Upregulates Angiotensin-Converting Enzyme-2 Receptor: A Potential Adhesion Site for Novel Coronavirus SARS-CoV-2 (Covid-19). J. Clin. Med. 2020, 9, 841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakovac, H. COVID-19-is the ACE2 just a foe? Am. J. Physiol. Cell. Mol. Physiol. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Z.; Yan, Y.; Shu, Y.; Gao, R.; Sun, Y.; Li, X.; Ju, X.; Liang, Z.; Liu, Q.; Zhao, Y.; et al. Angiotensin-converting enzyme 2 protects from lethal avian influenza A H5N1 infections. Nat. Commun. 2014, 5, 3594. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Huang, Z.; Lin, L.; Lv, J. Coronavirus Disease 2019 (COVID-19) and Cardiovascular Disease: A Viewpoint on the Potential Influence of Angiotensin-Converting Enzyme Inhibitors/Angiotensin Receptor Blockers on Onset and Severity of Severe Acute Respiratory Syndrome Coronavirus 2 Infection. J. Am. Heart Assoc. 2020, 9, e016219. [Google Scholar] [PubMed]
- Mourad, J.-J.; Levy, B.I. Interaction between RAAS inhibitors and ACE2 in the context of COVID-19. Nat. Rev. Cardiol. 2020, 17, 313. [Google Scholar] [CrossRef] [Green Version]
- South, A.M.; Diz, D.; Chappell, M.C. COVID-19, ACE2 and the Cardiovascular Consequences. Am. J. Physiol. Heart Circ. Physiol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [Green Version]
- Sommerstein, R.; Kochen, M.M.; Messerli, F.H.; Grani, C. Coronavirus Disease 2019 (COVID-19): Do Angiotensin-Converting Enzyme Inhibitors/Angiotensin Receptor Blockers Have a Biphasic Effect? J. Am. Heart Assoc. 2020, 9, e016509. [Google Scholar] [CrossRef]
- Danser, A.J.; Epstein, M.; Batlle, D. Renin-Angiotensin System Blockers and the COVID-19 Pandemic: At Present There Is No Evidence to Abandon Renin-Angiotensin System Blockers. Hypertension 2020, 12015082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Hao, H.; Leeper, N.J.; Zhu, L. Thrombotic Regulation from the Endothelial Cell Perspectives. Arter. Thromb. Vasc. Biol. 2018, 38, e90–e95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernfield, M.; Götte, M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J.; Zako, M. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 1999, 68, 729–777. [Google Scholar] [CrossRef] [PubMed]
- Mast, A.E. Tissue Factor Pathway Inhibitor: Multiple Anticoagulant Activities for a Single Protein. Arter. Thromb. Vasc. Biol. 2016, 36, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Martin, F.A.; Murphy, R.P.; Cummins, P.M. Thrombomodulin and the vascular endothelium: Insights into functional, regulatory, and therapeutic aspects. Am. J. Physiol. Circ. Physiol. 2013, 304, H1585–H1597. [Google Scholar] [CrossRef] [Green Version]
- Oliver, J.; Webb, D.J.; Newby, D.E. Stimulated tissue plasminogen activator release as a marker of endothelial function in humans. Arter. Thromb. Vasc. Biol. 2005, 25, 2470–2479. [Google Scholar] [CrossRef]
- Huber, D.; Cramer, E.M.; Kaufmann, J.E.; Meda, P.; Massé, J.-M.; Kruithof, E.K.O.; Vischer, U.M. Tissue-type plasminogen activator (t-PA) is stored in Weibel-Palade bodies in human endothelial cells both in vitro and in vivo. Blood 2002, 99, 3637–3645. [Google Scholar] [CrossRef] [Green Version]
- Godo, S.; Shimokawa, H. Endothelial Functions. Arter. Thromb. Vasc Biol. 2017, 37, e108–e114. [Google Scholar] [CrossRef] [Green Version]
- Vanhoutte, P.M.; Shimokawa, H.; Tang, E.H.; Feletou, M. Endothelial dysfunction and vascular disease. Acta Physiol. 2009, 196, 193–222. [Google Scholar] [CrossRef] [Green Version]
- Boyce, S.; Lwaleed, B.; Kazmi, R. Homeostasis of Hemostasis: The Role of Endothelium. Semin. Thromb. Hemost. 2015, 41, 549–555. [Google Scholar] [CrossRef]
- Loscalzo, J. Oxidative stress in endothelial cell dysfunction and thrombosis. Pathophysiol. Haemost. Thromb. 2002, 32, 359–360. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G. Endothelial cells: The heart attack of the Clones. Sci. Transl. Med. 2018, 10, eaar7529. [Google Scholar] [CrossRef] [PubMed]
- Avogaro, A.; Albiero, M.; Menegazzo, L.; De Kreutzenberg, S.; Fadini, G.P. Endothelial dysfunction in diabetes: The role of reparatory mechanisms. Diabetes Care 2011, 34 (Suppl. 2), S285–S290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goligorsky, M.S. Vascular endothelium in diabetes. Am. J. Physiol. Physiol. 2017, 312, F266–F275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, R.; Kaur, M.; Singh, J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: Molecular insights and therapeutic strategies. Cardiovasc. Diabetol. 2018, 17, 121. [Google Scholar] [CrossRef]
- Maamoun, H.; Abdelsalam, S.S.; Zeidan, A.; Korashy, H.M.; Agouni, A. Endoplasmic Reticulum Stress: A Critical Molecular Driver of Endothelial Dysfunction and Cardiovascular Disturbances Associated with Diabetes. Int. J. Mol. Sci. 2019, 20, 1658. [Google Scholar] [CrossRef] [Green Version]
- Eringa, E.C.; Serné, E.H.; Meijer, R.I.; Schalkwijk, C.G.; Houben, A.J.H.M.; Stehouwer, C.D.A.; Smulders, Y.M.; Van Hinsbergh, V.W.M. Endothelial dysfunction in (pre)diabetes: Characteristics, causative mechanisms and pathogenic role in type 2 diabetes. Rev. Endocr. Metab. Disord. 2013, 14, 39–48. [Google Scholar] [CrossRef]
- Jansson, P.A. Endothelial dysfunction in insulin resistance and type 2 diabetes. J. Intern. Med. 2007, 262, 173–183. [Google Scholar] [CrossRef]
- Gambardella, J.; Sardu, C.; Santulli, G. COVID-19 and endothelial dysfunction. JAMA 2020, in press. [Google Scholar]
- Esler, M.; Esler, D. Can angiotensin receptor-blocking drugs perhaps be harmful in the COVID-19 pandemic? J. Hypertens. 2020, 38, 781–782. [Google Scholar] [CrossRef]
- Fang, L.; Karakiulakis, G.; Roth, M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir. Med. 2020, 8, e21. [Google Scholar] [CrossRef]
- Nascimento, I.J.B.D.; Cacic, N.; Abdulazeem, H.M.; Von Groote, T.; Jayarajah, U.; Weerasekara, I.; Esfahani, M.A.; Civile, V.T.; Marusic, A.; Jeroncic, A.; et al. Novel Coronavirus Infection (COVID-19) in Humans: A Scoping Review and Meta-Analysis. Clin. Med. 2020, 9, 941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.T.; Leung, K.; Bushman, M.; Kishore, N.; Niehus, R.; De Salazar, P.M.; Cowling, B.J.; Lipsitch, M.; Leung, G.M. Estimating clinical severity of COVID-19 from the transmission dynamics in Wuhan, China. Nat. Med. 2020, 26, 506–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, W.-J.; Ni, Z.-Y.; Hu, Y.; Liang, W.-H.; Ou, C.-Q.; He, J.-X.; Liu, L.; Shan, H.; Lei, C.-L.; Hui, D.S.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061. [Google Scholar] [CrossRef]
- Zhang, J.-J.; Dong, X.; Cao, Y.-Y.; Yuan, Y.-D.; Yang, Y.-B.; Yan, Y.-Q.; Akdis, C.A.; Gao, Y.-D. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy 2020. [Google Scholar] [CrossRef]
- Williams, B.; Mancia, G.; Spiering, W.; Rosei, E.A.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; De Simone, G.; Dominiczak, A.F.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur. Heart J. 2018, 39, 3021–3104. [Google Scholar] [CrossRef]
- Vaduganathan, M.; Vardeny, O.; Michel, T.; McMurray, J.J.V.; Pfeffer, M.A.; Solomon, S. Renin-Angiotensin-Aldosterone System Inhibitors in Patients with Covid-19. N. Engl. J. Med. 2020, 382, 1653–1659. [Google Scholar] [CrossRef]
- Gurwitz, D. Angiotensin receptor blockers as tentative SARS-CoV-2 therapeutics. Drug Dev. Res. 2020. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, X.; Chen, J.; Zhang, H.; Deng, A. Association of Renin-Angiotensin System Inhibitors With Severity or Risk of Death in Patients With Hypertension Hospitalized for Coronavirus Disease 2019 (COVID-19) Infection in Wuhan, China. JAMA Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrario, C.M.; Jessup, J.; Chappell, M.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Diz, D.I.; Gallagher, P.E. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation 2005, 111, 2605–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jessup, J.A.; Gallagher, P.E.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Chappell, M.C.; Ferrario, C.M. Effect of angiotensin II blockade on a new congenic model of hypertension derived from transgenic Ren-2 rats. Am. J. Physiol. Circ. Physiol. 2006, 291, H2166–H2172. [Google Scholar] [CrossRef] [PubMed]
- Igase, M.; Strawn, W.B.; Gallagher, P.E.; Geary, R.L.; Ferrario, C.M. Angiotensin II AT1 receptors regulate ACE2 and angiotensin-(1-7) expression in the aorta of spontaneously hypertensive rats. Am. J. Physiol. Circ. Physiol. 2005, 289, H1013–H1019. [Google Scholar] [CrossRef]
- South, A.M.; Tomlinson, L.; Edmonston, D.; Hiremath, S.; Sparks, M.A. Controversies of renin-angiotensin system inhibition during the COVID-19 pandemic. Nat. Rev. Nephrol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Ferrario, C.M.; Ahmad, S.; Groban, L. Mechanisms by which angiotensin-receptor blockers increase ACE2 levels. Nat. Rev. Cardiol. 2020. [Google Scholar] [CrossRef]
- Ishiyama, Y.; Gallagher, P.E.; Averill, D.B.; Tallant, E.A.; Brosnihan, K.B.; Ferrario, C.M. Upregulation of angiotensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension 2004, 43, 970–976. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.-Y.; Song, B.; Oudit, G.Y.; Davidge, S.T.; Yu, H.-M.; Jiang, Y.-Y.; Gao, P.-J.; Zhu, D.-L.; Ning, G.; Kassiri, Z.; et al. ACE2 deficiency enhances angiotensin II-mediated aortic profilin-1 expression, inflammation and peroxynitrite production. PLoS ONE 2012, 7, e38502. [Google Scholar] [CrossRef]
- Soler, M.J.; Ye, M.; Wysocki, J.; William, J.; Lloveras, J.; Batlle, D. Localization of ACE2 in the renal vasculature: Amplification by angiotensin II type 1 receptor blockade using telmisartan. Am. J. Physiol. Physiol. 2009, 296, F398–F405. [Google Scholar] [CrossRef] [Green Version]
- Patel, V.B.; Clarke, N.; Wang, Z.; Fan, D.; Parajuli, N.; Basu, R.; Putko, B.; Kassiri, Z.; Turner, A.J.; Oudit, G.Y. Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: A positive feedback mechanism in the RAS. J. Mol. Cell. Cardiol. 2014, 66, 167–176. [Google Scholar] [CrossRef]
- Yamamuro, M.; Yoshimura, M.; Nakayama, M.; Abe, K.; Sumida, H.; Sugiyama, S.; Saito, Y.; Nakao, K.; Yasue, H.; Ogawa, H. Aldosterone, but not angiotensin II, reduces angiotensin converting enzyme 2 gene expression levels in cultured neonatal rat cardiomyocytes. Circ. J. 2008, 72, 1346–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keidar, S.; Gamliel-Lazarovich, A.; Kaplan, M.; Pavlotzky, E.; Hamoud, S.; Hayek, T.; Karry, R.; Abassi, Z. Mineralocorticoid Receptor Blocker Increases Angiotensin-Converting Enzyme 2 Activity in Congestive Heart Failure Patients. Circ. Res. 2005, 97, 946–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuhashi, M.; Moniwa, N.; Mita, T.; Fuseya, T.; Ishimura, S.; Ohno, K.; Shibata, S.; Tanaka, M.; Watanabe, Y.; Akasaka, H.; et al. Urinary angiotensin-converting enzyme 2 in hypertensive patients may be increased by olmesartan, an angiotensin II receptor blocker. Am. J. Hypertens. 2015, 28, 15–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, G.I.; Thomas, D.A.; Grant, P.J.; Turner, A.J.; Hooper, N.M. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem. J. 2004, 383, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, J.; Peiris, M. Good ACE, bad ACE do battle in lung injury, SARS. Nat. Med. 2005, 11, 821–822. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.A. Angiotensin-(1-7). Hypertension 2014, 63, 1138–1147. [Google Scholar] [CrossRef] [PubMed]
- El-Hashim, A.Z.; Renno, W.M.; Raghupathy, R.; Abduo, H.T.; Akhtar, S.; Benter, I.F. Angiotensin-(1-7) inhibits allergic inflammation, via the MAS1 receptor, through suppression of ERK1/2- and NF-kappaB-dependent pathways. Br. J. Pharmacol. 2012, 166, 1964–1976. [Google Scholar] [CrossRef] [Green Version]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Povlsen, A.L.; Grimm, D.; Wehland, M.; Infanger, M.; Kruger, M. The Vasoactive Mas Receptor in Essential Hypertension. J. Clin. Med. 2020, 9, 267. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.B.; Verma, A. COVID-19 and Angiotensin-Converting Enzyme Inhibitors and Angiotensin Receptor Blockers: What Is the Evidence? JAMA 2020. [Google Scholar] [CrossRef]
- de Simone, G.; Mancusi, C. Speculation is not evidence: Antihypertensive therapy and COVID-19. Eur. Heart J. Cardiovasc. Pharm. 2020. [Google Scholar] [CrossRef] [PubMed]
- Iaccarino, G.; Borghi, C.; Cicero, A.F.G.; Ferri, C.; Minuz, P.; Muiesan, M.L.; Mulatero, P.; Mule, G.; Pucci, G.; Salvetti, M.; et al. Renin-Angiotensin System Inhibition in Cardiovascular Patients at the Time of COVID19: Much Ado for Nothing? A Statement of Activity from the Directors of the Board and the Scientific Directors of the Italian Society of Hypertension. High Blood Press. Cardiovasc. Prev. 2020, 27, 105–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sultana, J.; Trotta, F.; Addis, A.; Brown, J.S.; Gil, M.; Menniti-Ippolito, F.; Milozzi, F.; Suissa, S.; Trifiro, G. Healthcare Database Networks for Drug Regulatory Policies: International Workshop on the Canadian, US and Spanish Experience and Future Steps for Italy. Drug Saf. 2020, 43, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talreja, H.; Tan, J.; Dawes, M.; Supershad, S.; Rabindranath, K.; Fisher, J.; Valappil, S.; van der Merwe, V.; Wong, L.; van der Merwe, W.; et al. A consensus statement on the use of angiotensin receptor blockers and angiotensin converting enzyme inhibitors in relation to COVID-19 (corona virus disease 2019). N. Z. Med. J. 2020, 133, 85–87. [Google Scholar]
- Zhang, P.; Zhu, L.; Cai, J.; Lei, F.; Qin, J.J.; Xie, J.; Liu, Y.M.; Zhao, Y.C.; Huang, X.; Lin, L.; et al. Association of Inpatient Use of Angiotensin Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers with Mortality Among Patients With Hypertension Hospitalized With COVID-19. Circ. Res. 2020. [Google Scholar] [CrossRef]
- Yang, G.; Tan, Z.; Zhou, L.; Yang, M.; Peng, L.; Liu, J.; Cai, J.; Yang, R.; Han, J.; Huang, Y.; et al. Effects Of ARBs And ACEIs On Virus Infection, Inflammatory Status And Clinical Outcomes In COVID-19 Patients With Hypertension: A Single Center Retrospective Study. Hypertension 2020. [Google Scholar] [CrossRef]
- Mehra, M.; Desai, S.; Kuy, S.; Henry, T.; Patel, A. Cardiovascular Disease, Drug Therapy, and Mortality in Covid-19. NEJM 2019, in press. [Google Scholar] [CrossRef]
- Reynolds, H.; Adhikari, S.; Pulgarin, C.; Troxel, A.; Iturrate, E.; Johnson, S.; Hausvater, A.; Newman, J.; Berger, J.; Bangalore, S.; et al. Renin–Angiotensin–Aldosterone System Inhibitors and Risk of Covid-19. NEJM 2019, in press. [Google Scholar] [CrossRef]
- Mancia, G.; Rea, F.; Ludergnani, M.; Apolone, G.; Corrao, G. Renin–Angiotensin–Aldosterone System Inhibitors and Risk of Covid-19. NEJM 2019, in press. [Google Scholar]
- Rajagopalan, S.; Harrison, D.G. Reversing endothelial dysfunction with ACE inhibitors. A new trend. Circulation 1996, 94, 240–243. [Google Scholar] [CrossRef]
- Beckman, J.A.; Creager, M.A. The nonlipid effects of statins on endothelial function. Trends Cardiovasc. Med. 2006, 16, 156–162. [Google Scholar] [CrossRef]
- Ruszkowski, P.; Masajtis-Zagajewska, A.; Nowicki, M. Effects of combined statin and ACE inhibitor therapy on endothelial function and blood pressure in essential hypertension—A randomised double-blind, placebo controlled crossover study. J. Renin Angiotensin Aldosterone Syst. 2019, 20, 1470320319868890. [Google Scholar] [CrossRef] [Green Version]
- Blum, A.; Shamburek, R. The pleiotropic effects of statins on endothelial function, vascular inflammation, immunomodulation and thrombogenesis. Atherosclerosis 2009, 203, 325–330. [Google Scholar] [CrossRef]
- Glowacka, I.; Bertram, S.; Herzog, P.; Pfefferle, S.; Steffen, I.; Muench, M.O.; Simmons, G.; Hofmann, H.; Kuri, T.; Weber, F.; et al. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J. Virol. 2010, 84, 1198–1205. [Google Scholar] [CrossRef] [Green Version]
- Luque, M.; Martin, P.; Martell, N.; Fernandez, C.; Brosnihan, K.B.; Ferrario, C.M. Effects of captopril related to increased levels of prostacyclin and angiotensin-(1-7) in essential hypertension. J. Hypertens. 1996, 14, 799–805. [Google Scholar] [CrossRef]
- Chen, L.; Hao, G. The role of angiotensin-converting enzyme 2 in coronaviruses/influenza viruses and cardiovascular disease. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef]
- Murray, E.; Tomaszewski, M.; Guzik, T.J. Binding of SARS-CoV-2 and angiotensin-converting enzyme 2: Clinical implications. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef]
- Sunden-Cullberg, J. Chronic Use of Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers Is High Among Intensive Care Unit Patients With Non-COVID-19 Sepsis but Carry a Moderately Increased Risk of Death. Hypertension 2020. [Google Scholar] [CrossRef]
- Fan, Z.; Wu, G.; Yue, M.; Ye, J.; Chen, Y.; Xu, B.; Shu, Z.; Zhu, J.; Lu, N.; Tan, X. Hypertension and hypertensive left ventricular hypertrophy are associated with ACE2 genetic polymorphism. Life Sci. 2019, 225, 39–45. [Google Scholar] [CrossRef]
- Pinheiro, D.S.; Santos, R.S.; Jardim, P.; Silva, E.G.; Reis, A.A.S.; Pedrino, G.R.; Ulhoa, C.J. The combination of ACE I/D and ACE2 G8790A polymorphisms revels susceptibility to hypertension: A genetic association study in Brazilian patients. PLoS ONE 2019, 14, e0221248. [Google Scholar] [CrossRef] [Green Version]
- Lackland, D.T. Racial differences in hypertension: Implications for high blood pressure management. Am. J. Med. Sci. 2014, 348, 135–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonow, R.O.; Fonarow, G.C.; O’Gara, P.T.; Yancy, C.W. Association of Coronavirus Disease 2019 (COVID-19) With Myocardial Injury and Mortality. JAMA Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020. [Google Scholar] [CrossRef] [Green Version]
- De Filippo, O.; D’Ascenzo, F.; Angelini, F.; Bocchino, P.P.; Conrotto, F.; Saglietto, A.; Secco, G.G.; Campo, G.; Gallone, G.; Verardi, R.; et al. Reduced Rate of Hospital Admissions for ACS during Covid-19 Outbreak in Northern Italy. New Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Garcia, S.; Albaghdadi, M.S.; Meraj, P.M.; Schmidt, C.; Garberich, R.; Jaffer, F.A.; Dixon, S.; Rade, J.J.; Tannenbaum, M.; Chambers, J.; et al. Reduction in ST-Segment Elevation Cardiac Catheterization Laboratory Activations in the United States during COVID-19 Pandemic. J. Am. Coll Cardiol. 2020. [Google Scholar] [CrossRef]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Fanelli, V.; Fiorentino, M.; Cantaluppi, V.; Gesualdo, L.; Stallone, G.; Ronco, C.; Castellano, G. Acute kidney injury in SARS-CoV-2 infected patients. Crit. Care 2020, 24, 155. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Luo, R.; Wang, K.; Zhang, M.; Wang, Z.; Dong, L.; Li, J.; Yao, Y.; Ge, S.; Xu, G. Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney Int. 2020. [Google Scholar] [CrossRef]
- Henry, B.M.; Lippi, G. Chronic kidney disease is associated with severe coronavirus disease 2019 (COVID-19) infection. Int. Urol. Nephrol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Ye, M.; Wysocki, J.; William, J.; Soler, M.J.; Cokic, I.; Batlle, D. Glomerular localization and expression of Angiotensin-converting enzyme 2 and Angiotensin-converting enzyme: Implications for albuminuria in diabetes. J. Am. Soc. Nephrol. 2006, 17, 3067–3075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.; We, X.; Liu, C.; Volpe, G.; Wang, Z. Single-cell atlas of a non-human primate reveals new pathogenic mechanisms of COVID-19. bioRXiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Su, H.; Yang, M.; Wan, C.; Yi, L.X.; Tang, F.; Zhu, H.Y.; Yi, F.; Yang, H.C.; Fogo, A.B.; Nie, X.; et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 2020. [Google Scholar] [CrossRef] [PubMed]
- Gentile, S.; Strollo, F.; Ceriello, A. COVID-19 Infection in italian people with diabetes: Lessons learned for our future (an experience to be used). Diabetes Res. Clin. Pract. 2020, 108137. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.C.W.; Holt, R.I.G. COVID-19 and diabetes. Diabet. Med. 2020, 37, 723–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muniyappa, R.; Gubbi, S. COVID-19 Pandemic, Corona Viruses, and Diabetes Mellitus. Am. J. Physiol. Endocrinol. Metab. 2020. [Google Scholar] [CrossRef] [Green Version]
- Arentz, M.; Yim, E.; Klaff, L.; Lokhandwala, S.; Riedo, F.X.; Chong, M.; Lee, M. Characteristics and Outcomes of 21 Critically Ill Patients With COVID-19 in Washington State. JAMA 2020, 323, 1612. [Google Scholar] [CrossRef] [Green Version]
- Bornstein, S.R.; Dalan, R.; Hopkins, D.; Mingrone, G.; Boehm, B.O. Endocrine and metabolic link to coronavirus infection. Nat. Rev. Endocrinol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Remuzzi, A.; Remuzzi, G. COVID-19 and Italy: What next? Lancet 2020, 395, 1225–1228. [Google Scholar] [CrossRef]
- Fadini, G.P.; Morieri, M.L.; Longato, E.; Avogaro, A. Prevalence and impact of diabetes among people infected with SARS-CoV-2. J. Endocrinol. Investig. 2020. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Yang, J.; Zhao, F.; Zhi, L.; Wang, X.; Liu, L.; Bi, Z.; Zhao, Y. Prevalence and impact of cardiovascular metabolic diseases on COVID-19 in China. Clin. Res. Cardiol. 2020, 109, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.K.; Feng, Y.; Yuan, M.Y.; Yuan, S.Y.; Fu, H.J.; Wu, B.Y.; Sun, G.Z.; Yang, G.R.; Zhang, X.L.; Wang, L.; et al. Plasma glucose levels and diabetes are independent predictors for mortality and morbidity in patients with SARS. Diabet. Med. 2006, 23, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Simonnet, A.; Chetboun, M.; Poissy, J.; Raverdy, V.; Noulette, J.; Duhamel, A.; Labreuche, J.; Mathieu, D.; Pattou, F.; Jourdain, M. High prevalence of obesity in severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) requiring invasive mechanical ventilation. Obesity (Silver Spring) 2020. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Zhao, W.; Xu, Z.; Gu, J. Timely blood glucose management for the outbreak of 2019 novel coronavirus disease (COVID-19) is urgently needed. Diabetes Res. Clin. Pract. 2020, 162, 108118. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.K.; Lin, S.S.; Ji, X.J.; Guo, L.M. Binding of SARS coronavirus to its receptor damages islets and causes acute diabetes. Acta Diabetol. 2010, 47, 193–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.L.; Wang, Y.; Yuan, L.; Li, Y.; Li, X.Y. The angiotensin-converting enzyme 2/angiotensin (1-7)/Mas axis protects the function of pancreatic beta cells by improving the function of islet microvascular endothelial cells. Int. J. Mol. Med. 2014, 34, 1293–1300. [Google Scholar] [CrossRef] [Green Version]
- Xuan, X.; Gao, F.; Ma, X.; Huang, C.; Wang, Y.; Deng, H.; Wang, S.; Li, W.; Yuan, L. Activation of ACE2/angiotensin (1-7) attenuates pancreatic beta cell dedifferentiation in a high-fat-diet mouse model. Metabolism 2018, 81, 83–96. [Google Scholar] [CrossRef]
- Shoemaker, R.; Yiannikouris, F.; Thatcher, S.; Cassis, L. ACE2 deficiency reduces beta-cell mass and impairs beta-cell proliferation in obese C57BL/6 mice. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E621–E631. [Google Scholar] [CrossRef] [Green Version]
- Bindom, S.M.; Lazartigues, E. The sweeter side of ACE2: Physiological evidence for a role in diabetes. Mol. Cell Endocrinol. 2009, 302, 193–202. [Google Scholar] [CrossRef]
- Roca-Ho, H.; Riera, M.; Palau, V.; Pascual, J.; Soler, M.J. Characterization of ACE and ACE2 Expression within Different Organs of the NOD Mouse. Int. J. Mol. Sci. 2017, 18, 563. [Google Scholar] [CrossRef] [Green Version]
- Blodgett, D.M.; Nowosielska, A.; Afik, S.; Pechhold, S.; Cura, A.J.; Kennedy, N.J.; Kim, S.; Kucukural, A.; Davis, R.J.; Kent, S.C.; et al. Novel Observations From Next-Generation RNA Sequencing of Highly Purified Human Adult and Fetal Islet Cell Subsets. Diabetes 2015, 64, 3172–3181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Bender, A.; Wang, P.; Karakose, E.; Inabnet, W.B.; Libutti, S.K.; Arnold, A.; Lambertini, L.; Stang, M.; Chen, H.; et al. Insights into beta cell regeneration for diabetes via integration of molecular landscapes in human insulinomas. Nat. Commun. 2017, 8, 767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Wang, X.; Chen, J.; Zuo, X.; Zhang, H.; Deng, A. COVID-19 infection may cause ketosis and ketoacidosis. Diabetes Obes. Metab. 2020. [Google Scholar] [CrossRef] [PubMed]
- Stoian, A.P.; Banerjee, Y.; Rizvi, A.A.; Rizzo, M. Diabetes and the COVID-19 Pandemic: How Insights from Recent Experience Might Guide Future Management. Metab. Syndr. Relat. Disord. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.D.; Xiao, M.; Zhang, S.; Xia, P.; Caio, W.; Jiang, W. Coagulopathy and Antiphospholipid Antibodies in Patients with Covid-19. NEJM 2020, 382, e38. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; She, J.; Wang, Y.; Ma, X. Venous thrombosis and arteriosclerosis obliterans of lower extremities in a very severe patient with 2019 novel coronavirus disease: A case report. J. Thromb. Thrombolysis 2020. [Google Scholar] [CrossRef]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Lu, L.; Cao, W.; Li, T. Hypothesis for potential pathogenesis of SARS-CoV-2 infection-a review of immune changes in patients with viral pneumonia. Emerg. Microbes Infect. 2020, 9, 727–732. [Google Scholar] [CrossRef] [Green Version]
- Iba, T.; Levy, J.H.; Warkentin, T.E.; Thachil, J.; van der Poll, T.; Levi, M. Scientific, Standardization Committee on DIC, the S, Standardization Committee on P, Critical Care of the International Society on T and Haemostasis. Diagnosis and management of sepsis-induced coagulopathy and disseminated intravascular coagulation. J. Thromb. Haemost. 2019, 17, 1989–1994. [Google Scholar] [CrossRef] [Green Version]
- Escher, R.; Breakey, N.; Lammle, B. Severe COVID-19 infection associated with endothelial activation. Thromb. Res. 2020, 190, 62. [Google Scholar] [CrossRef]
- McCormack, J.J.; Lopes da Silva, M.; Ferraro, F.; Patella, F.; Cutler, D.F. Weibel-Palade bodies at a glance. J. Cell Sci. 2017, 130, 3611–3617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, B.; Baker, A.Q.; Gallacher, B.; Lodwick, D. Angiotensin II increases vascular permeability factor gene expression by human vascular smooth muscle cells. Hypertension 1995, 25, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Victorino, G.P.; Newton, C.R.; Curran, B. Effect of angiotensin II on microvascular permeability. J. Surg. Res. 2002, 104, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Dielis, A.W.; Smid, M.; Spronk, H.M.; Hamulyak, K.; Kroon, A.A.; ten Cate, H.; de Leeuw, P.W. The prothrombotic paradox of hypertension: Role of the renin-angiotensin and kallikrein-kinin systems. Hypertension 2005, 46, 1236–1242. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Barker, T.A.; Berk, B.C. Angiotensin II and the endothelium: Diverse signals and effects. Hypertension 2005, 45, 163–169. [Google Scholar] [CrossRef]
- Celi, A.; Cianchetti, S.; Dell’Omo, G.; Pedrinelli, R. Angiotensin II, tissue factor and the thrombotic paradox of hypertension. Expert Rev. Cardiovasc. Ther. 2010, 8, 1723–1729. [Google Scholar] [CrossRef]
- Jagroop, I.A.; Mikhailidis, D.P. Angiotensin II can induce and potentiate shape change in human platelets: Effect of losartan. J. Hum. Hypertens. 2000, 14, 581–585. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.A.; MacIntyre, D.E.; Kenyon, C.J.; Semple, P.F. Angiotensin II effects on platelet function. J. Hypertens. Suppl. 1985, 3, S251–S253. [Google Scholar] [CrossRef]
- Larsson, P.T.; Schwieler, J.H.; Wallen, N.H. Platelet activation during angiotensin II infusion in healthy volunteers. Blood Coagul. Fibrinolysis 2000, 11, 61–69. [Google Scholar] [CrossRef]
- Langeggen, H.; Berge, K.E.; Macor, P.; Fischetti, F.; Tedesco, F.; Hetland, G.; Berg, K.; Johnson, E. Detection of mRNA for the terminal complement components C5, C6, C8 and C9 in human umbilical vein endothelial cells in vitro. Apmis 2001, 109, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Langeggen, H.; Pausa, M.; Johnson, E.; Casarsa, C.; Tedesco, F. The endothelium is an extrahepatic site of synthesis of the seventh component of the complement system. Clin. Exp. Immunol. 2000, 121, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Dauchel, H.; Julen, N.; Lemercier, C.; Daveau, M.; Ozanne, D.; Fontaine, M.; Ripoche, J. Expression of complement alternative pathway proteins by endothelial cells. Differential regulation by interleukin 1 and glucocorticoids. Eur. J. Immunol. 1990, 20, 1669–1675. [Google Scholar] [CrossRef] [PubMed]
- Warren, H.B.; Pantazis, P.; Davies, P.F. The third component of complement is transcribed and secreted by cultured human endothelial cells. Am. J. Pathol. 1987, 129, 9–13. [Google Scholar] [PubMed]
- Johnson, E.; Hetland, G. Human umbilical vein endothelial cells synthesize functional C3, C5, C6, C8 and C9 in vitro. Scand J. Immunol. 1991, 33, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Shagdarsuren, E.; Wellner, M.; Braesen, J.H.; Park, J.K.; Fiebeler, A.; Henke, N.; Dechend, R.; Gratze, P.; Luft, F.C.; Muller, D.N. Complement activation in angiotensin II-induced organ damage. Circ. Res. 2005, 97, 716–724. [Google Scholar] [CrossRef] [Green Version]
- Ruan, C.C.; Gao, P.J. Role of Complement-Related Inflammation and Vascular Dysfunction in Hypertension. Hypertension 2019, 73, 965–971. [Google Scholar] [CrossRef]
- Fischetti, F.; Tedesco, F. Cross-talk between the complement system and endothelial cells in physiologic conditions and in vascular diseases. Autoimmunity 2006, 39, 417–428. [Google Scholar] [CrossRef]
- Risitano, A.M.; Mastellos, D.C.; Huber-Lang, M.; Yancopoulou, D.; Garlanda, C.; Ciceri, F.; Lambris, J.D. Complement as a target in COVID-19? Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Schulz, C.; Engelmann, B.; Massberg, S. Crossroads of coagulation and innate immunity: The case of deep vein thrombosis. J. Thromb. Haemost. 2013, 11 (Suppl. 1), 233–241. [Google Scholar] [CrossRef]
- abret, N.; Britton, G.J.; Gruber, C.; Hegde, S.; Kim, J.; Kuksin, M.; Levantovsky, R.; Malle, L.; Moreira, A.; Park, M.D.; et al. The Sinai Immunology Review Project. Immunology of COVID-19: Current state of the science. Immunity 2020. [Google Scholar] [CrossRef]
- Steinberg, B.E.; Goldenberg, N.M.; Lee, W.L. Do viral infections mimic bacterial sepsis? The role of microvascular permeability: A review of mechanisms and methods. Antiviral. Res. 2012, 93, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Pappas, C.; Belser, J.A.; Houser, K.V.; Zhong, W.; Wadford, D.A.; Stevens, T.; Balczon, R.; Katz, J.M.; Tumpey, T.M. Human pulmonary microvascular endothelial cells support productive replication of highly pathogenic avian influenza viruses: Possible involvement in the pathogenesis of human H5N1 virus infection. J. Virol. 2012, 86, 667–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maniatis, N.A.; Orfanos, S.E. The endothelium in acute lung injury/acute respiratory distress syndrome. Curr. Opin. Crit. Care 2008, 14, 22–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polverino, F.; Celli, B.R.; Owen, C.A. COPD as an endothelial disorder: Endothelial injury linking lesions in the lungs and other organs? (2017 Grover Conference Series). Pulm. Circ. 2018, 8, 2045894018758528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millar, F.R.; Summers, C.; Griffiths, M.J.; Toshner, M.R.; Proudfoot, A.G. The pulmonary endothelium in acute respiratory distress syndrome: Insights and therapeutic opportunities. Thorax 2016, 71, 462–473. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, S.M.; Wang, C.; Tigdi, J.; Si, X.; Dumpit, C.; Charles, S.; Gamage, A.; Moraes, T.J.; Lee, W.L. Influenza infects lung microvascular endothelium leading to microvascular leak: Role of apoptosis and claudin-5. PLoS ONE 2012, 7, e47323. [Google Scholar] [CrossRef] [Green Version]
- Chan, M.C.; Chan, R.W.; Yu, W.C.; Ho, C.C.; Chui, W.H.; Lo, C.K.; Yuen, K.M.; Guan, Y.I.; Nicholls, J.M.; Peiris, J.S. Influenza H5N1 virus infection of polarized human alveolar epithelial cells and lung microvascular endothelial cells. Respir. Res. 2009, 10, 102. [Google Scholar] [CrossRef]
- Lee, N.; Hui, D.; Wu, A.; Chan, P.; Cameron, P.; Joynt, G.M.; Ahuja, A.; Yung, M.Y.; Leung, C.B.; To, K.F.; et al. A major outbreak of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 2003, 348, 1986–1994. [Google Scholar] [CrossRef]
- Wong, R.S.; Wu, A.; To, K.F.; Lee, N.; Lam, C.W.; Wong, C.K.; Chan, P.K.; Ng, M.H.; Yu, L.M.; Hui, D.S.; et al. Haematological manifestations in patients with severe acute respiratory syndrome: Retrospective analysis. BMJ 2003, 326, 1358–1362. [Google Scholar] [CrossRef] [Green Version]
- Xiang-Hua, Y.; Le-Min, W.; Ai-Bin, L.; Zhu, G.; Riquan, L.; Xu-You, Z.; Wei-Wei, R.; Ye-Nan, W. Severe acute respiratory syndrome and venous thromboembolism in multiple organs. Am. J. Respir. Crit. Care Med. 2010, 182, 436–437. [Google Scholar] [CrossRef] [PubMed]
- de Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Bunce, P.E.; High, S.M.; Nadjafi, M.; Stanley, K.; Liles, W.C.; Christian, M.D. Pandemic H1N1 influenza infection and vascular thrombosis. Clin. Infect. Dis. 2011, 52, e14–e17. [Google Scholar] [CrossRef] [Green Version]
- Huzmeli, C.; Saglam, M.; Arikan, A.; Doner, B.; Akinci, G.; Candan, F. Infrarenal Aorta Thrombosis Associated with H1N1 Influenza A Virus Infection. Case Rep. Infect. Dis. 2016, 2016, 9567495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiguro, T.; Matsuo, K.; Fujii, S.; Takayanagi, N. Acute thrombotic vascular events complicating influenza-associated pneumonia. Respir. Med. Case Rep. 2019, 28, 100884. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. A new shield for a cytokine storm. Cell 2011, 146, 861–862. [Google Scholar] [CrossRef] [Green Version]
- Tscherne, D.M.; Garcia-Sastre, A. Virulence determinants of pandemic influenza viruses. J. Clin. Investig. 2011, 121, 6–13. [Google Scholar] [CrossRef]
- Canna, S.W.; Behrens, E.M. Making sense of the cytokine storm: A conceptual framework for understanding, diagnosing, and treating hemophagocytic syndromes. Pediatr. Clin. N. Am. 2012, 59, 329–344. [Google Scholar] [CrossRef] [Green Version]
- Chaudhry, H.; Zhou, J.; Zhong, Y.; Ali, M.M.; McGuire, F.; Nagarkatti, P.S.; Nagarkatti, M. Role of cytokines as a double-edged sword in sepsis. In Vivo 2013, 27, 669–684. [Google Scholar]
- Bracaglia, C.; Prencipe, G.; De Benedetti, F. Macrophage Activation Syndrome: Different mechanisms leading to a one clinical syndrome. Pediatr. Rheumatol. Online J. 2017, 15, 5. [Google Scholar] [CrossRef] [Green Version]
- Hayden, A.; Park, S.; Giustini, D.; Lee, A.Y.; Chen, L.Y. Hemophagocytic syndromes (HPSs) including hemophagocytic lymphohistiocytosis (HLH) in adults: A systematic scoping review. Blood Rev. 2016, 30, 411–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; Hlh Across Speciality Collaboration UK. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Gattinoni, L.; Coppola, S.; Cressoni, M.; Busana, M.; Rossi, S.; Chiumello, D. Covid-19 Does Not Lead to a “Typical” Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, S.F.; Ho, Y.C. SARS-CoV-2: A storm is raging. J. Clin. Investig. 2020, 130, 2202–2205. [Google Scholar] [CrossRef] [PubMed]
- Staedtke, V.; Bai, R.Y.; Kim, K.; Darvas, M.; Davila, M.L.; Riggins, G.J.; Rothman, P.B.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Disruption of a self-amplifying catecholamine loop reduces cytokine release syndrome. Nature 2018, 564, 273–277. [Google Scholar] [CrossRef]
- Sinha, P.; Delucchi, K.L.; McAuley, D.F.; O’Kane, C.M.; Matthay, M.A.; Calfee, C.S. Development and validation of parsimonious algorithms to classify acute respiratory distress syndrome phenotypes: A secondary analysis of randomised controlled trials. Lancet Respir. Med. 2020, 8, 247–257. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Walsh, K.B.; Cahalan, S.; Fremgen, D.M.; Roberts, E.; Scott, F.; Martinborough, E.; Peach, R.; Oldstone, M.B.; Rosen, H. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 2011, 146, 980–991. [Google Scholar] [CrossRef] [Green Version]
- Sorriento, D.; Santulli, G.; Del Giudice, C.; Anastasio, A.; Trimarco, B.; Iaccarino, G. Endothelial cells are able to synthesize and release catecholamines both in vitro and in vivo. Hypertension 2012, 60, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Wang, X.; Yang, P.; Shutong, Z. COVID-19 complicated by acute pulmonary embolism. Radiology 2020, 2, e200067. [Google Scholar] [CrossRef] [Green Version]
- Danzi, G.B.; Loffi, M.; Galeazzi, G.; Gherbesi, E. Acute pulmonary embolism and COVID-19 pneumonia: A random association? Eur. Heart J. 2020. [Google Scholar] [CrossRef] [Green Version]
- Leonard-Lorant, I.; Delabranche, X.; Severac, F.; Helms, J.; Pauzet, C.; Collange, O.; Schneider, F.; Labani, A.; Bilbault, P.; Moliere, S.; et al. Acute Pulmonary Embolism in COVID-19 Patients on CT Angiography and Relationship to D-Dimer Levels. Radiology 2020, 201561. [Google Scholar] [CrossRef] [Green Version]
- Jolobe, O.M.P. Similarities Between Community-Acquired Pneumonia and Pulmonary Embolism. Am. J. Med. 2019, 132, e863. [Google Scholar] [CrossRef] [Green Version]
- Santulli, G. MicroRNAs and Endothelial (Dys) Function. J. Cell Physiol. 2016, 231, 1638–1644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Q.; Yang, J.; Santulli, G.; Reiken, S.R.; Wronska, A.; Kim, M.M.; Osborne, B.W.; Lacampagne, A.; Yin, Y.; Marks, A.R. Maintenance of normal blood pressure is dependent on IP3R1-mediated regulation of eNOS. Proc. Natl. Acad. Sci. USA 2016, 113, 8532–8537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gando, S.; Levi, M.; Toh, C.H. Disseminated intravascular coagulation. Nat. Rev. Dis. Primers. 2016, 2, 16037. [Google Scholar] [CrossRef] [PubMed]
- Walborn, A.; Rondina, M.; Mosier, M.; Fareed, J.; Hoppensteadt, D. Endothelial Dysfunction Is Associated with Mortality and Severity of Coagulopathy in Patients with Sepsis and Disseminated Intravascular Coagulation. Clin. Appl. Thromb. Hemost. 2019, 25, 1076029619852163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, N.; Bai, H.; Chen, X.; Gong, J.; Li, D.; Sun, Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 2020. [Google Scholar] [CrossRef] [PubMed]
- Rubin, E.J.; Baden, L.R.; Morrissey, S. Audio Interview: New Research on Possible Treatments for Covid-19. New Engl. J. Med. 2020, 382, e30. [Google Scholar] [CrossRef]
- Ahn, D.G.; Shin, H.J.; Kim, M.H.; Lee, S.; Kim, H.S.; Myoung, J.; Kim, B.T.; Kim, S.J. Current Status of Epidemiology, Diagnosis, Therapeutics, and Vaccines for Novel Coronavirus Disease 2019 (COVID-19). J. Microbiol. Biotechnol. 2020, 30, 313–324. [Google Scholar] [CrossRef]
- Xu, X.; Han, M.; Li, T.; Sun, W.; Wang, D.; Fu, B.; Zhou, Y.; Zheng, X.; Yang, Y.; Li, X.; et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc. Natl. Acad. Sci. USA 2020, 202005615. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Limon, P.; Ortega, R.; Arias de la Rosa, I.; Abalos-Aguilera, M.D.C.; Perez-Sanchez, C.; Jimenez-Gomez, Y.; Peralbo-Santaella, E.; Font, P.; Ruiz-Vilches, D.; Ferrin, G.; et al. Tocilizumab improves the proatherothrombotic profile of rheumatoid arthritis patients modulating endothelial dysfunction, NETosis, and inflammation. Transl. Res. 2017, 183, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Kajikawa, M.; Higashi, Y.; Tomiyama, H.; Maruhashi, T.; Kurisu, S.; Kihara, Y.; Mutoh, A.; Ueda, S.I. Effect of short-term colchicine treatment on endothelial function in patients with coronary artery disease. Int. J. Cardiol. 2019, 281, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Parchure, N.; Zouridakis, E.G.; Kaski, J.C. Effect of azithromycin treatment on endothelial function in patients with coronary artery disease and evidence of Chlamydia pneumoniae infection. Circulation 2002, 105, 1298–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, T.; Chen, B.; Zhao, Z.; He, N.; Zeng, Z.; Wu, B.; Fukushima, Y.; Dai, M.; Huang, Q.; Xu, D.; et al. Histamine H2 receptor activation exacerbates myocardial ischemia/reperfusion injury by disturbing mitochondrial and endothelial function. Basic Res. Cardiol. 2013, 108, 342. [Google Scholar] [CrossRef] [PubMed]
- Yazdany, J.; Kim, A.H.J. Use of Hydroxychloroquine and Chloroquine During the COVID-19 Pandemic: What Every Clinician Should Know. Ann. Intern. Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- Le, N.T.; Takei, Y.; Izawa-Ishizawa, Y.; Heo, K.S.; Lee, H.; Smrcka, A.V.; Miller, B.L.; Ko, K.A.; Ture, S.; Morrell, C.; et al. Identification of activators of ERK5 transcriptional activity by high-throughput screening and the role of endothelial ERK5 in vasoprotective effects induced by statins and antimalarial agents. J. Immunol. 2014, 193, 3803–3815. [Google Scholar] [CrossRef]
- Rahman, R.; Murthi, P.; Singh, H.; Gurusinghe, S.; Mockler, J.C.; Lim, R.; Wallace, E.M. The effects of hydroxychloroquine on endothelial dysfunction. Pregnancy Hypertens. 2016, 6, 259–262. [Google Scholar] [CrossRef]
- Gambardella, J.; Morelli, M.; Sardu, C.; Santulli, G. Targeting Endothelial Dysfunction in COVID-19. Nature 2020, in press. [Google Scholar]
- Ciccarelli, M.; Santulli, G.; Campanile, A.; Galasso, G.; Cervero, P.; Altobelli, G.G.; Cimini, V.; Pastore, L.; Piscione, F.; Trimarco, B.; et al. Endothelial alpha1-adrenoceptors regulate neo-angiogenesis. Br. J. Pharmacol. 2008, 153, 936–946. [Google Scholar] [CrossRef]
- Wilbert-Lampen, U.; Seliger, C.; Zilker, T.; Arendt, R.M. Cocaine increases the endothelial release of immunoreactive endothelin and its concentrations in human plasma and urine: Reversal by coincubation with sigma-receptor antagonists. Circulation 1998, 98, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Amer, M.S.; McKeown, L.; Tumova, S.; Liu, R.; Seymour, V.A.; Wilson, L.A.; Naylor, J.; Greenhalgh, K.; Hou, B.; Majeed, Y.; et al. Inhibition of endothelial cell Ca(2)(+) entry and transient receptor potential channels by Sigma-1 receptor ligands. Br. J. Pharmacol. 2013, 168, 1445–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massamiri, T.; Duckles, S.P. Sigma receptor ligands inhibit rat tail artery contractile responses by multiple mechanisms. J. Pharmacol. Exp. Ther. 1991, 259, 22–29. [Google Scholar] [PubMed]
- Hamidi Shishavan, M.; Henning, R.H.; van Buiten, A.; Goris, M.; Deelman, L.E.; Buikema, H. Metformin Improves Endothelial Function and Reduces Blood Pressure in Diabetic Spontaneously Hypertensive Rats Independent from Glycemia Control: Comparison to Vildagliptin. Sci. Rep. 2017, 7, 10975. [Google Scholar] [CrossRef] [Green Version]
- Bolz, S.S.; Pohl, U. Indomethacin enhances endothelial NO release--evidence for a role of PGI2 in the autocrine control of calcium-dependent autacoid production. Cardiovasc. Res. 1997, 36, 437–444. [Google Scholar] [CrossRef] [Green Version]
- Sfikakis, P.P.; Papamichael, C.; Stamatelopoulos, K.S.; Tousoulis, D.; Fragiadaki, K.G.; Katsichti, P.; Stefanadis, C.; Mavrikakis, M. Improvement of vascular endothelial function using the oral endothelin receptor antagonist bosentan in patients with systemic sclerosis. Arthritis Rheum. 2007, 56, 1985–1993. [Google Scholar] [CrossRef]
- Gupta, N.; Zhao, Y.Y.; Evans, C.E. The stimulation of thrombosis by hypoxia. Thromb. Res. 2019, 181, 77–83. [Google Scholar] [CrossRef]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Paranjpe, I.; Fuster, V.; Lala, A.; Russak, A.; Glicksberg, B.S.; Levin, M.A.; Charney, A.W.; Narula, J.; Fayad, Z.A.; Bagiella, E.; et al. Association of Treatment Dose Anticoagulation with In-Hospital Survival Among Hospitalized Patients with COVID-19. J. Am. Coll. Cardiol. 2020. [Google Scholar] [CrossRef]
- Basu, A.; Kanda, T.; Beyene, A.; Saito, K.; Meyer, K.; Ray, R. Sulfated homologues of heparin inhibit hepatitis C virus entry into mammalian cells. J. Virol. 2007, 81, 3933–3941. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Pavy, M.; Young, N.; Freeman, C.; Lobigs, M. Antiviral effect of the heparan sulfate mimetic, PI-88, against dengue and encephalitic flaviviruses. Antiviral. Res. 2006, 69, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.J.; Pizzato, M.; Takeuchi, Y.; Devereux, S. Heparin binds to murine leukemia virus and inhibits Env-independent attachment and infection. J. Virol. 2002, 76, 6909–6918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connell, B.J.; Lortat-Jacob, H. Human immunodeficiency virus and heparan sulfate: From attachment to entry inhibition. Front. Immunol. 2013, 4, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahmias, A.J.; Kibrick, S. Inhibitory effect of heparin on herpes simplex virus. J. Bacteriol. 1964, 87, 1060–1066. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sardu, C.; Gambardella, J.; Morelli, M.B.; Wang, X.; Marfella, R.; Santulli, G. Hypertension, Thrombosis, Kidney Failure, and Diabetes: Is COVID-19 an Endothelial Disease? A Comprehensive Evaluation of Clinical and Basic Evidence. J. Clin. Med. 2020, 9, 1417. https://doi.org/10.3390/jcm9051417
Sardu C, Gambardella J, Morelli MB, Wang X, Marfella R, Santulli G. Hypertension, Thrombosis, Kidney Failure, and Diabetes: Is COVID-19 an Endothelial Disease? A Comprehensive Evaluation of Clinical and Basic Evidence. Journal of Clinical Medicine. 2020; 9(5):1417. https://doi.org/10.3390/jcm9051417
Chicago/Turabian StyleSardu, Celestino, Jessica Gambardella, Marco Bruno Morelli, Xujun Wang, Raffaele Marfella, and Gaetano Santulli. 2020. "Hypertension, Thrombosis, Kidney Failure, and Diabetes: Is COVID-19 an Endothelial Disease? A Comprehensive Evaluation of Clinical and Basic Evidence" Journal of Clinical Medicine 9, no. 5: 1417. https://doi.org/10.3390/jcm9051417