Genotype-Related Clinical Characteristics and Myocardial Fibrosis and Their Association with Prognosis in Hypertrophic Cardiomyopathy

 , , , ,
, , , ,
Abstract
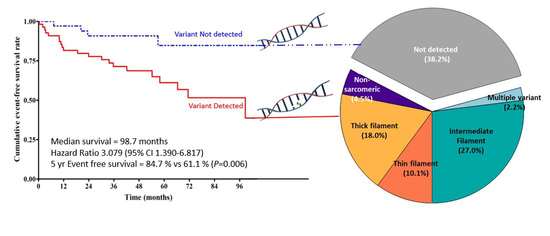
:
1. Introduction
2. Materials and Methods
2.1. Subjects and Design of the Study
2.2. Genetic Testing and Data Analysis
2.3. Criteria for Evidence-Based Classification of Candidate Variants
2.4. Data Collection
2.5. 2D Echocardiography
2.6. Acquisition and Analysis of CMR Images
2.7. Outcomes
2.8. Statistical Analysis
3. Results
3.1. Study Population and Baseline Clinical Characteristics
3.2. Variant Profile of HCM Patients
3.3. Differences in Baseline Clinical Characteristics between HCM Patients in Detected vs. Not-Detected Groups
3.4. Imaging Phenotypes of HCM Patients in Detected vs. Not Detected Groups
3.5. Clinical Outcomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maron, B.J.; McKenna, W.J.; Danielson, G.K.; Kappenberger, L.J.; Kuhn, H.J.; Seidman, C.E.; Shah, P.M.; Spencer, W.H., 3rd; Spirito, P.; Ten Cate, F.J.; et al. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J. Am. Coll. Cardiol. 2003, 42, 1687–1713. [Google Scholar] [PubMed] [Green Version]
- Bos, J.M.; Towbin, J.A.; Ackerman, M.J. Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2009, 54, 201–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, C.; Bagnall, R.D.; Lam, L.; Semsarian, C.; Ingles, J. Multiple Gene Variants in Hypertrophic Cardiomyopathy in the Era of Next-Generation Sequencing. Circ. Cardiovasc. Genet. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Walsh, R.; Thomson, K.L.; Ware, J.S.; Funke, B.H.; Woodley, J.; McGuire, K.J.; Mazzarotto, F.; Blair, E.; Seller, A.; Taylor, J.C.; et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. Off. J. Am. Coll. Med Genet. 2017, 19, 192–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, M.A.; Cook, S.A.; Seidman, J.G.; Seidman, C.E. Clinical and Mechanistic Insights into the Genetics of Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 68, 2871–2886. [Google Scholar] [CrossRef] [PubMed]
- Alfares, A.A.; Kelly, M.A.; McDermott, G.; Funke, B.H.; Lebo, M.S.; Baxter, S.B.; Shen, J.; McLaughlin, H.M.; Clark, E.H.; Babb, L.J.; et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: Expanded panels offer limited additional sensitivity. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 880–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.; Hamilton, R.; et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 2011, 8, 1308–1339. [Google Scholar] [CrossRef]
- Briasoulis, A.; Mallikethi-Reddy, S.; Palla, M.; Alesh, I.; Afonso, L. Myocardial fibrosis on cardiac magnetic resonance and cardiac outcomes in hypertrophic cardiomyopathy: A meta-analysis. Heart 2015, 101, 1406–1411. [Google Scholar] [CrossRef]
- Weng, Z.; Yao, J.; Chan, R.H.; He, J.; Yang, X.; Zhou, Y.; He, Y. Prognostic Value of LGE-CMR in HCM: A Meta-Analysis. JACC Cardiovasc. Imaging 2016, 9, 1392–1402. [Google Scholar] [CrossRef]
- Williams, L.K.; Frenneaux, M.P.; Steeds, R.P. Echocardiography in hypertrophic cardiomyopathy diagnosis, prognosis, and role in management. Eur. J. Echocardiogr. J. Work. Group Echocardiogr. Eur. Soc. Cardiol. 2009, 10, iii9-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gersh, B.J.; Maron, B.J.; Bonow, R.O.; Dearani, J.A.; Fifer, M.A.; Link, M.S.; Naidu, S.S.; Nishimura, R.A.; Ommen, S.R.; Rakowski, H.; et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: Executive summary: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Thorac. Cardiovasc. Surg. 2011, 142, 1303–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, M.; Teraoka, K.; Kawade, M.; Hirano, M.; Yamashina, A. Frequency and distribution of late gadolinium enhancement in magnetic resonance imaging of patients with apical hypertrophic cardiomyopathy and patients with asymmetrical hypertrophic cardiomyopathy: A comparative study. Int. J. Cardiovasc. Imaging 2009, 25 (Suppl. 1), 131–138. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://gnomad.broadinstitute.org/ (accessed on 5 May 2018).
- Available online: http://coda.nih.go.kr/coda/KRGDB/index.jsp/ (accessed on 5 May 2018).
- Viswanathan, S.K.; Sanders, H.K.; McNamara, J.W.; Jagadeesan, A.; Jahangir, A.; Tajik, A.J.; Sadayappan, S. Hypertrophic cardiomyopathy clinical phenotype is independent of gene mutation and mutation dosage. PLoS ONE 2017, 12, e0187948. [Google Scholar] [CrossRef] [Green Version]
- Marian, A.J.; Braunwald, E. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Available online: http://www.hgmd.org/ (accessed on 5 May 2018).
- Available online: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 5 May 2018).
- Available online: http://sift.jcvi.org/ (accessed on 5 May 2018).
- Available online: http://genetics.bwh.harvard.edu/pph2/ (accessed on 5 May 2018).
- Frenneaux, M.P.; Counihan, P.J.; Caforio, A.L.; Chikamori, T.; McKenna, W.J. Abnormal blood pressure response during exercise in hypertrophic cardiomyopathy. Circulation 1990, 82, 1995–2002. [Google Scholar] [CrossRef] [Green Version]
- Ciampi, Q.; Betocchi, S.; Lombardi, R.; Manganelli, F.; Storto, G.; Losi, M.A.; Pezzella, E.; Finizio, F.; Cuocolo, A.; Chiariello, M. Hemodynamic determinants of exercise-induced abnormal blood pressure response in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2002, 40, 278–284. [Google Scholar] [CrossRef] [Green Version]
- Olivotto, I.; Maron, B.J.; Montereggi, A.; Mazzuoli, F.; Dolara, A.; Cecchi, F. Prognostic value of systemic blood pressure response during exercise in a community-based patient population with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 1999, 33, 2044–2051. [Google Scholar] [CrossRef] [Green Version]
- Cerqueira, M.D.; Weissman, N.J.; Dilsizian, V.; Jacobs, A.K.; Kaul, S.; Laskey, W.K.; Pennell, D.J.; Rumberger, J.A.; Ryan, T.; Verani, M.S. Standardized myocardial segmentation and nomenclature for tomographic imaging of the heart. A statement for healthcare professionals from the Cardiac Imaging Committee of the Council on Clinical Cardiology of the American Heart Association. Circulation 2002, 105, 539–542. [Google Scholar] [PubMed] [Green Version]
- Lang, R.M.; Bierig, M.; Devereux, R.B.; Flachskampf, F.A.; Foster, E.; Pellikka, P.A.; Picard, M.H.; Roman, M.J.; Seward, J.; Shanewise, J.S.; et al. Recommendations for chamber quantification: A report from the American Society of Echocardiography’s Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J. Am. Soc. Echocardiogr. Off. Publ. Am. Soc. Echocardiogr. 2005, 18, 1440–1463. [Google Scholar] [CrossRef]
- Maron, B.J.; Maron, M.S.; Semsarian, C. Genetics of hypertrophic cardiomyopathy after 20 years: Clinical perspectives. J. Am. Coll. Cardiol. 2012, 60, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Olivotto, I.; Girolami, F.; Ackerman, M.J.; Nistri, S.; Bos, J.M.; Zachara, E.; Ommen, S.R.; Theis, J.L.; Vaubel, R.A.; Re, F.; et al. Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clin. Proc. 2008, 83, 630–638. [Google Scholar] [CrossRef]
- Jarcho, J.A.; McKenna, W.; Pare, J.A.; Solomon, S.D.; Holcombe, R.F.; Dickie, S.; Levi, T.; Donis-Keller, H.; Seidman, J.G.; Seidman, C.E. Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. N. Engl. J. Med. 1989, 321, 1372–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, R.; Buchan, R.; Wilk, A.; John, S.; Felkin, L.E.; Thomson, K.L.; Chiaw, T.H.; Loong, C.C.W.; Pua, C.J.; Raphael, C.; et al. Defining the genetic architecture of hypertrophic cardiomyopathy: Re-evaluating the role of non-sarcomeric genes. Eur. Heart J. 2017, 38, 3461–3468. [Google Scholar] [CrossRef] [PubMed]
- Bottillo, I.; D’Angelantonio, D.; Caputo, V.; Paiardini, A.; Lipari, M.; De Bernardo, C.; Giannarelli, D.; Pizzuti, A.; Majore, S.; Castori, M.; et al. Molecular analysis of sarcomeric and non-sarcomeric genes in patients with hypertrophic cardiomyopathy. Gene 2016, 577, 227–235. [Google Scholar] [CrossRef] [Green Version]
- O’Mahony, C.; Jichi, F.; Pavlou, M.; Monserrat, L.; Anastasakis, A.; Rapezzi, C.; Biagini, E.; Gimeno, J.R.; Limongelli, G.; McKenna, W.J.; et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur. Heart J. 2014, 35, 2010–2020. [Google Scholar] [CrossRef]
- Sedaghat-Hamedani, F.; Kayvanpour, E.; Tugrul, O.F.; Lai, A.; Amr, A.; Haas, J.; Proctor, T.; Ehlermann, P.; Jensen, K.; Katus, H.A.; et al. Clinical outcomes associated with sarcomere mutations in hypertrophic cardiomyopathy: A meta-analysis on 7675 individuals. Clin. Res. Cardiol. Off. J. Ger. Card. Soc. 2018, 107, 30–41. [Google Scholar] [CrossRef]
- Olivotto, I.; Girolami, F.; Sciagra, R.; Ackerman, M.J.; Sotgia, B.; Bos, J.M.; Nistri, S.; Sgalambro, A.; Grifoni, C.; Torricelli, F.; et al. Microvascular function is selectively impaired in patients with hypertrophic cardiomyopathy and sarcomere myofilament gene mutations. J. Am. Coll. Cardiol. 2011, 58, 839–848. [Google Scholar] [CrossRef] [Green Version]
- Van Driest, S.L.; Jaeger, M.A.; Ommen, S.R.; Will, M.L.; Gersh, B.J.; Tajik, A.J.; Ackerman, M.J. Comprehensive analysis of the beta-myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2004, 44, 602–610. [Google Scholar] [CrossRef] [Green Version]
- Gruner, C.; Ivanov, J.; Care, M.; Williams, L.; Moravsky, G.; Yang, H.; Laczay, B.; Siminovitch, K.; Woo, A.; Rakowski, H. Toronto hypertrophic cardiomyopathy genotype score for prediction of a positive genotype in hypertrophic cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girolami, F.; Ho, C.Y.; Semsarian, C.; Baldi, M.; Will, M.L.; Baldini, K.; Torricelli, F.; Yeates, L.; Cecchi, F.; Ackerman, M.J.; et al. Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. J. Am. Coll. Cardiol. 2010, 55, 1444–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alejandra Restrepo-Cordoba, M.; Campuzano, O.; Ripoll-Vera, T.; Cobo-Marcos, M.; Mademont-Soler, I.; Gámez, J.M.; Dominguez, F.; Gonzalez-Lopez, E.; Padron-Barthe, L.; Lara-Pezzi, E.; et al. Usefulness of Genetic Testing in Hypertrophic Cardiomyopathy: An Analysis Using Real-World Data. J. Cardiovasc. Transl. Res. 2017, 10, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Chida, A.; Inai, K.; Sato, H.; Shimada, E.; Nishizawa, T.; Shimada, M.; Furutani, M.; Furutani, Y.; Kawamura, Y.; Sugimoto, M.; et al. Prognostic predictive value of gene mutations in Japanese patients with hypertrophic cardiomyopathy. Heart Vessel. 2017, 32, 700–707. [Google Scholar] [CrossRef]
- Mc, L.A.; Ellims, A.H.; Prabhu, S.; Voskoboinik, A.; Iles, L.M.; Hare, J.L.; Kaye, D.M.; Macciocca, I.; Mariani, J.A.; Kalman, J.M.; et al. Diffuse Ventricular Fibrosis on Cardiac Magnetic Resonance Imaging Associates With Ventricular Tachycardia in Patients With Hypertrophic Cardiomyopathy. J. Cardiovasc. Electrophysiol. 2016, 27, 571–580. [Google Scholar] [CrossRef]
- Dimitrow, P.P.; Klimeczek, P.; Vliegenthart, R.; Pasowicz, M.; Oudkerk, M.; Podolec, P.; Tracz, W.; Dubiel, J.S. Late hyperenhancement in gadolinium-enhanced magnetic resonance imaging: Comparison of hypertrophic cardiomyopathy patients with and without nonsustained ventricular tachycardia. Int. J. Cardiovasc. Imaging 2008, 24, 77–83. [Google Scholar] [CrossRef]
- Weissler-Snir, A.; Hindieh, W.; Gruner, C.; Fourey, D.; Appelbaum, E.; Rowin, E.; Care, M.; Lesser, J.R.; Haas, T.S.; Udelson, J.E.; et al. Lack of Phenotypic Differences by Cardiovascular Magnetic Resonance Imaging in MYH7 (β-Myosin Heavy Chain)- Versus MYBPC3 (Myosin-Binding Protein C)-Related Hypertrophic Cardiomyopathy. Circ. Cardiovasc. Imaging 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.J.H.; Heidary, S.; Pavlovic, A.; Schlachter, A.; Dash, R.; Fleischmann, D.; Ashley, E.A.; Wheeler, M.T.; Yang, P.C. Defining genotype-phenotype relationships in patients with hypertrophic cardiomyopathy using cardiovascular magnetic resonance imaging. PLoS ONE 2019, 14, e0217612. [Google Scholar] [CrossRef] [Green Version]
- Rowin, E.J.; Hausvater, A.; Link, M.S.; Abt, P.; Gionfriddo, W.; Wang, W.; Rastegar, H.; Estes, N.A.M.; Maron, M.S.; Maron, B.J. Clinical Profile and Consequences of Atrial Fibrillation in Hypertrophic Cardiomyopathy. Circulation 2017, 136, 2420–2436. [Google Scholar] [CrossRef]
- Garg, L.; Gupta, M.; Sabzwari, S.R.A.; Agrawal, S.; Agarwal, M.; Nazir, T.; Gordon, J.; Bozorgnia, B.; Martinez, M.W. Atrial fibrillation in hypertrophic cardiomyopathy: Prevalence, clinical impact, and management. Heart Fail. Rev. 2019, 24, 189–197. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Protein | Location or Function | Chromosomal Position | MIM Number |
|---|---|---|---|---|
| ACTC1 | Actin, alpha, cardiac muscle 1 | Sarcomere | 15q14 | 102540 |
| ACTN2 | Actinin alpha 2 | Z-disk | 1q43 | 102573 |
| ANKRD1 | Ankyrin repeat domain 1 | Z-disk | 10q23.31 | 609599 |
| CAV3 | Caveolin 3 | Sarcolemma | 3p25.3 | 601253 |
| CRYAB | Crystallin alpha B | Z-disk | 11q23.1 | 123590 |
| CSRP3 | Cysteine and glycine rich protein 3 | Z-disk | 11p15.1 | 600824 |
| JPH2 | Junctophilin 2 | Intracellular calcium signaling | 20q13.12 | 605267 |
| LDB3 | LIM domain binding 3 | Z-disk | 10q23.2 | 605906 |
| MYBPC3 | Myosin binding protein C, cardiac | Sarcomere | 11p11.2 | 600958 |
| MYH6 | Myosin heavy chain 6 | Sarcomere | 14q11.2 | 160710 |
| MYH7 | Myosin heavy chain 7 | Sarcomere | 14q11.2 | 160760 |
| MYL2 | Myosin light chain 2 | Sarcomere | 12q24.11 | 160781 |
| MYL3 | Myosin light chain 3 | Sarcomere | 3p21.31 | 160790 |
| MYLK2 | Myosin light chain kinase 2 | Phosphorylate myosin light chain 2 | 20q11.21 | 606566 |
| MYOZ2 | Myozenin 2 | Z-disk | 4q26 | 605602 |
| NEXN | Nexilin F-actin binding protein | Z-disk | 1p31.1 | 613121 |
| PLN | Phospholamban | Regulator of sarcoplasmic reticulum calcium | 6q22.31 | 172405 |
| TCAP | Titin-cap | Z-disk | 17q12 | 604488 |
| TNNC1 | Troponin C1, slow skeletal and cardiac type | Sarcomere | 3p21.1 | 191040 |
| TNNI3 | Troponin I3, cardiac type | Sarcomere | 19q13.4 | 191044 |
| TNNT2 | Troponin T2, cardiac type | Sarcomere | 1q32.1 | 191045 |
| TPM1 | Tropomyosin 1 | Sarcomere | 15q22.2 | 191010 |
| VCL | Vinculin | Z-disk | 10q22.2 | 193065 |
| Variables | Detected (n = 55) | Not Detected (n = 34) | p-Value |
|---|---|---|---|
| Mean ± SD or Number (%) | |||
| Age at diagnosis | 48.69 ± 10.8 | 48.34 ± 10.3 | 0.880 |
| Sex, male | 45 (81.8) | 31 (91.2) | 0.225 |
| Hypertension | 12 (21.8) | 11 (32.4) | 0.270 |
| Diabetes mellitus | 5 (9.1) | 1 (2.9) | 0.261 |
| Smoking | 0.102 | ||
| Current smoker | 11 (20.0) | 13 (38.2) | |
| Ex-smoker | 15 (27.3) | 10 (29.4) | |
| Familial history of HCM | 9 (16.4) | 5 (14.7) | 0.835 |
| Familial history of SCD | 14 (25.5) | 5 (14.7) | 0.229 |
| Chest discomfort/pain | 13 (23.6) | 15 (44.1) | 0.060 |
| Syncope | 5 (9.1) | 4 (11.8) | 0.684 |
| Palpitation | 19 (34.5) | 5 (14.7) | 0.040 * |
| Dyspnea, NYHA class | 0.457 | ||
| I | 43 (78.2) | 24 (70.6) | |
| II | 11 (20.0) | 10 (29.4) | |
| III | 1 (1.8) | 0 (0.0) | |
| NT-proBNP (pg/mL) | 568.9 ± 619.6 | 449.8 ± 493.2 | 0.188 |
| ECG findings | |||
| Atrial fibrillation | 5 (9.1) | 1 (2.9) | 0.261 |
| T-wave inversion | 45 (81.8) | 29 (85.3) | 0.670 |
| LBBB | 0 | 0 | |
| RBBB | 2 (3.6) | 2 (5.9) | 0.619 |
| LVH | 27 (49.1) | 21 (61.8) | 0.244 |
| PR interval | 170.9 ± 24.7 | 161.7 ± 19.6 | 0.078 |
| QRS duration | 94.0 ± 11.5 | 102.1 ± 11.9 | 0.002 * |
| QTc interval | 445.4 ± 31.2 | 458.4 ± 23.5 | 0.041 * |
| NSVT on Holter ECG | 13 (24.5) | 2 (5.9) | 0.030 * |
| Abnormal BP response | 23 (41.8) | 12 (38.2) | 0.546 |
| Case No. | Gene | Refseq | Nucleotide Change | Protein Change | ACMG Interpretation a | gnomAD ALL | gnomAD East Asian | KRGDB | Polyphen-2 | SIFT |
|---|---|---|---|---|---|---|---|---|---|---|
| 01-001-017 | MYBPC3 | NM_000256.3 | c.1000G>A | p.(Glu334Lys) | LPV | 2.54 × 10−4 | 3.55 × 10−3 | 4.00 × 10−3 | Possibly damaging | Deleterious |
| 01-001-018 | TNNT2 | NM_001001430.2 | c.785A>G | p.(Asn262Ser) | VUS | 4.76 × 10−6 | 0 | N/A | Benign | Tolerated |
| 01-001-026 | MYBPC3 | NM_000256.3 | c.86delT | p.(Phe29Serfs*10) | LPV | N/A | N/A | N/A | N/A | N/A |
| 01-001-041 | TNNI3 | NM_000363.4 | c.433C>T | p.(Arg145Trp) | PV | 4.07 × 10−6 | 0 | N/A | Probably damaging | Deleterious |
| 01-001-043 | MYBPC3 | NM_000256.3 | c.2833_2834delCG | p.(Arg945Glyfs*105) | PV | 4.10 × 10−6 | 5.86 × 10−5 | N/A | N/A | N/A |
| 01-001-046 | MYBPC3 | NM_000256.3 | c.1483C>T | p.(Arg495Trp) | VUS | N/A | N/A | N/A | Possibly damaging | Deleterious |
| 01-001-047 | MYBPC3 | NM_000256.3 | c.2164G>T | p.(Glu722*) | LPV | N/A | N/A | N/A | N/A | N/A |
| 01-001-049 | MYOZ2 | NM_016599.4 | c.147T>A | p.(His49Gln) | VUS | N/A | N/A | N/A | Possibly damaging | Tolerated |
| 01-001-061 | ACTN2 | NM_001103.3 | c.440C>T | p.(Ser147Leu) | VUS | 4.06 × 10−6 | 5.80 × 10−5 | N/A | Probably damaging | Deleterious |
| 01-001-062 | MYH7 | NM_000257.2 | c.2069T>A | p.(Met690Lys) | VUS | N/A | N/A | N/A | Possibly damaging | Deleterious |
| 01-001-064 | MYH7 | NM_000257.2 | c.2608C>T | p.(Arg870Cys) | LPV | 2.03 × 10−5 | 5.80 × 10−5 | 8.00 × 10−4 | Possibly damaging | Deleterious |
| 01-001-064 | TNNI3 | NM_000363.4 | c.434G>A | p.(Arg145Gln) | LPV | 1.63 × 10−5 | 1.16 × 10−4 | N/A | Probably damaging | Tolerated |
| 01-001-068 | MYH7 | NM_000257.2 | c.2189T>C | p.(Ile730Thr) | VUS | N/A | N/A | N/A | Possibly damaging | Deleterious |
| 01-001-069 | MYBPC3 | NM_000256.3 | c.2833_2834delCG | p.(Arg945Glyfs*105) | PV | 4.10 × 10−6 | 5.86 × 10−5 | N/A | N/A | N/A |
| 01-001-069 | MYH6 | NM_002471.3 | c.5476_5477delinsAA | p.(Gly1826Asn) | VUS | N/A | N/A | 1.60 × 10−3 | Benign | Tolerated |
| 01-001-071 | MYL3 | NM_000258.2 | c.505G>C | p.(Val169Leu) | VUS | N/A | N/A | N/A | Benign | Deleterious |
| 01-001-072 | MYH7 | NM_000257.2 | c.400T>C | p.(Tyr134His) | VUS | N/A | N/A | N/A | Possibly damaging | Deleterious |
| 01-001-074 | MYH7 | NM_000257.2 | c.2608C>T | p.(Arg870Cys) | LPV | 2.03 × 10−5 | 5.80 × 10−5 | 8.00 × 10−4 | Possibly damaging | Deleterious |
| 01-001-083 | MYBPC3 | NM_000256.3 | c.2273G>A | p.(Gly758Asp) | VUS | 3.23 × 10−5 | 6.17 × 10−4 | N/A | Possibly damaging | Deleterious |
| 01-001-085 | MYH6 | NM_002471.3 | c.3413G>A | p.(Arg1138His) | VUS | 4.31 × 10−5 | 6.10 × 10−5 | N/A | Possibly damaging | Deleterious |
| 01-001-086 | MYBPC3 | NM_000256.3 | c.1358_1359delCT | p.(Pro453Argfs*2) | LPV | N/A | N/A | N/A | N/A | N/A |
| 01-001-088 | MYBPC3 | NM_000256.3 | c.2164G>T | p.(Glu722*) | LPV | N/A | N/A | N/A | N/A | N/A |
| 01-001-090 | MYBPC3 | NM_000256.3 | c.2441_2443delAGA | p.(Lys814del) | LPV | 6.52 × 10−5 | 0 | N/A | N/A | N/A |
| 01-001-091 | TNNI3 | NM_000363.4 | c.434G>A | p.(Arg145Gln) | LPV | 1.63 × 10−5 | 1.16 × 10−4 | N/A | Probably damaging | Tolerated |
| 01-001-092 | MYBPC3 | NM_000256.3 | c.3316G>A | p.(Asp1106Asn) | VUS | 2.69 × 10−5 | 1.86 × 10−4 | 1.60 × 10−3 | Possibly damaging | Deleterious |
| 01-001-093 | MYBPC3 | NM_000256.3 | c.2459G>A | p.(Arg820Gln) | PV | 1.27 × 10−5 | 0 | N/A | Possibly damaging | Deleterious |
| 01-001-096 | MYBPC3 | NM_000256.3 | c.3799delC | p.(Arg1267Alafs*64) | LPV | N/A | N/A | N/A | N/A | N/A |
| 01-001-097 | MYH7 | NM_000257.2 | c.1350G>T | p.(Lys450Asn) | VUS | N/A | N/A | N/A | Possibly damaging | Deleterious |
| 01-001-099 | MYH7 | NM_000257.2 | c.1988G>A | p.(Arg663His) | PV | 8.12 × 10−6 | 0 | N/A | Possibly damaging | Deleterious |
| 01-001-102 | TPM1 | NM_001018005.1 | c.376G>A | p.(Gly126Ser) | VUS | N/A | N/A | N/A | Possibly damaging | Tolerated |
| 01-001-104 | CSRP3 | NM_003476.4 | c.229G>A | p.(Ala77Thr) | VUS | 2.03 × 10−5 | 1.74 × 10−4 | N/A | Probably damaging | Deleterious |
| 01-001-111 | MYBPC3 | NM_000256.3 | c.1091-8G>A | p.(?) | VUS | 8.49 × 10−6 | 1.18 × 10−4 | 8.00 × 10−4 | N/A | N/A |
| 01-001-115 | MYBPC3 | NM_000256.3 | c.1624_1624+1delinsAGCTCAT | p.(Glu542_Gln1274delinsSerSer) | VUS | N/A | N/A | N/A | N/A | N/A |
| 01-001-118 | JPH2 | NM_020433.4 | c.1306C>T | p.(Arg436Cys) | VUS | 1.64 × 10−5 | 2.34 × 10−4 | 2.40 × 10−3 | Probably damaging | Deleterious |
| 01-001-119 | MYBPC3 | NM_000256.3 | c.2833_2834delCG | p.(Arg945Glyfs*105) | PV | 4.10 × 10−6 | 5.86 × 10−5 | N/A | N/A | N/A |
| 01-001-125 | TNNI3 | NM_000363.4 | c.434G>A | p.(Arg145Gln) | LPV | 1.63 × 10−5 | 1.16 × 10−4 | N/A | Probably damaging | Tolerated |
| 01-001-126 | MYBPC3 | NM_000256.3 | c.2067+1G>A | p.(?) | PV | N/A | N/A | 1.60 × 10−3 | N/A | N/A |
| 01-001-128 | TNNI3 | NM_000363.4 | c.434G>A | p.(Arg145Gln) | LPV | 1.63 × 10−5 | 1.16 × 10−4 | N/A | Probably damaging | Tolerated |
| 01-001-129 | MYBPC3 | NM_000256.3 | c.2459G>A | p.(Arg820Gln) | PV | 1.63 × 10−5 | 5.80 × 10−5 | N/A | Possibly damaging | Deleterious |
| 01-001-130 | MYH6 | NM_002471.3 | c.5026G>A | p.(Val1676Met) | VUS | 2.03 × 10−5 | 5.80 × 10−5 | N/A | Possibly damaging | Deleterious |
| 01-001-131 | TNNT2 | NM_001001430.2 | c.853G>A | p.(Gly285Arg) | VUS | 2.93 × 10−5 | 5.91 × 10−5 | N/A | Possibly damaging | Deleterious |
| 01-001-137 | MYBPC3 | NM_000256.3 | c.2459G>A | p.(Arg820Gln) | PV | 1.63 × 10−5 | 5.80 × 10−5 | N/A | Possibly damaging | Deleterious |
| 01-001-139 | MYBPC3 | NM_000256.3 | c.3626A>G | p.(Lys1209Arg) | VUS | N/A | N/A | N/A | Possibly damaging | Deleterious |
| 01-001-141 | MYBPC3 | NM_000256.3 | c.2273G>A | p.(Gly758Asp) | VUS | 3.23 × 10−5 | 6.17 × 10−4 | N/A | Possibly damaging | Deleterious |
| 01-001-144 | MYH7 | NM_000257.2 | c.1477A>G | p.(Met493Val) | VUS | N/A | N/A | N/A | Benign | Deleterious |
| 01-001-145 | MYH7 | NM_000257.2 | c.4130C>T | p.(Thr1377Met) | LPV | 4.06 × 10−6 | 0 | N/A | Probably damaging | Deleterious |
| 01-001-148 | TNNI3 | NM_000363.4 | c.434G>A | p.(Arg145Gln) | LPV | 1.63 × 10−5 | 1.16 × 10−4 | N/A | Probably damaging | Tolerated |
| 01-001-155 | MYL3 | NM_000258.2 | c.170C>G | p.(Ala57Gly) | LPV | 7.31 × 10−5 | 2.90 × 10−4 | 1.60 × 10−3 | Benign | Deleterious |
| 01-001-159 | TPM1 | NM_001018005.1 | c.791A>G | p.(Lys264Arg) | VUS | N/A | N/A | N/A | Benign | Tolerated |
| 01-001-180 | MYBPC3 | NM_000256.3 | c.2067+1G>A | p.(?) | PV | N/A | N/A | N/A | N/A | N/A |
| 01-001-180 | MYH7 | NM_000257.2 | c.2972A>G | p.(Lys991Arg) | VUS | 4.06 × 10−6 | 0 | N/A | Benign | Deleterious |
| 01-001-181 | MYBPC3 | NM_000256.3 | c.2833_2834delCG | p.(Arg945Glyfs*105) | PV | 4.10 × 10−6 | 5.86 × 10−5 | N/A | N/A | N/A |
| 01-001-181 | MYH6 | NM_002471.3 | c.2079C>A | p.(His693Gln) | VUS | 4.06 × 10−6 | 0 | N/A | Probably damaging | Deleterious |
| 01-001-184 | MYH7 | NM_000257.2 | c.1350G>T | p.(Lys450Asn) | VUS | N/A | N/A | N/A | Probably damaging | Deleterious |
| 01-001-187 | TPM1 | NM_001018005.1 | c.791A>G | p.(Lys264Arg) | VUS | N/A | N/A | N/A | Benign | Tolerated |
| 01-001-190 | VCL | NM_014000.2 | c.3247G>A | p.(Glu1083Lys) | VUS | 4.70 × 10−6 | 0 | N/A | Probably damaging | Tolerated |
| 01-001-190 | MYH6 | NM_002471.3 | c.5071C>T | p.(Arg1691Cys) | VUS | 1.22 × 10−5 | 1.74 × 10−4 | 5.80 × 10−4 | Probably damaging | Deleterious |
| 01-001-200 | MYH7 | NM_000257.2 | c.1426C>T | p.(Leu476Phe) | VUS | N/A | N/A | N/A | Probably damaging | Deleterious |
| 01-001-207 | MYBPC3 | NM_000256.3 | c.3313_3314insGG | p.(Ala1105Glyfs*85) | LPV | N/A | N/A | N/A | N/A | N/A |
| 01-001-223 | MYH7 | NM_000257.2 | c.5560-8G>A | p.(?) | VUS | 0.00 × 1000 | 0 | N/A | N/A | N/A |
| 01-001-228 | TNNI3 | NM_000363.4 | c.235C>T | p.(Arg79Cys) | VUS | 4.46 × 10−4 | 5.81 × 10−3 | 5.00 × 10−3 | Probably damaging | Deleterious |
| Variables | Detected (n = 55) | Not Detected (n = 34) | p-Value |
|---|---|---|---|
| Mean ± SD or Number (%) | |||
| Echocardiographic findings | |||
| Maximal wall thickness (mm) | 18.4 ± 4.3 | 17.5 ± 4.2 | 0.328 |
| LVEDD (mm) | 47.9 ± 5.4 | 49.0 ± 5.7 | 0.380 |
| LVESD (mm) | 26.7 ± 6.8 | 28.5 ± 3.5 | 0.102 |
| IVSd (mm) | 15.8 ± 4.9 | 14.7 ± 4.6 | 0.299 |
| LVPWd (mm) | 9.1 ± 2.3 | 9.6 ± 2.5 | 0.351 |
| LV mass index (g/m2) | 125.9 ± 31.9 | 118.2 ± 46.1 | 0.367 |
| Relative wall thickness | 0.385 ± 0.10 | 0.385 ± 0.12 | 0.604 |
| LVEF (%) | 66.67 ± 6.5 | 65.31 ± 6.76 | 0.353 |
| LAVI (ml/m2) | 42.5 ± 17.2 | 37.8 ± 10.3 | 0.119 |
| E (m/sec) | 0.62 ± 0.25 | 0.63 ± 0.23 | 0.822 |
| A (m/sec) | 0.62 ± 0.26 | 0.60 ± 0.17 | 0.717 |
| Septal E’ (m/sec) | 0.061 ± 0.022 | 0.057 ± 0.018 | 0.448 |
| E/septal E’ | 15.6 ± 28.1 | 12.1 ± 4.31 | 0.659 |
| RV involvement | 9 (16.4) | 5 (14.7) | 0.543 |
| LVOT obstruction | 10 (18.2) | 6 (17.6) | 0.592 |
| MV obstruction | 8 (14.5) | 5 (14.7) | 0.606 |
| LV gradient (mmHg) | 22.1 ± 37.0 | 21.8 ± 26.2 | 0.263 |
| SAM | 10 (18.2) | 6 (17.6) | 0.592 |
| HCM subtype | 0.547 | ||
| Septal | 24 (43.6) | 12 (35.3) | |
| Apical | 7 (12.7) | 8 (23.5) | |
| Concentric | 3 (5.5) | 2 (5.9) | |
| Septal/Apical | 19 (34.5) | 12 (35.3) | |
| Other | 2 (3.6) | 0 | |
| CMR findings | |||
| LVEDV (mL) | 156.6 ± 185.7 | 186.1 ± 225.5 | 0.034 |
| LVESV (mL) | 41.4 ± 12.0 | 48.5 ± 22.1 | 0.052 |
| LVEF (%) | 68.4 ± 8.3 | 68.2 ± 7.6 | 0.891 |
| Stroke volume (mL) | 90.9 ± 23.7 | 99.4 ± 16.8 | 0.070 |
| Cardiac output (L) | 6.16 ± 1.64 | 6.58 ± 1.21 | 0.197 |
| LV mass (g) | 161.1 ± 48.4 | 183.4 ± 64.6 | 0.138 |
| LV mass index (g/m2) | 90.0 ± 23.5 | 100.1 ± 35.3 | 0.145 |
| Presence of LGE | 51 (92.7) | 26 (76.5) | 0.029 * |
| LGE volume (mL) | 13.4 ± 12.5 | 11.0 ± 11.7 | 0.159 |
| LGE volume percent (%) | 9.12 ± 6.7 | 6.06 ± 4.9 | 0.021 * |
| Variables | Detected (n = 55) | Not detected (n = 34) | p-Value |
|---|---|---|---|
| Mean ± SD or Number (%) | |||
| MACCE | 21 (38.2) | 4 (11.8) | 0.008 * |
| All cause death | 0 | 0 | |
| Sudden cardiac death | 0 | 0 | |
| Sustained VT/VF | 4 (7.3) | 0 | |
| Worsening HF driven hospitalization | 5 (9.1) | 2 (5.9) | |
| New onset AF driven hospitalization | 7 (12.7) | 0 | |
| Stroke | 1 (1.8) | 1 (2.9) | |
| Syncope | 3 (5.5) | 1 (2.9) | |
| AMI | 1 (1.8) | 0 | |
| Interventions | 9 (16.4) | 1 (2.9) | 0.082 |
| ICD implantation | 4 (7.2) | 0 | |
| Septal myectomy | 6 (10.9) | 1 (2.9) | |
| Heart transplantation | 1 (1.8) | 0 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.Y.; Park, J.E.; Lee, S.-C.; Jeon, E.-S.; On, Y.K.; Kim, S.M.; Choe, Y.H.; Ki, C.-S.; Kim, J.-W.; Kim, K.H. Genotype-Related Clinical Characteristics and Myocardial Fibrosis and Their Association with Prognosis in Hypertrophic Cardiomyopathy. J. Clin. Med. 2020, 9, 1671. https://doi.org/10.3390/jcm9061671
Kim HY, Park JE, Lee S-C, Jeon E-S, On YK, Kim SM, Choe YH, Ki C-S, Kim J-W, Kim KH. Genotype-Related Clinical Characteristics and Myocardial Fibrosis and Their Association with Prognosis in Hypertrophic Cardiomyopathy. Journal of Clinical Medicine. 2020; 9(6):1671. https://doi.org/10.3390/jcm9061671
Chicago/Turabian StyleKim, Hyung Yoon, Jong Eun Park, Sang-Chol Lee, Eun-Seok Jeon, Young Keun On, Sung Mok Kim, Yeon Hyeon Choe, Chang-Seok Ki, Jong-Won Kim, and Kye Hun Kim. 2020. "Genotype-Related Clinical Characteristics and Myocardial Fibrosis and Their Association with Prognosis in Hypertrophic Cardiomyopathy" Journal of Clinical Medicine 9, no. 6: 1671. https://doi.org/10.3390/jcm9061671