Genetic Factors Involved in Cardiomyopathies and in Cancer

 , , and
, , and
Abstract
:1. Genetic Causes of Cardiomyopathies
2. Cancer Therapy-Related Myocardial Dysfunction and Heart Failure
2.1. Anthracyclines
2.2. Other Conventional Chemotherapies
2.3. Radiotherapy
3. Evaluation of Cardiac Injury from Chemotherapy
4. Impact of Chemotherapy on Development of Systolic Impairment in Patients with Germinal Mutations in Cardiac Associated Gene Loci
5. Predisposition to Cancer in Patients with Cardiac Associated Genetic Variants
5.1. Arrhythmogenic Cardiomyopathy: Disease of the Desmosome
5.2. Inherited Cardiac Disease and Cancer
5.3. Desmosome and Its Association with Invasiveness of Cancer Cells
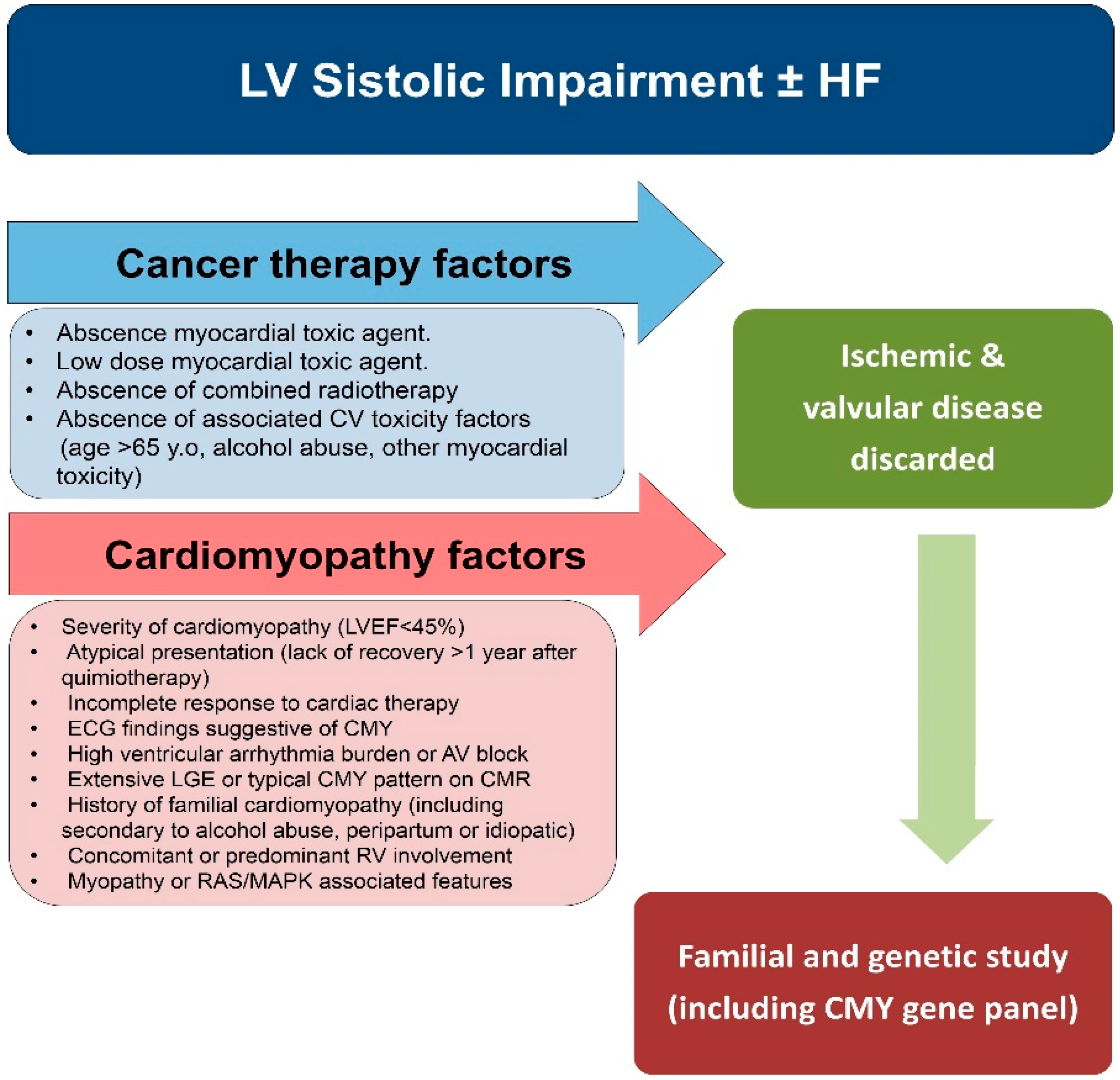
6. Importance of Genetic and Family Study in Patients with Severe Cardiotoxicity
Supplementary Materials
Funding
Conflicts of Interest
References
- Braunwald, E. Cardiomyopathies: An overview. Circ. Res. 2017, 121, 711–721. [Google Scholar] [CrossRef]
- McKenna, W.J.; Maron, B.J.; Thiene, G. Classification, epidemiology, and global burden of cardiomyopathies. Circ. Res. 2017, 121, 722–730. [Google Scholar] [CrossRef] [Green Version]
- Ezekowitz, JA.; O’Meara, E.; McDonald, M.A.; Abrams, H.; Chan, M.; Ducharme, A.; Giannetti, N.; Grzeslo, A.; Hamilton, P.G.; Heckman, G.A.; et al. 2017 comprehensive update of the canadian cardiovascular society guidelines for the management of heart failure. Can. J. Cardiol. 2017, 33, 1342–1433. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Sánchez, I.; Romero-Puche, A.J.; Sáez, E.G.-M.; Sabater-Molina, M.; López-Ayala, J.M.; Esparza, C.M.; López-Cuenca, D.; De La Morena, G.; Castro-García, F.J.; Gimeno-Blanes, J.R.; et al. Factors influencing the phenotypic expression of hypertrophic cardiomyopathy in genetic carriers. Rev. Española Cardiol. (Engl. Ed.) 2018, 71, 146–154. [Google Scholar] [CrossRef]
- Carbone, A.; D’Andrea, A.; Riegler, L.; Scarafile, R.; Pezzullo, E.; Martone, F.; America, R.; Liccardo, B.; Galderisi, M.; Bossone, E.; et al. Cardiac damage in athlete’s heart: When the “supernormal” heart fails! World J. Cardiol. 2017, 9, 470–480. [Google Scholar] [CrossRef]
- Teare, D. Asymmetrical hypertrophy of the heart in young adults. Heart 1958, 20, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, J.; Frese, K.; Peil, B.; Kloos, W.; Keller, A.; Nietsch, R.; Feng, Z.; Müller, S.; Kayvanpour, E.; Vogel, B.; et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J. 2014, 36, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- Amor-Salamanca, A.; Guzzo-Merello, G.; González-López, E.; Domínguez, F.; Restrepo-Córdoba, A.; Cobo-Marcos, M.; Gomez-Bueno, M.; Segovia-Cubero, J.; Alonso-Pulpón, L.; Garcia-Pavia, P.; et al. Prognostic impact and predictors of ejection fraction recovery in patients with alcoholic cardiomyopathy. Rev. Española Cardiol. (Engl. Ed.) 2018, 71, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Ware, J.S.; Amor-Salamanca, A.; Tayal, U.; Govind, R.; Serrano, I.; Salazar-Mendiguchía, J.; García-Pinilla, J.M.; Pascual-Figal, D.A.; Núñez, J.; Guzzo-Merello, G.; et al. Genetic etiology for alcohol-induced cardiac toxicity. J. Am. Coll. Cardiol. 2018, 71, 2293–2302. [Google Scholar] [CrossRef]
- Fang, W.; Luo, R.; Tang, Y.; Hua, W.; Fu, M.; Chen, W.; Lai, L.; Li, X. The prognostic factors of alcoholic cardiomyopathy: A single-center cohort study. Medicine 2018, 97, e11744. [Google Scholar] [CrossRef] [PubMed]
- Guzzo-Merello, G.; Segovia, J.; Domínguez, F.; Cobo-Marcos, M.; Gómez-Bueno, M.; Avellana, P.; Millán, I.; Alonso-Pulpón, L.; Garcia-Pavia, P. Natural history and prognosticfactors in alcoholiccardiomyopathy. JACC Heart Fail. 2015, 3, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Ware, J.S.; Li, J.; Mazaika, E.; Yasso, C.M.; DeSouza, T.; Cappola, T.P.; Tsai, E.J.; Hilfiker-Kleiner, D.; Kamiya, C.A; Mazzarotto, F.; et al. Shared genetic predisposition in peripartum and dilated cardiomyopathies. N. Engl. J. Med. 2016, 374, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, B.; Colvin, M.; Cook, J.; Cooper, L.T.; Deswal, A.; Fonarow, G.C.; Francis, G.S.; Lenihan, D.; Lewis, E.F.; McNamara, D.M.; et al. Current Diagnostic and treatment strategies for specific dilated cardiomyopathies: A scientific statement from the american heart association. Circulation 2016, 134, e579–e646. [Google Scholar] [CrossRef] [PubMed]
- Bauersachs, J.; König, T.; Van Der Meer, P.; Petrie, M.C.; Hilfiker-Kleiner, D.; Mbakwem, A.; Hamdan, R.; Jackson, A.M.; Forsyth, P.; De Boer, R.A.; et al. Pathophysiology, diagnosis and management of peripartum cardiomyopathy: A position statement from the Heart Failure Association of the European Society of Cardiology Study Group on peripartum cardiomyopathy. Eur. J. Heart Fail. 2019, 21, 827–843. [Google Scholar] [CrossRef] [PubMed]
- Huizar, J.F.; Ellenbogen, K.A.; Tan, A.Y.; Kaszala, K. Arrhythmia-induced cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 2328–2344. [Google Scholar] [CrossRef] [PubMed]
- Martin, CA.; Lambiase, P. Pathophysiology, diagnosis and treatment of tachycardiomyopathy. Heart 2017, 103, 1543–1552. [Google Scholar] [CrossRef]
- Cardinale, D.; Colombo, A.; LaMantia, G.; Colombo, N.; Civelli, M.; De Giacomi, G.; Rubino, M.; Veglia, F.; Fiorentini, C.; Cipolla, C.M.; et al. Anthracycline-induced cardiomyopathy: Clinical relevance and response to pharmacologic therapy. J. Am. Coll. Cardiol. 2010, 55, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Van Spaendonck-Zwarts, K.Y.; Van Rijsingen, I.A.; Berg, M.P.V.D.; Deprez, R.H.L.; Post, J.G.; Van Mil, A.M.; Asselbergs, F.W.; Christiaans, I.; Van Langen, I.M.; Wilde, A.A.M.; et al. Genetic analysis in 418 index patients with idiopathic dilated cardiomyopathy: Overview of 10 years’ experience. Eur. J. Heart Fail. 2013, 15, 628–636. [Google Scholar] [CrossRef]
- Drafts, B.C.; Twomley, K.M.; D’Agostino, R.; Lawrence, J.; Avis, N.; Ellis, L.R.; Thohan, V.; Jordan, J.; Melin, S.A.; Torti, F.M.; et al. Low to moderate dose anthracycline-based chemotherapy is associated with early noninvasive imaging evidence of subclinical cardiovascular disease. JACC Cardiovasc. Imaging 2013, 6, 877–885. [Google Scholar] [CrossRef] [Green Version]
- Tayal, U.; Newsome, S.; Buchan, R.; Whiffin, N.; Halliday, B.; Lota, A.; Roberts, A.; Baksi, A.J.; Voges, I.; Midwinter, W.; et al. Phenotype and clinical outcomes of Titin cardiomyopathy. J. Am. Coll. Cardiol. 2017, 70, 2264–2274. [Google Scholar] [CrossRef]
- Holmström, M.; Kivistö, S.; Heliö, T.; Jurkko, R.; Kaartinen, M.; Antila, M.; Reissell, E.; Kuusisto, J.; Kärkkäinen, S.; Peuhkurinen, K.; et al. Late gadolinium enhanced cardiovascular magnetic resonance of lamin A/C gene mutation related dilated cardiomyopathy. J. Cardiovasc. Magn. Reson. 2011, 13, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gräni, C.; Eichhorn, C.; Bière, L.; Murthy, V.L.; Agarwal, V.; Kaneko, K.; Cuddy, S.; Aghayev, A.; Steigner, M.; Blankstein, R.; et al. Prognostic value of cardiac magnetic resonance tissue characterization in risk stratifying patients with suspected myocarditis. J. Am. Coll. Cardiol. 2017, 70, 1964–1976. [Google Scholar] [CrossRef] [PubMed]
- Aquaro, G.D.; Perfetti, M.; Camastra, G.; Monti, L.; Dellegrottaglie, S.; Moro, C.; Pepe, A.; Todiere, G.; Lanzillo, C.; Scatteia, A.; et al. Faculty Opinions recommendation of Cardiac MR with late gadolinium enhancement in acute myocarditis with preserved systolic function: ITAMY study. J. Am. Coll. Cardiol. 2018, 70. [Google Scholar] [CrossRef]
- Fallah-Rad, N.; Lytwyn, M.; Fang, T.; Kirkpatrick, I.; Jassal, D.S. Delayed contrast enhancement cardiac magnetic resonance imaging in trastuzumab induced cardiomyopathy. J. Cardiovasc. Magn. Reson. 2008, 10, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wadhwa, D.; Fallah-Rad, N.; Grenier, D.; Krahn, M.; Fang, T.; Ahmadie, R.; Walker, J.; Lister, D.; Arora, R.C.; Barac, I.; et al. Trastuzumab mediated cardiotoxicity in the setting of adjuvant chemotherapy for breast cancer: A retrospective study. Breast Cancer Res. Treat. 2008, 117, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Lawley, C.; Wainwright, C.; Segelov, E.; Lynch, J.; Beith, J.; McCrohon, J. Pilot study evaluating the role of cardiac magnetic resonance imaging in monitoring adjuvant trastuzumab therapy for breast cancer. Asia-Pac. J. Clin. Oncol. 2012, 8, 95–100. [Google Scholar] [CrossRef]
- Garcia-Pavia, P.; Kim, Y.; Restrepo-Cordoba, M.A.; Lunde, I.G.; Wakimoto, H.; Smith, A.M.; Toepfer, C.N.; Getz, K.; Gorham, J.; Patel, P.; et al. Genetic variants associated with cancer therapy-induced cardiomyopathy. Circulation 2019, 140, 31–41. [Google Scholar] [CrossRef]
- Neilan, T.G.; Coelho-Filho, O.R.; Pena-Herrera, D.; Shah, R.V.; Jerosch-Herold, M.; Francis, S.A.; Moslehi, J.; Kwong, R.Y. Left ventricular mass in patients with a cardiomyopathy after treatment with anthracyclines. Am. J. Cardiol. 2012, 110, 1679–1686. [Google Scholar] [CrossRef] [Green Version]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.M.; Ware, J.S.; Herman, D.S.; Schafer, S.; Baksi, J.; Bick, A.G.; Buchan, R.; Walsh, R.; John, S.; Wilkinson, S.; et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci. Transl. Med. 2015, 7, 270ra6. [Google Scholar] [CrossRef] [Green Version]
- Bondue, A.; Arbustini, E.; Bianco, A.M.; Ciccarelli, M.; Dawson, D.K.; De Rosa, M.; Hamdani, N.; Hilfiker-Kleiner, D.; Meder, B.; Leite-Moreira, A.F.; et al. Complex roads from genotype to phenotype in dilated cardiomyopathy: Scientific update from the Working Group of Myocardial Function of the European Society of Cardiology. Cardiovasc. Res. 2018, 114, 1287–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protonotarios, A.; Elliott, P.M. Arrhythmogenic cardiomyopathies (ACs): Diagnosis, risk stratification and management. Heart 2019, 105, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Sen-Chowdhry, S.; Syrris, P.; Ward, D.; Asimaki, A.; Sevdalis, E.; McKenna, W.J. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation 2007, 115, 1710–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caforio, A.L.P.; Re, F.; Avella, A.; Marcolongo, R.; Baratta, P.; Seguso, M.; Gallo, N.; Plebani, M.; Izquierdo-Bajo, A.; Cheng, C.-Y.; et al. Evidence from family studies for autoimmunity in arrhythmogenic right ventricular cardiomyopathy: Associations of circulating anti-heart and anti-intercalated disk autoantibodies with disease severity and family history. Circulation 2020, 141, 1238–1248. [Google Scholar] [CrossRef]
- Mogensen, J.; Kubo, T.; Duque, M.; Uribe, W.; Shaw, A.; Murphy, R.; Gimeno, J.R.; Elliott, P.; McKenna, W.J. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J. Clin. Investig. 2003, 111, 209–216. [Google Scholar] [CrossRef] [Green Version]
- Witteles, R.M.; Bokhari, S.; Damy, T.; Elliott, P.M.; Falk, R.H.; Fine, N.M.; Gospodinova, M.; Obici, L.; Rapezzi, C.; Garcia-Pavia, P.; et al. Screening for transthyretin amyloid cardiomyopathy in everyday practice. JACC: Heart Fail. 2019, 7, 709–716. [Google Scholar] [CrossRef]
- Zamorano, J.L.; Lancellotti, P.; Rodriguez Munoz, D.; Aboyans, V.; Asteggiano, R.; Galderisi, M.; Habib, G.; Lenihan, D.J.; Lip, G.Y.H.; Lyon, A.R.; et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur. Heart J. 2016, 37, 2768–2801. [Google Scholar] [CrossRef]
- Ferlay, J.; Steliarova-Foucher, E.; Lortet-Tieulent, J.; Rosso, S.; Coebergh, J.; Comber, H.; Forman, D.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur. J. Cancer 2013, 49, 1374–1403. [Google Scholar] [CrossRef] [Green Version]
- Carver, J.R.; Shapiro, C.L.; Ng, A.; Jacobs, L.; Schwartz, C.; Virgo, K.S.; Hagerty, K.L.; Somerfield, M.R.; Vaughn, D.J. American society of clinical oncology clinical evidence review on the ongoing care of adult cancer survivors: Cardiac and pulmonary late effects. J. Clin. Oncol. 2007, 25, 3991–4008. [Google Scholar] [CrossRef] [Green Version]
- Silber, J.H.; Cnaan, A.; Clark, B.J.; Paridon, S.M.; Chin, A.; Rychik, J.; Hogarty, A.N.; Cohen, M.; Barber, G.; Rutkowski, M.; et al. Enalapril to prevent cardiac function decline in long-term survivors of pediatric cancer exposed to anthracyclines. J. Clin. Oncol. 2004, 22, 820–828. [Google Scholar] [CrossRef]
- Bloom, M.W.; Hamo, C.E.; Cardinale, D.; Ky, B.; Nohria, A.; Baer, L.; Skopicki, H.; Lenihan, D.J.; Gheorghiade, M.; Lyon, A.R.; et al. Cancer therapy-related cardiac dysfunction and heart failure: Part 1: Definitions, pathophysiology, risk factors, and imaging. Circ. Heart Fail. 2016, 9, e002661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewer, M.S.; Ewer, S.M. Cardiotoxicity of anticancer treatments. Nat. Rev. Cardiol. 2015, 12, 547–558. [Google Scholar] [CrossRef]
- Echaburu, J.A.V.; García, A.M.; Peñas, R.D.L.; Beltrán, A.S.; Andrés, R.; Beato, C.; De La Cruz, S.; Gavilá, J.; González-Santiago, S.; Lopez-Fernandez, T.; et al. SEOM clinical guidelines on cardiovascular toxicity (2018). Clin. Transl. Oncol. 2019, 21, 94–105. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.-S.; Lyu, Y.L.; Liu, L.; Yeh, E.T.H. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef]
- Steinherz, L.J.; Steinherz, P.G.; Tan, C.T.; Heller, G.; Murphy, M.L. Cardiac toxicity 4 to 20 years after completing anthracycline therapy. JAMA 1991, 266, 1672–1677. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Layard, M.W.; Basa, P.; Davis, H.L.; Von Hoff, A.L.; Rozencweig, M.; Muggia, F.M. Risk factors for doxorubicin-induced congestive heart failure. Ann. Intern. Med. 1979, 91, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Volkova, M.; Russell, R. Anthracycline cardiotoxicity: Prevalence, pathogenesis and treatment. Curr. Cardiol. Rev. 2011, 7, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Swain, S.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef]
- Herrmann, J.; Lerman, A.; Sandhu, N.P.; Villarraga, H.R.; Mulvagh, S.L.; Kohli, M. Evaluation and management of patients with heart disease and cancer: Cardio-oncology. Mayo Clin. Proc. 2014, 89, 1287–1306. [Google Scholar] [CrossRef] [Green Version]
- Cardinale, D.; Colombo, A.; Bacchiani, G.; Tedeschi, I.; Meroni, C.A.; Veglia, F.; Civelli, M.; LaMantia, G.; Colombo, N.; Curigliano, G.; et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation 2015, 131, 1981–1988. [Google Scholar] [CrossRef] [Green Version]
- Braverman, A.C.; Antin, J.H.; Plappert, M.T.; Cook, E.F.; Lee, R.T. Cyclophosphamide cardiotoxicity in bone marrow transplantation: A prospective evaluation of new dosing regimens. J. Clin. Oncol. 1991, 9, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Gottdiener, J.S.; Appelbaum, F.R.; Ferrans, V.J.; Deisseroth, A.; Ziegler, J. Cardiotoxicity associated with high-dose cyclophosphamide therapy. Arch. Intern. Med. 1981, 141, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Mackey, J.; Martin, M.; Pienkowski, T.; Rolski, J.; Guastalla, J.-P.; Sami, A.; Glaspy, J.; Juhos, E.; Wardley, A.; Fornander, T.; et al. Adjuvant docetaxel, doxorubicin, and cyclophosphamide in node-positive breast cancer: 10-year follow-up of the phase 3 randomised BCIRG 001 trial. Lancet Oncol. 2013, 14, 72–80. [Google Scholar] [CrossRef]
- Gollerkeri, A.; Harrold, L.; Rose, M.; Jain, D.; Burtness, B. Use of paclitaxel in patients with pre-existing cardiomyopathy: A review of our experience. Int. J. Cancer 2001, 93, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Moja, L.; Tagliabue, L.; Balduzzi, S.; Parmelli, E.; Pistotti, V.; Guarneri, V.; D’Amico, R. Trastuzumab containing regimens for early breast cancer. Cochrane Database Syst. Rev. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Suter, T.; Procter, M.; Van Veldhuisen, D.J.; Muscholl, M.; Bergh, J.; Carlomagno, C.; Perren, T.; Passalacqua, R.; Bighin, C.; Klijn, J.G.; et al. Trastuzumab-associated cardiac adverse effects in the herceptin adjuvant trial. J. Clin. Oncol. 2007, 25, 3859–3865. [Google Scholar] [CrossRef] [PubMed]
- Cote, G.M.; Sawyer, D.B.; Chabner, B.A. ERBB2 inhibition and heart failure. N. Engl. J. Med. 2012, 367, 2150–2153. [Google Scholar] [CrossRef]
- De Lorenzo, C.; Paciello, R.; Riccio, G.; Rea, D.; Barbieri, A.; Coppola, C.; Maurea, N. Cardiotoxic effects of the novel approved anti-ErbB2 agents and reverse cardioprotective effects of ranolazine. OncoTargets Ther. 2018, 11, 2241–2250. [Google Scholar] [CrossRef] [Green Version]
- Cameron, D.; Brown, J.; Dent, R.; Jackisch, C.; Mackey, J.; Pivot, X.; Steger, G.G.; Suter, T.M.; Toi, M.; Parmar, M.; et al. Adjuvant bevacizumab-containing therapy in triple-negative breast cancer (BEATRICE): Primary results of a randomised, phase 3 trial. Lancet Oncol. 2013, 14, 933–942. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Cella, D.; Reeves, J.; Hawkins, R.; Guo, J.; Nathan, P.; Staehler, M.; De Souza, P.; Merchan, J.R.; et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N. Engl. J. Med. 2013, 369, 722–731. [Google Scholar] [CrossRef] [Green Version]
- Qi, W.-X.; Shen, Z.; Tang, L.-N.; Yao, Y. Congestive heart failure risk in cancer patients treated with vascular endothelial growth factor tyrosine kinase inhibitors: A systematic review and meta-analysis of 36 clinical trials. Br. J. Clin. Pharmacol. 2014, 78, 748–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filopei, J.; Frishman, W. Radiation-induced heart disease. Cardiol. Rev. 2012, 20, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Van Nimwegen, F.A.; Schaapveld, M.; Janus, C.P.M.; Krol, S.; Petersen, E.J.; Raemaekers, J.M.M.; Kok, W.E.M.; Aleman, B.M.P.; Van Leeuwen, F.E. Cardiovascular disease after hodgkin lymphoma treatment: 40-year disease risk. JAMA Intern. Med. 2015, 175, 1007. [Google Scholar] [CrossRef] [PubMed]
- Plana, J.C.; Galderisi, M.; Barac, A.; Ewer, M.S.; Ky, B.; Scherrer-Crosbie, M.; Ganame, J.; Sebag, I.A.; Agler, D.A.; Badano, L.P.; et al. Expert consensus for multimodality imaging evaluation of adult patients during and after cancer therapy: A report from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J. Am. Soc. Echocardiogr. 2014, 27, 911–939. [Google Scholar] [CrossRef]
- Thavendiranathan, P.; Grant, A.; Negishi, T.; Plana, J.C.; Popović, Z.B.; Marwick, T.H. Reproducibility of echocardiographic techniques for sequential assessment of left ventricular ejection fraction and volumes: Application to patients undergoing cancer chemotherapy. J. Am. Coll. Cardiol. 2013, 61, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Armenian, S.H.; Hudson, M.M.; Mulder, R.L.; Chen, M.H.; Constine, L.S.; Dwyer, M.; Nathan, P.C.; Tissing, W.J.; Shankar, S.; Sieswerda, E.; et al. Recommendations for cardiomyopathy surveillance for survivors of childhood cancer: A report from the International Late Effects of Childhood Cancer Guideline Harmonization Group. Lancet Oncol. 2015, 16, e123–e136. [Google Scholar] [CrossRef] [Green Version]
- Sawaya, H.; Sebag, I.A.; Plana, J.C.; Januzzi, J.L.; Ky, B.; Tan, T.C.; Cohen, V.; Banchs, J.; Carver, J.R.; Wiegers, S.E.; et al. Assessment of echocardiography and biomarkers for the extended prediction of cardiotoxicity in patients treated with anthracyclines, taxanes, and trastuzumab. Circ. Cardiovasc. Imaging 2012, 5, 596–603. [Google Scholar] [CrossRef] [Green Version]
- Negishi, K.; Negishi, T.; Hare, J.L.; Haluska, B.A.; Plana, J.C.; Marwick, T.H. Independent and incremental value of deformation indices for prediction of trastuzumab-induced cardiotoxicity. J. Am. Soc. Echocardiogr. 2013, 26, 493–498. [Google Scholar] [CrossRef]
- Grothues, F.; Smith, G.C.; Moon, J.C.C.; Bellenger, N.G.; Collins, P.; Klein, H.U.; Pennell, D.J. Comparison of interstudy reproducibility of cardiovascular magnetic resonance with two-dimensional echocardiography in normal subjects and in patients with heart failure or left ventricular hypertrophy. Am. J. Cardiol. 2002, 90, 29–34. [Google Scholar] [CrossRef]
- Sandner, T.A.; Houck, P.; Runge, V.M.; Sincleair, S.; Huber, A.M.; Theisen, D.; Reiser, M.F.; Wintersperger, B.J. Accuracy of accelerated cine MR imaging at 3 Tesla in longitudinal follow-up of cardiac function. Eur. Radiol. 2008, 18, 2095–2101. [Google Scholar] [CrossRef]
- Lightfoot, J.C.; D’Agostino, R.B.; Hamilton, C.A.; Jordan, J.; Torti, F.M.; Kock, N.D.; Jordan, J.; Workman, S.; Hundley, W.G. Novel approach to early detection of doxorubicin cardiotoxicity by gadolinium-enhanced cardiovascular magnetic resonance imaging in an experimental model. Circ. Cardiovasc. Imaging 2010, 3, 550–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, G.T.; Plana, J.C.; Zhang, N.; Srivastava, D.; Green, D.M.; Ness, K.K.; Donovan, F.D.; Metzger, M.L.; Arevalo, A.; Durand, J.-B.; et al. Screening adult survivors of childhood cancer for cardiomyopathy: Comparison of echocardiography and cardiac magnetic resonance imaging. J. Clin. Oncol. 2012, 30, 2876–2884. [Google Scholar] [CrossRef] [PubMed]
- Chaosuwannakit, N.; D’Agostino, R.; Hamilton, C.A.; Lane, K.S.; Ntim, W.O.; Lawrence, J.; Melin, S.A.; Ellis, L.R.; Torti, F.M.; Little, W.C.; et al. Aortic stiffness increases upon receipt of anthracycline chemotherapy. J. Clin. Oncol. 2010, 28, 166–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, M.A.; Cornel, J.H.; Van De Ven, P.M.; Van Rossum, A.C.; Allaart, C.P.; Germans, T. The prognostic value of late gadolinium-enhanced cardiac magnetic resonance imaging in nonischemic dilated cardiomyopathy: A Review and Meta-Analysis. JACC Cardiovasc. Imaging 2018, 11, 1274–1284. [Google Scholar] [CrossRef]
- Fallah-Rad, N.; Walker, J.; Wassef, A.; Lytwyn, M.; Bohonis, S.; Fang, T.; Tian, G.; Kirkpatrick, I.D.; Singal, P.K.; Krahn, M.; et al. The utility of cardiac biomarkers, tissue velocity and strain imaging, and cardiac magnetic resonance imaging in predicting early left ventricular dysfunction in patients with human epidermal growth factor receptor II–positive breast cancer treated with adjuvant trastuzumab therapy. J. Am. Coll. Cardiol. 2011, 57, 2263–2270. [Google Scholar] [CrossRef]
- Bernaba, B.N.; Chan, J.B.; Lai, C.K.; Fishbein, M.C. Pathology of late-onset anthracycline cardiomyopathy. Cardiovasc. Pathol. 2010, 19, 308–311. [Google Scholar] [CrossRef]
- Tham, E.B.; Haykowsky, M.J.; Chow, K.; Spavor, M.; Kaneko, S.; Khoo, N.S.; Pagano, J.J.; Mackie, A.S.; Thompson, R. Diffuse myocardial fibrosis by T1-mapping in children with subclinical anthracycline cardiotoxicity: Relationship to exercise capacity, cumulative dose and remodeling. J. Cardiovasc. Magn. Reson. 2013, 15, 48. [Google Scholar] [CrossRef] [Green Version]
- Neilan, T.G.; Coelho-Filho, O.R.; Shah, R.V.; Feng, J.; Pena-Herrera, D.; Mandry, D.; Pierre-Mongeon, F.; Heydari, B.; Francis, S.A.; Moslehi, J.; et al. Myocardial extracellular volume by cardiac magnetic resonance imaging in patients treated with anthracycline-based chemotherapy. Am. J. Cardiol. 2012, 111, 717–722. [Google Scholar] [CrossRef] [Green Version]
- Galán-Arriola, C.; Lobo, M.; Vílchez-Tschischke, J.P.; López, G.J.; De Molina-Iracheta, A.; Perez-Martinez, C.; Aguero, J.; Fernández-Jiménez, R.; Martín-García, A.; Oliver, E.; et al. Serial magnetic resonance imaging to identify early stages of anthracycline-induced cardiotoxicity. J. Am. Coll. Cardiol. 2019, 73, 779–791. [Google Scholar] [CrossRef]
- Pinto, Y.M.; Elliott, P.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Böhm, M.; Duboc, D.; Gimeno, J.; De Groote, P.; Imazio, M.; et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef] [Green Version]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kühl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNally, E.; Mestroni, L. Dilated cardiomyopathy: Genetic determinants and mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef] [PubMed]
- Garfinkel, A.C.; Seidman, J.G.; Seidman, C.E. Genetic pathogenesis of hypertrophic and dilated cardiomyopathy. Heart Fail. Clin. 2018, 14, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Sweet, M.; Taylor, M.R.; Mestroni, L. Diagnosis, prevalence, and screening of familial dilated cardiomyopathy. Expert Opin. Orphan Drugs 2015, 3, 869–876. [Google Scholar] [CrossRef] [Green Version]
- Ganesh, S.K.; Arnett, N.K.; Assimes, T.L.; Basson, C.T.; Chakravarti, A.; Ellinor, P.T.; Engler, M.B.; Goldmuntz, E.; Herrington, D.M.; Hershberger, R.E.; et al. Genetics and genomics for the prevention and treatment of cardiovascular disease: Update: A scientific statement from the American Heart Association. Circulation 2013, 128, 2813–2851. [Google Scholar] [CrossRef]
- Berg, M.P.V.D.; Van Spaendonck-Zwarts, K.Y.; Van Veldhuisen, D.J.; Gietema, J.; Postma, A.; Van Tintelen, J.P.; Spaendonck-Zwarts, K.Y. Familial dilated cardiomyopathy: Another risk factor for anthracycline-induced cardiotoxicity? Eur. J. Heart Fail. 2010, 12, 1297–1299. [Google Scholar] [CrossRef]
- Shipman, K.; Arnold, I. Case of epirubicin-induced cardiomyopathy in familial cardiomyopathy. J. Clin. Oncol. 2011, 29, e537–e538. [Google Scholar] [CrossRef]
- Linschoten, M.; Teske, A.; Baas, A.; Vink, A.; Dooijes, D.; Baars, H.; Asselbergs, F.W. Truncating Titin ( TTN) variants in chemotherapy-induced cardiomyopathy. J. Card. Fail. 2017, 23, 476–479. [Google Scholar] [CrossRef] [Green Version]
- Wasielewski, M.; Van Spaendonck, K.Y.; Westerink, N.-D.L.; Jongbloed, J.D.H.; Postma, A.; Gietema, J.; Van Tintelen, J.P.; Berg, M.P.V.D. Potential genetic predisposition for anthracycline-associated cardiomyopathy in families with dilated cardiomyopathy. Open Heart 2014, 1, e000116. [Google Scholar] [CrossRef] [Green Version]
- Gardner, L.; Shen, F.; Radovich, M.; Li, L.; Miller, K.; Jiang, G.; Lai, D.; O’Neill, A.M.; A Sparano, J.; Davidson, N.E.; et al. Genome wide association study for anthracycline-induced congestive heart failure. J. Clin. Oncol. 2016, 34, 1017. [Google Scholar] [CrossRef] [Green Version]
- Wells, Q.S.; Veatch, O.J.; Fessel, J.P.; Joon, A.Y.; Levinson, R.T.; Mosley, J.D.; Held, E.P.; Lindsay, C.S.; Shaffer, C.M.; Weeke, P.E.; et al. Genome-wide association and pathway analysis of left ventricular function after anthracycline exposure in adults. Pharm. Genom. 2017, 27, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Serie, D.J.; Crook, J.; Necela, B.M.; Axenfeld, B.C.; Dockter, T.J.; Colon-Otero, G.; Perez, E.A.; Thompson, E.A.; Norton, N. Breast cancer clinical trial of chemotherapy and trastuzumab: Potential tool to identify cardiac modifying variants of dilated cardiomyopathy. J. Cardiovasc. Dev. Dis. 2017, 4, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrado, D.; Basso, C.; Rizzoli, G.; Schiavon, M.; Thiene, G. Does sports activity enhance the risk of sudden death in adolescents and young adults? J. Am. Coll. Cardiol. 2003, 42, 1959–1963. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Manchón, J.; Fernandez, E.; Igual, B.; Asimaki, A.; Syrris, P.; Osca, J.; Salvador, A.; Zorio, E. Left dominant arrhythmogenic cardiomyopathy caused by a novel nonsense mutation in desmoplakin. Rev. Española Cardiol. (Engl. Ed.) 2011, 64, 530–534. [Google Scholar] [CrossRef]
- Sen-Chowdhry, S.; Syrris, P.; McKenna, W.J. Role of genetic analysis in the management of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J. Am. Coll. Cardiol. 2007, 50, 1813–1821. [Google Scholar] [CrossRef] [Green Version]
- Asimaki, A.; Tandri, H.; Huang, H.; Halushka, M.K.; Gautam, S.; Basso, C.; Thiene, G.; Tsatsopoulou, A.; Protonotarios, N.; McKenna, W.J.; et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N. Engl. J. Med. 2009, 360, 1075–1084. [Google Scholar] [CrossRef]
- Garcia-Gras, E.; Lombardi, R.; Giocondo, M.J.; Willerson, J.T.; Schneider, M.D.; Khoury, D.S.; Marian, A.J. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J. Clin. Investig. 2006, 116, 2012–2021. [Google Scholar] [CrossRef] [Green Version]
- Beauchamp, P.; Desplantez, T.; McCain, M.L.; Li, W.; Asimaki, A.; Rigoli, G.; Parker, K.K.; Saffitz, J.E.; Kléber, A.G. Electrical coupling and propagation in engineered ventricular myocardium with heterogeneous expression of connexin43. Circ. Res. 2012, 110, 1445–1453. [Google Scholar] [CrossRef] [Green Version]
- Asimaki, A.; Tandri, H.; Duffy, E.R.; Winterfield, J.R.; Mackey-Bojack, S.; Picken, M.M.; Cooper, L.T.; Wilber, D.J.; Marcus, F.I.; Basso, C.; et al. Altered desmosomal proteins in granulomatous myocarditis and potential pathogenic links to arrhythmogenic right ventricular cardiomyopathy. Circ. Arrhythmia Electrophysiol. 2011, 4, 743–752. [Google Scholar] [CrossRef] [Green Version]
- Denayer, E.; De Ravel, T.; Legius, E. Clinical and molecular aspects of RAS related disorders. J. Med. Genet. 2008, 45, 695–703. [Google Scholar] [CrossRef]
- Quarta, G.; Syrris, P.; Ashworth, M.; Jenkins, S.; Alapi, K.Z.; Morgan, J.; Muir, A.; Pantazis, A.; McKenna, W.J.; Elliott, P.M.; et al. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2011, 33, 1128–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosmic. COSMIC—Catalogue of Somatic Mutations in Cancer [Internet]. [Cited 2020 May 24]. Available online: https://cancer.sanger.ac.uk/cosmic (accessed on 2 March 2020).
- Akhtar, M.M.; Elliott, P. The genetics of hypertrophic cardiomyopathy. Glob. Cardiol. Sci. Pr. 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Gandjbakhch, E.; Redheuil, A.; Pousset, F.; Charron, P.; Frank, R. Clinical Diagnosis, Imaging, and Genetics of arrhythmogenic right ventricular cardiomyopathy/dysplasia: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 784–804. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; A James, C.; Calkins, H. Diagnostic and therapeutic strategies for arrhythmogenic right ventricular dysplasia/cardiomyopathy patient. Europace 2018, 21, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The task force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar]
- Tadros, H.J.; Life, C.S.; Garcia, G.; Pirozzi, E.; Jones, E.G.; Datta, S.; Parvatiyar, M.S.; Chase, P.B.; Allen, H.D.; Kim, J.J.; et al. Meta-analysis of cardiomyopathy-associated variants in troponin genes identifies loci and intragenic hotspots that are associated with worse clinical outcomes. J. Mol. Cell. Cardiol. 2020, 142, 118–125. [Google Scholar] [CrossRef]
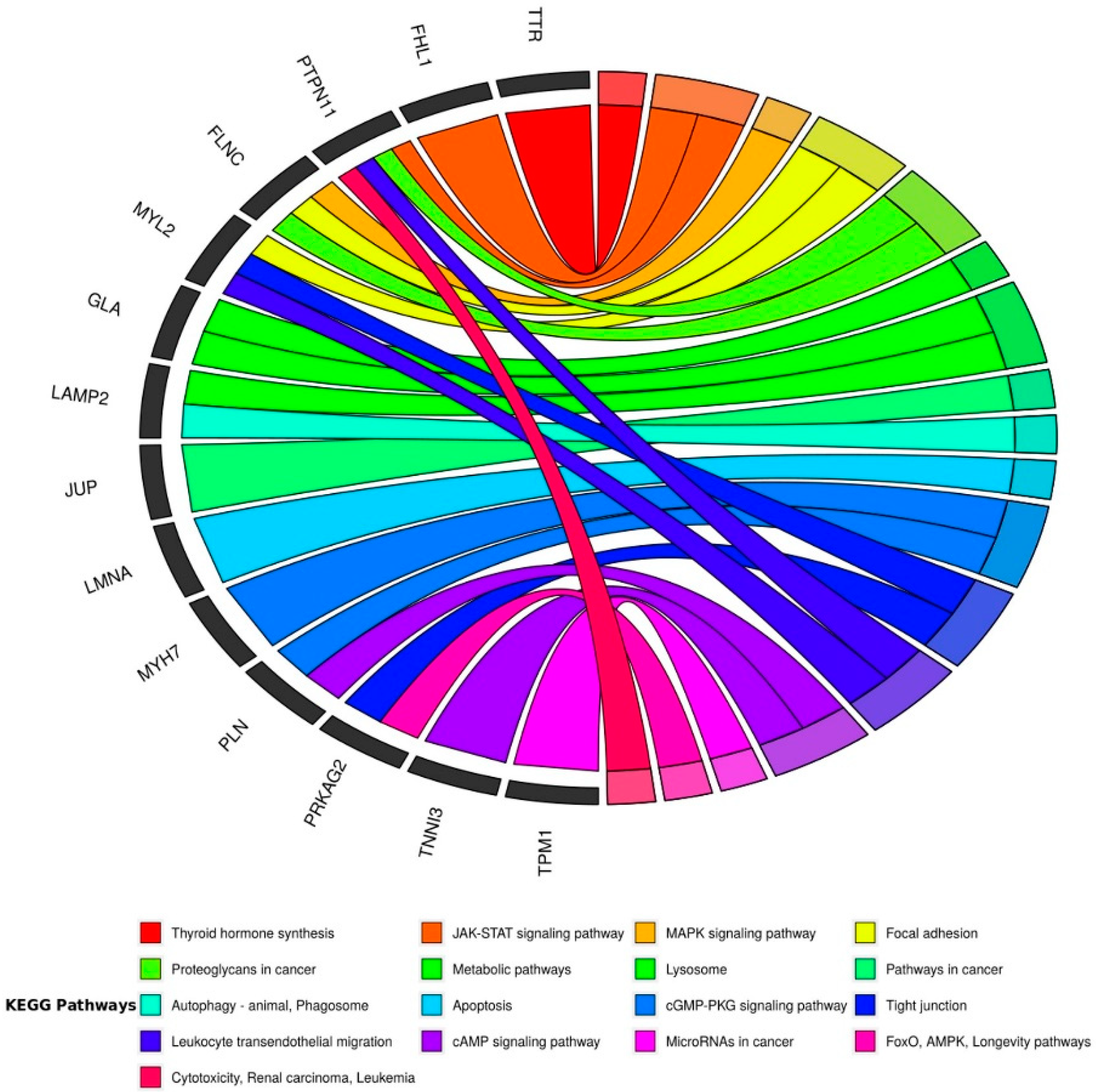
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Leon, E.Z.; Carrasco-Navarro, U.; Fierro, F. NeVOmics: An enrichment tool for gene ontology and functional network analysis and visualization of data from OMICs technologies. Genes 2018, 9, 569. [Google Scholar] [CrossRef] [Green Version]
- Walter, W.; Sánchez-Cabo, F.; Ricote, M. GOplot: An R package for visually combining expression data with functional analysis. Bioinformatics 2015, 31, 2912–2914. [Google Scholar] [CrossRef]
- Al-Jassar, C.; Bikker, H.; Overduin, M.; Chidgey, M. Mechanistic basis of desmosome-targeted diseases. J. Mol. Biol. 2013, 425, 4006–4022. [Google Scholar] [CrossRef] [Green Version]
- Gallicano, G.I.; Kouklis, P.; Bauer, C.; Yin, M.; Vasioukhin, V.; Degenstein, L.; Fuchs, E. Desmoplakin Is required early in development for assembly of desmosomes and cytoskeletal linkage. J. Cell Biol. 1998, 143, 2009–2022. [Google Scholar] [CrossRef]
- Leung, C.L.; Green, K.J.; Liem, R.K.H. Plakins: A family of versatile cytolinker proteins. Trends Cell Biol. 2002, 12, 37–45. [Google Scholar] [CrossRef]
- Jonkman, M.F.; Pasmooij, A.M.G.; Pasmans, S.G.M.A.; Berg, M.P.V.D.; Ter Horst, H.J.; Timmer, A.; Pas, H.H. Loss of desmoplakin tail causes lethal acantholytic epidermolysis bullosa. Am. J. Hum. Genet. 2005, 77, 653–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalczyk, A.P.; Bornslaeger, E.A.; Borgwardt, J.E.; Palka, H.L.; Dhaliwal, A.S.; Corcoran, C.M.; Denning, M.F.; Green, K.J. The amino-terminal domain of desmoplakin binds to plakoglobin and clusters desmosomal cadherin-plakoglobin complexes. J. Cell Biol. 1997, 139, 773–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, M.G.H.; Hanahan, U. Genetic deletion of the desmosomal component desmoplakin promotes tumor microinvasion in a mouse model of pancreatic neuroendocrine carcinogenesis. PLoS Genet. 2010, 6, e1001120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrod, D.; Chidgey, M. Desmosome structure, composition and function. Biochim. Biophys. Acta 2008, 1778, 572–587. [Google Scholar] [CrossRef]
- Tselepis, C.; Chidgey, M.; North, A.; Garrod, D. Desmosomal adhesion inhibits invasive behavior. Proc. Natl. Acad. Sci. USA 1998, 95, 8064–8069. [Google Scholar] [CrossRef] [Green Version]
- Oshiro, M.M.; Kim, C.J.; Wozniak, R.J.; Junk, D.; Muñoz-Rodríguez, J.L.; A Burr, J.; Fitzgerald, M.; Pawar, S.C.; Cress, A.E.; Domann, F.; et al. Epigenetic silencing of DSC3 is a common event in human breast cancer. Breast Cancer Res. 2005, 7, R669–R680. [Google Scholar] [CrossRef] [Green Version]
- Papagerakis, S.; Shabana, A.-H.; Pollock, B.H.; Papagerakis, P.; Depondt, J.; Berdal, A. Altered desmoplakin expression at transcriptional and protein levels provides prognostic information in human oropharyngeal cancer. Hum. Pathol. 2009, 40, 1320–1329. [Google Scholar] [CrossRef]
- Alazawi, W.O.F.; Morris, L.S.; Stanley, M.A.; Garrod, D.R.; Coleman, N. Altered expression of desmosomal components in high-grade squamous intraepithelial lesions of the cervix. Virchows Arch. 2003, 443, 51–56. [Google Scholar] [CrossRef]
- Cui, T.; Chen, Y.; Yang, L.; Knösel, T.; Zöller, K.; Huber, O.; Petersen, D.I. DSC3 expression is regulated by p53, and methylation of DSC3 DNA is a prognostic marker in human colorectal cancer. Br. J. Cancer 2011, 104, 1013–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamidov, Z.; Altendorf-Hofmann, A.; Chen, Y.; Settmacher, U.; Petersen, D.I.; Knösel, T. Reduced expression of desmocollin 2 is an independent prognostic biomarker for shorter patients survival in pancreatic ductal adenocarcinoma. J. Clin. Pathol. 2011, 64, 990–994. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Wang, L.; Yang, X.-X.; Jiang, Y.-H.; Guo, X.-L. The aberrant expression or disruption of desmocollin2 in human diseases. Int. J. Biol. Macromol. 2019, 131, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.; Hardy, R.; Haq, A.; Ogunbiyi, O.; Morton, D.; Chidgey, M. Desmocollin switching in colorectal cancer. Br. J. Cancer 2006, 95, 1367–1370. [Google Scholar] [CrossRef] [Green Version]
- Davies, E.; Cochrane, R.; Hiscox, S.; Jiang, W.; Sweetland, H.; Mansel, R. The role of desmoglein 2 and E-cadherin in the invasion and motility of human breast cancer cells. Int. J. Oncol. 1997, 11, 415–419. [Google Scholar] [CrossRef]
- Sun, R.; Ma, C.; Wang, W.; Yang, S. Upregulation of desmoglein 2 and its clinical value in lung adenocarcinoma: A comprehensive analysis by multiple bioinformatics methods. PeerJ 2020, 8, e8420. [Google Scholar] [CrossRef]
- Hofmann, I. Plakophilins and their roles in diseased states. Cell Tissue Res. 2020, 379, 5–12. [Google Scholar] [CrossRef]
- Beaudry, V.G.; Jiang, D.; Dusek, R.L.; Park, E.J.; Knezevich, S.; Ridd, K.; Vogel, H.; Bastian, B.C.; Attardi, L.D. Loss of the p53/p63 regulated desmosomal protein Perp promotes tumorigenesis. PLoS Genet. 2010, 6, e1001168. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Chen, Y.; Cui, T.; Knosel, T.; Zhang, Q.; Albring, K.F.; Huber, O.; Petersen, I. Desmoplakin acts as a tumor suppressor by inhibition of the Wnt/beta-catenin signaling pathway in human lung cancer. Carcinogenesis 2012, 33, 1863–1870. [Google Scholar] [CrossRef] [Green Version]
- Dawson, K.; Aflaki, M.; Nattel, S. Role of the Wnt-Frizzled system in cardiac pathophysiology: A rapidly developing, poorly understood area with enormous potential. J. Physiol. 2012, 591, 1409–1432. [Google Scholar] [CrossRef]
- Chen, S.N.; Gurha, P.; Lombardi, R.; Ruggiero, A.; Willerson, J.T.; Marian, A.J. The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ. Res. 2013, 114, 454–468. [Google Scholar] [CrossRef]
- Morin, P.J. Beta-catenin signaling and cancer. Bioessays 1999, 21, 1021–1030. [Google Scholar] [CrossRef]
- Hidalgo, M. Pancreatic cancer. N. Engl. J Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2016, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Tung, B.; Schade, B.; Cardiff, R.D.; Aina, O.H.; Sanguin-Gendreau, V.; Muller, W.J.; Bui, T. β-Catenin haploinsufficiency promotes mammary tumorigenesis in an ErbB2-positive basal breast cancer model. Proc. Natl. Acad. Sci. USA 2017, 114, E707–E716. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Hruban, R.H.; Klein, A.P. Familial pancreatic cancer. Arch. Pathol. Lab Med. 2009, 133, 365–374. [Google Scholar]
- Biedermann, K.; Vogelsang, H.; Becker, I.; Plaschke, S.; Siewert, J.R.; Höfler, H.; Keller, G. Desmoglein 2 is expressed abnormally rather than mutated in familial and sporadic gastric cancer. J. Pathol. 2005, 207, 199–206. [Google Scholar] [CrossRef]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC cancer gene census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- Michels, V.V.; Moll, P.P.; Miller, F.A.; Tajik, A.J.; Chu, J.S.; Driscoll, D.J.; Burnett, J.C.; Rodeheffer, R.J.; Chesebro, J.H.; Tazelaar, H.D.; et al. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N. Engl. J. Med. 1992, 326, 77–82. [Google Scholar] [CrossRef]
- Baig, M.K.; Goldman, J.H.; Caforio, A.L.; Coonar, A.S.; Keeling, P.J.; McKenna, W.J. Familial dilated cardiomyopathy: Cardiac abnormalities are common in asymptomatic relatives and may represent early disease. J. Am. Coll. Cardiol. 1998, 31, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Grünig, E.; Tasman, J.A.; Kücherer, H.; Franz, W.; Kübler, W.; Katus, H.A. Frequency and phenotypes of familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 1998, 31, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Hershberger, R.E.; Siegfried, J.D. Update 2011: Clinical and genetic issues in familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 2011, 57, 1641–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, E.L.; Hershberger, R.E. Genetic counseling and screening issues in familial dilated cardiomyopathy. J. Genet. Couns. 2001, 10, 397–415. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.H.; Hamilton, R.; et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: This document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011, 13, 1077–1109. [Google Scholar] [CrossRef] [PubMed]
- López-Ayala, J.M.; Oliva-Sandoval, M.J.; Sánchez-Muñoz, J.J.; Gimeno, J.R. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2015, 385, 662. [Google Scholar] [CrossRef]
- Barriales-Villa, R.; Gimeno-Blanes, J.R.; Zorio-Grima, E.; Ripoll-Vera, T.; Evangelista-Masip, A.; Moya-Mitjans, A.; Serratosa-Fernández, L.; Albert-Brotons, D.C.; García-Pinilla, J.M.; Garcia-Pavia, P.; et al. Plan of action for inherited cardiovascular diseases: Synthesis of recommendations and action algorithms. Rev. Española Cardiol. (Engl. Ed.) 2016, 69, 300–309. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.; Morales, A.; Taylor, M.R.G.; Vatta, M.; Ware, S.M.; et al. Genetic evaluation of cardiomyopathy: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2018, 20, 899–909. [Google Scholar] [CrossRef] [Green Version]
- Merlo, M.; Cannatà, A.; Gobbo, M.; Stolfo, D.; Elliott, P.M.; Sinagra, G. Evolving concepts in dilated cardiomyopathy. Eur. J. Heart Fail. 2017, 20, 228–239. [Google Scholar] [CrossRef] [Green Version]
- Masarone, D.; Kaski, J.P.; Pacileo, G.; Elliott, P.; Bossone, E.; Day, S.M.; Limongelli, G. Epidemiology and clinical aspects of genetic cardiomyopathies. Heart Fail. Clin. 2018, 14, 119–128. [Google Scholar] [CrossRef]
- Aminkeng, F.; Ross, C.; Rassekh, S.R.; Hwang, S.; Rieder, M.J.; Bhavsar, A.; Smith, A.; Sanatani, S.; Gelmon, K.A.; Bernstein, D.; et al. Recommendations for genetic testing to reduce the incidence of anthracycline-induced cardiotoxicity. Br. J. Clin. Pharmacol. 2016, 82, 683–695. [Google Scholar] [CrossRef] [Green Version]
- Curigliano, G.; Lenihan, D.; Fradley, M.; Ganatra, S.; Barac, A.; Blaes, A.; Herrmann, J.; Porter, C.; Lyon, A.; Lancellotti, P.; et al. Management of cardiac disease in cancer patients throughout oncological treatment: ESMO consensus recommendations. Ann. Oncol. 2020, 31, 171–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armenian, S.H.; Lacchetti, C.; Barac, A.; Carver, J.; Constine, L.S.; Denduluri, N.; Dent, S.; Douglas, P.S.; Durand, J.B.; Ewer, M.; et al. Prevention and monitoring of cardiac dysfunction in survivors of adult cancers: American society of clinical oncology clinical practice guideline. J. Clin. Oncol. 2017, 35, 893–911. [Google Scholar] [CrossRef] [PubMed]
- Charron, P.; Arad, M.; Arbustini, E.; Basso, C.; Bilinska, Z.T.; Elliott, P.; Helio, T.; Keren, A.; McKenna, W.J.; Monserrat, L.; et al. Genetic counselling and testing in cardiomyopathies: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2010, 31, 2715–2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]

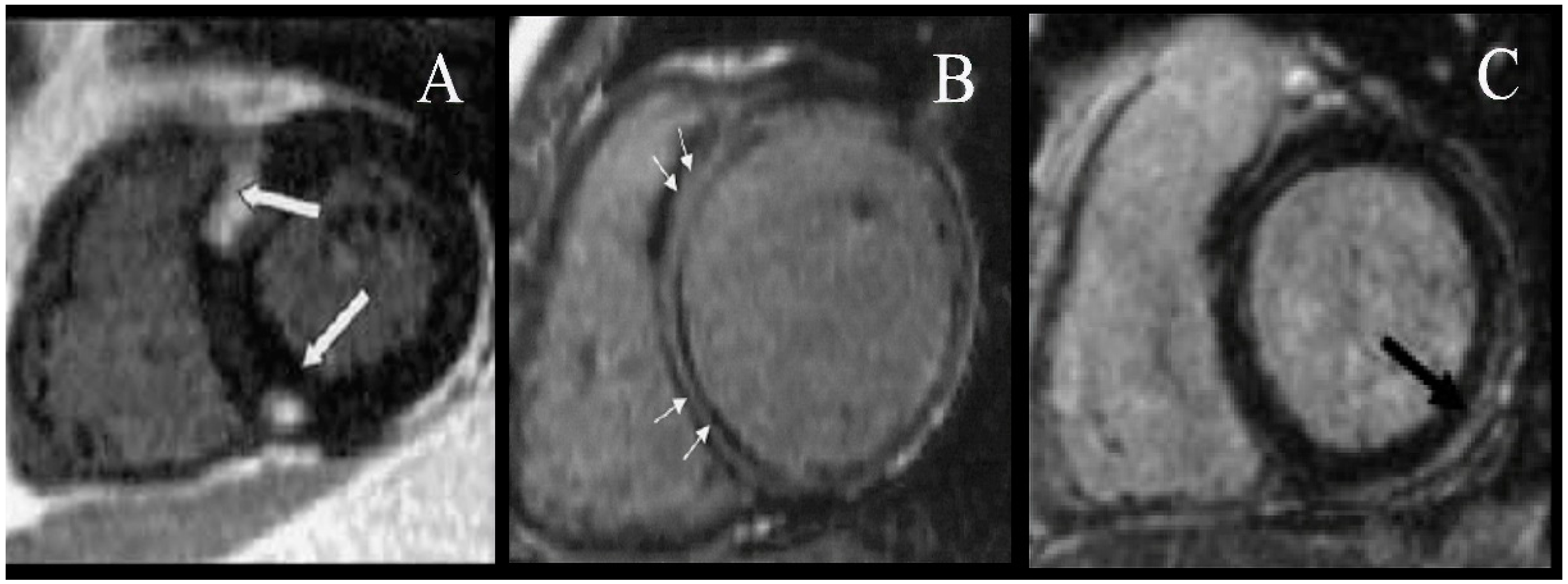
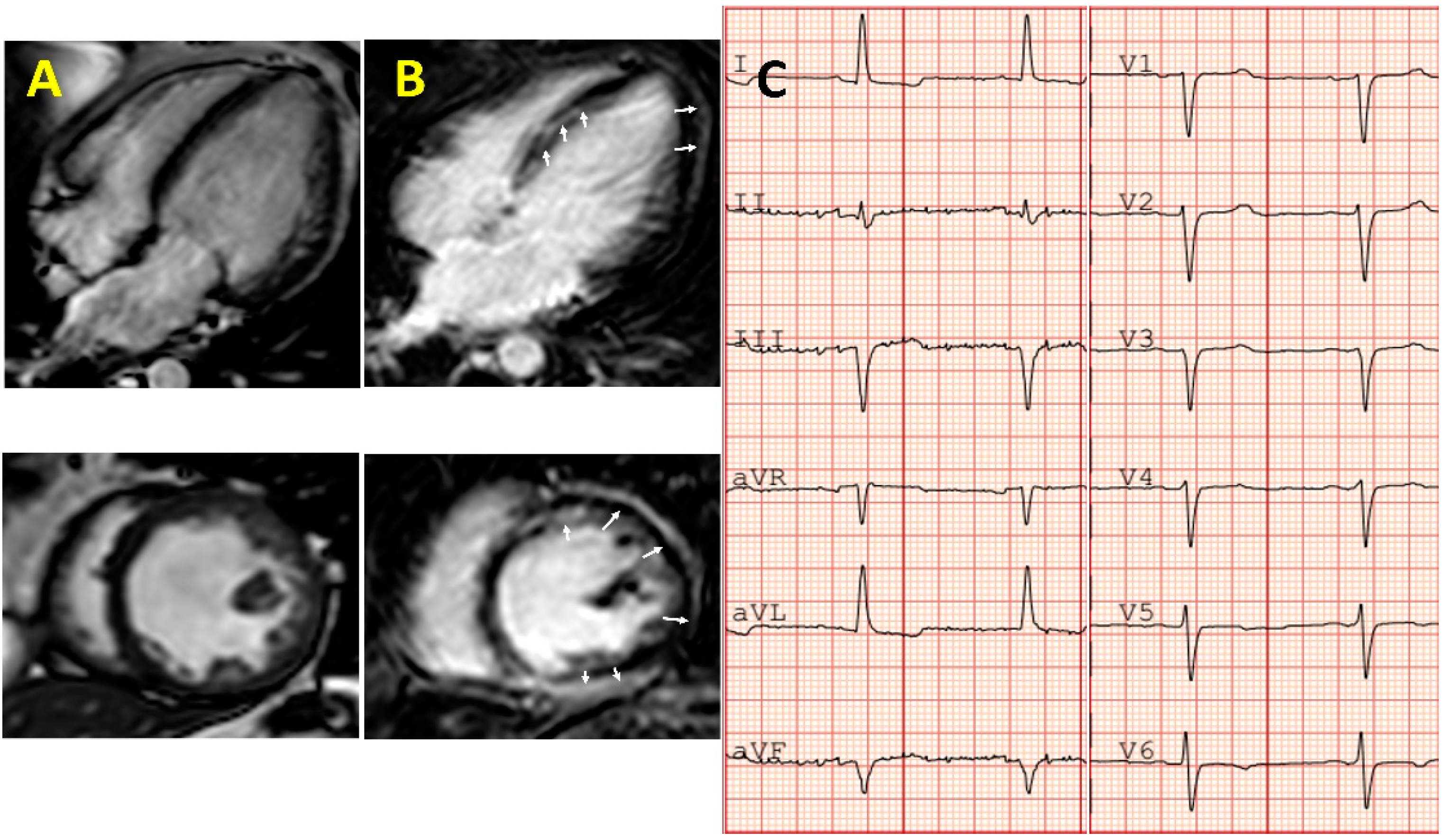


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Cardiomyopathy | Prevalence | DCM (%) | Family History of CMY (%) | LVEF Recovery (%) | LGE Prevalence (%) | Most Common LGE Distribution/LGE Pattern* | Other CMR Features | Death or HTx (%) | Gene | Genetic Variants (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| Idiopathic Dilated Cardiomyopathy [7,19,20,21] | 1:250–1:1000 | 30–50 | 30–50 | 30–65 | Interventricular septum/Linear, | - ↑LVEDV, ↑LVESV, ↓LVEF - Diffuse LV wall thinning -Shortened postcontrast T1 | BAG3 DES DMD FLNC LMNAMYBPC3 MYH7 PLNRBM20SCN5A TNNT2 TTN | 40–50 | ||
| 35 TTN mutation | Mid-wallInterventricular septum/Linear | |||||||||
| 88 LMNA mutation | Mid-wallBasal or mid-ventricular septal wall/Predominantly Epicardial | |||||||||
| NA PLN mutation | Posterolateral LV wall/Predominantly epicardial | |||||||||
| Alcoholic Cardiomyopathy [8,9,11] | 21–47 | 42 | 37–50 | 8 | 21–33 | BAG3 LMNA MYH7 TTN | 13.5 (9.9TTN) | |||
| Peripartum Cardiomyopathy [12,13,14] | 1:100–1:4000 | 6 | 10–15 | 45–78 | 7–21 | MYBPC3 MYH7 SCN5A TNNT2 TTN | 15–20 (9.9 TTN) | |||
| Tachycardia-Induced Cardiomyopathy [13,15,16] | 10–82 | 8–12 | SCN5A | |||||||
| Myocarditis [13,22,23] | 44–93 | LV inferolateral wall (PBV19)- anteroseptal(HHV6)/Subepicardial patchy or mid-wall | Myocardial edema in T2-STIR, EGE, pericardial effusion | |||||||
| Cancer therapy-related myocardial dysfunction [17,24,25,26,27,28] | 42–68 | <10 (higher if trastuzumab adjuvancy) | Inferolateral wall/Subepicardial | - Early injury: ↑LVESV, ↓LVEF, ↓ LVMI, EGE +, T1 mapping +/-, ECV +/-, T2 mapping + | MYH7 TTN | 7.5 | ||||
| - During/early post-therapy:↑LVESV, ↓LVEF, LGE + | ||||||||||
| - Late cardiotoxicity: ↑LVESV, LVEF, LGE +, ECV+ |
| Cardiovascular Toxicity | Chemotherapy Agents |
|---|---|
| Heart failure | • Anthracyclines (dose dependent): doxorubicin, daunorubicin, idarubicin, epirubicin, mitoxantrone |
| • Alkylating agents: cyclophosphamide, ifosfamide | |
| • Antimicrotubule agents: docetaxel | |
| • Monoclonal antibodies: trastuzumab, bevacizumab | |
| • Small molecule tyrosine kinase inhibitors: sunitinib, pazopanib, sorafenib, imatinib, dasatinib, lapatinib, nilotinib | |
| • Proteasome inhibitors: carfilzomib, Bortezomib | |
| Myopericarditis | • Alkylating agents: Cyclophosphamide |
| • Antimetabolites: 5-fuorouracil, cytarabine | |
| • Monoclonal antibodies: Trastuzumab, rituximab | |
| • Cytokines: Interleukin-2 | |
| • Immune-checkpoint inhibitors |
| Gene | Protein | Locus | Main Cardiac Disease | Frequency (%) | Ref. | Somatic Mutations in Cancer (%) | Main Tissues | % of Mutated Samples per Specific Tissue * | Comments | Ref. OMIM |
|---|---|---|---|---|---|---|---|---|---|---|
| ACTC1 | Actin α-cardiacmuscle 1 | NG_007553 | HCM, DCM | 1 | [103] | 1.08 | Skin | 6.47 | * 102540 | |
| BAG3 | BAG cochaperone 3 | NG_016125 | DCM | - | 0.91 | * 603883 | ||||
| DES | Desmin | NG_008043 | HCM, DCM, ACM | <1 | [82,104,105] | 0.91 | Skin | 3.07 | * 125660 | |
| DMD | Dystrophin | NG_012232 | DCM | 13.09 | Cervix | 10.5 | * 300377 | |||
| Endometrium | 10.84 | |||||||||
| Large intestine | 6.89 | |||||||||
| Lung | 6.56 | |||||||||
| Parathyroid | 6.9 | |||||||||
| Skin | 11 | |||||||||
| Soft tissue | 6.62 | |||||||||
| Stomach | 7.83 | |||||||||
| Urinary tract | 7.73 | |||||||||
| DSC2 | Desmocollin 2 | NG_008208 | ACM | 1–8 | [104,105] | 1.57 | * 125645 | |||
| DSG2 | Desmoglein 2 | NG_007072 | ACM | 3–20 | [104,105] | 17.16 | Skin | 4.37 | * 125671 | |
| DSP | Desmoplakin | NG_008803 | ACM, DCM | 1–15, 2 | [82,104,105] | 3.55 | Cervix Skin | 5 10.03 | * 125647 | |
| EMD | Emerin | NG_008677 | DCM | - | 0.34 | * 300384 | ||||
| FHL1 | Four and a half LIM domains 1 | NG_015895 | HCM | - | 1.30 | * 300163 | ||||
| FHOD3 | Formin homology 2 domain containing 3 | NG_042837 | HCM | - | 6.14 | Skin | 3.56 | * 609691 | ||
| FLNC | Filamin C | NG_011807 | ACM, DCM | 1 | [82,103] | 4.20 | Cervix Peritoneum Skin | 5.5 10.53 12.14 | * 102565 | |
| GLA | Galactosidase alpha | NG_007119 | HCM | 0.55 | Fabry disease | * 300644 | ||||
| JUP | Junction plakoglobin | NG_009090 | ACM | 0–1 | [104,105] | 1.39 | Stomach | 7.63 (CNV) | Naxos disease | * 173325 |
| LAMP2 | Lysosomal associated membrane protein 2 | NG_007995 | HCM | 0.7–2.7 | [106] | 1.06 | Danondisease | * 309060 | ||
| LMNA | Lamin A/C | NG_008692 | DCM, ACM | 4–8, 3–4 | [82,104,105] | 1.16 | Thyroid Breast | 3.18 3.59 (CNV) | Charcot-Marie-Tooth disease, Emery-Dreifuss muscular dystrophy, Hutchinson-Gilford progeria, Malouf syndrome | * 150330 |
| MYBPC3 | Myosin binding protein C cardiac | NG_007667 | HCM, DCM | 35–40 | [82,103] | 1.75 | Thymus | 3.57 | * 600958 | |
| MYH7 | Myosin heavy chain 7 | NG_007884 | HCM, DCM | 40–44, 3–4 | [82,103] | 3.67 | Skin | 11.09 | * 160760 | |
| MYL2 | Myosin light chain 2 | NG_007554 | HCM | 1–2 | [103] | 0.71 | * 160781 | |||
| MYL3 | Myosin light chain 3 | NG_007555 | HCM | 1 | [103] | 0.31 | * 160790 | |||
| PKP2 | Plakophilin 2 | NG_009000 | ACM, DCM | 20–45, | [104,105] | 2.54 | Skin | 4.69 | * 602861 | |
| PLN | Phospholamban | NG_009082 | ACM, DCM | 0–12 | [104,105] | 0.33 | * 172405 | |||
| PRKAG2 | AMP-activated protein kinase subunit-γ-2 | NG_007486 | HCM | 1 | [103] | 3.96 | Skin Stomach | 3.01 (CNV) 3.05 (CNV) | Wolff-Parkinson-White syndrome, Glycogen storage disease of heart | * 602743 |
| PTPN11 | Protein tyrosine phosphatase non-receptor type 11 | NG_007459 | HCM | - | 2.09 | Penis | 4.76 | LEOPARD syndrome, Leukemia, juvenile myelomonocytic somatic, Noonan syndrome | * 176876 | |
| RBM20 | RNA binding motif protein 20 | NG_021177 | DCM | 2 | [82] | 2.70 | CNS Skin | 3.89 (CNV) 3.88 | * 613171 | |
| SCN5A | Sodium voltage-gated channel alpha subunit 5 | NG_008934 | DCM | 2–3 | [82] | 4.78 | Skin | 15.37 | Brugada syndrome, Long QT syndrome | * 600163 |
| TMEM43 | Transmembrane protein 43 | NG_008975 | ACM | <2 | [104,105] | 0.60 | Urinary tract | 3.02 (CNV) | Emery-Dreifuss muscular dystrophy | * 612048 |
| TNNC1 | Troponin C1. slow skeletal and cardiac type | NG_008963 | HCM, DCM | 0.61, 0.24 | [107] | 0.26 | * 191040 | |||
| TNNI3 | Troponin I3 cardiac type | NG_007866 | HCM, DCM | 1.35–5, 0.57 | [103,107] | 0.57 | * 191044 | |||
| TNNT2 | Troponin T2 cardiac type | NG_007556 | HCM, DCM | 5–15, 3 | [82,103] | 0.89 | Breast Thyroid | 6.65 (CNV) 5.26 (CNV) | * 191045 | |
| TPM1 | Tropomyosin 1 | NG_007557 | HCM, DCM | 3, 1–2 | [82,103] | 0.94 | * 191010 | |||
| TRIM63 | Tripartite motif containing 63 | NG_033268 | HCM | - | 0.72 | * 606131 | ||||
| TTN | Titin | NG_011618 | DCM | 12–25 | [82] | 24.49 | Biliary tract | 14.26 | * 188840 | |
| Breast | 14.34 | |||||||||
| CNS | 9.74 | |||||||||
| Cervix | 26.5 | |||||||||
| Endometrium | 25.49 | |||||||||
| GT | 100 | |||||||||
| Large intestine | 26.49 | |||||||||
| Liver | 19.67 | |||||||||
| Lung | 25.55 | |||||||||
| Meninges | 7.02 | |||||||||
| Esophagus | 16.92 | |||||||||
| Ovary | 18.1 | |||||||||
| Pancreas | 9.6 | |||||||||
| parathyroid | 8.33 | |||||||||
| Peritoneum | 5.56 | |||||||||
| Pleura | 6.94 | |||||||||
| Prostate | 8.5 | |||||||||
| Salivary gland | 5.26 | |||||||||
| Skin | 42.31 | |||||||||
| Stomach | 33.64 | |||||||||
| Testis | 5.62 | |||||||||
| Thyroid | 10.54 | |||||||||
| UAT | 20.76 | |||||||||
| Urinary tract | 36.14 | |||||||||
| TTR | Transthyretin | NG_009490 | HCM | - | 0.37 | Amyloidosis | * 176300 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabater-Molina, M.; Navarro-Peñalver, M.; Muñoz-Esparza, C.; Esteban-Gil, Á.; Santos-Mateo, J.J.; Gimeno, J.R. Genetic Factors Involved in Cardiomyopathies and in Cancer. J. Clin. Med. 2020, 9, 1702. https://doi.org/10.3390/jcm9061702
Sabater-Molina M, Navarro-Peñalver M, Muñoz-Esparza C, Esteban-Gil Á, Santos-Mateo JJ, Gimeno JR. Genetic Factors Involved in Cardiomyopathies and in Cancer. Journal of Clinical Medicine. 2020; 9(6):1702. https://doi.org/10.3390/jcm9061702
Chicago/Turabian StyleSabater-Molina, María, Marina Navarro-Peñalver, Carmen Muñoz-Esparza, Ángel Esteban-Gil, Juan Jose Santos-Mateo, and Juan R. Gimeno. 2020. "Genetic Factors Involved in Cardiomyopathies and in Cancer" Journal of Clinical Medicine 9, no. 6: 1702. https://doi.org/10.3390/jcm9061702