Temperature-Responsive Biocompatible Copolymers Incorporating Hyperbranched Polyglycerols for Adjustable Functionality


Abstract
:1. Introduction
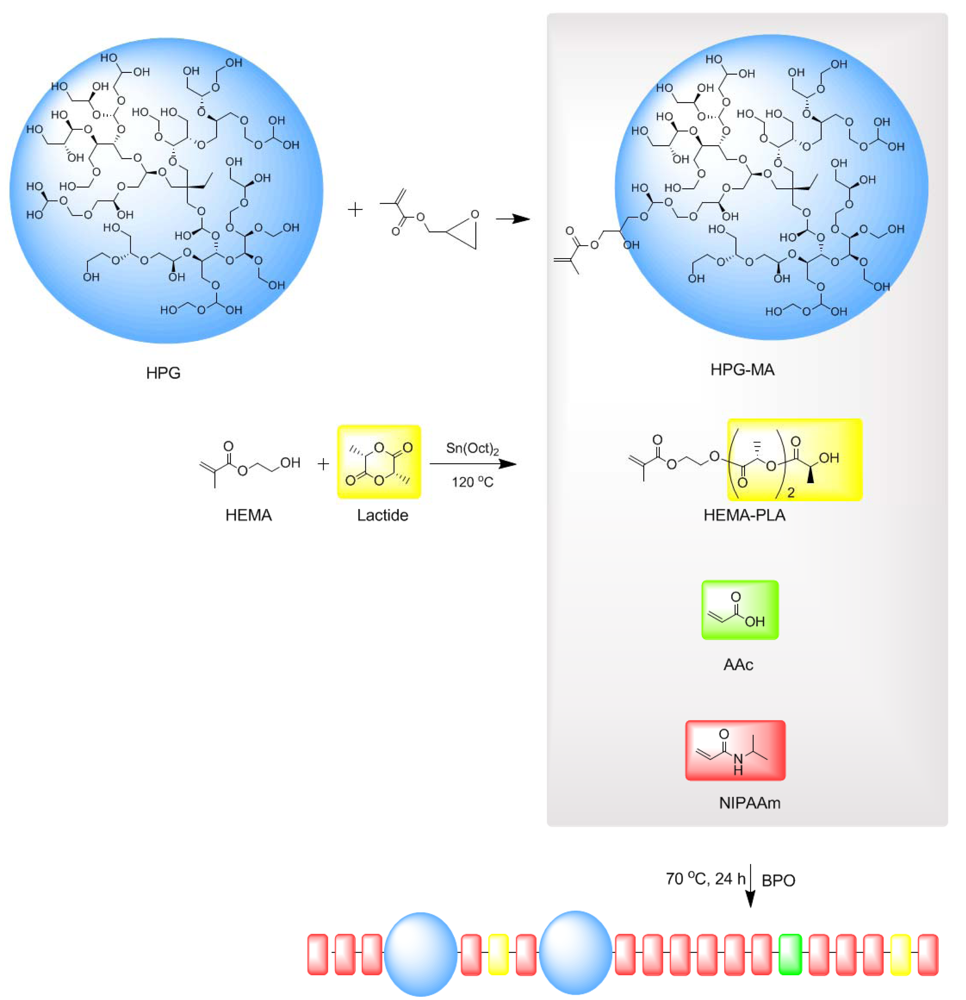
2. Experimental Section
2.1. Materials
2.2. Synthesis
Synthesis of HPG-MA
Synthesis of HEMAPLA
Synthesis of Poly(NIPAAm-co-HEMAPLA-co-AAc-co-HPG-MA)
2.3. Characterization
Nuclear Magnetic Resonance
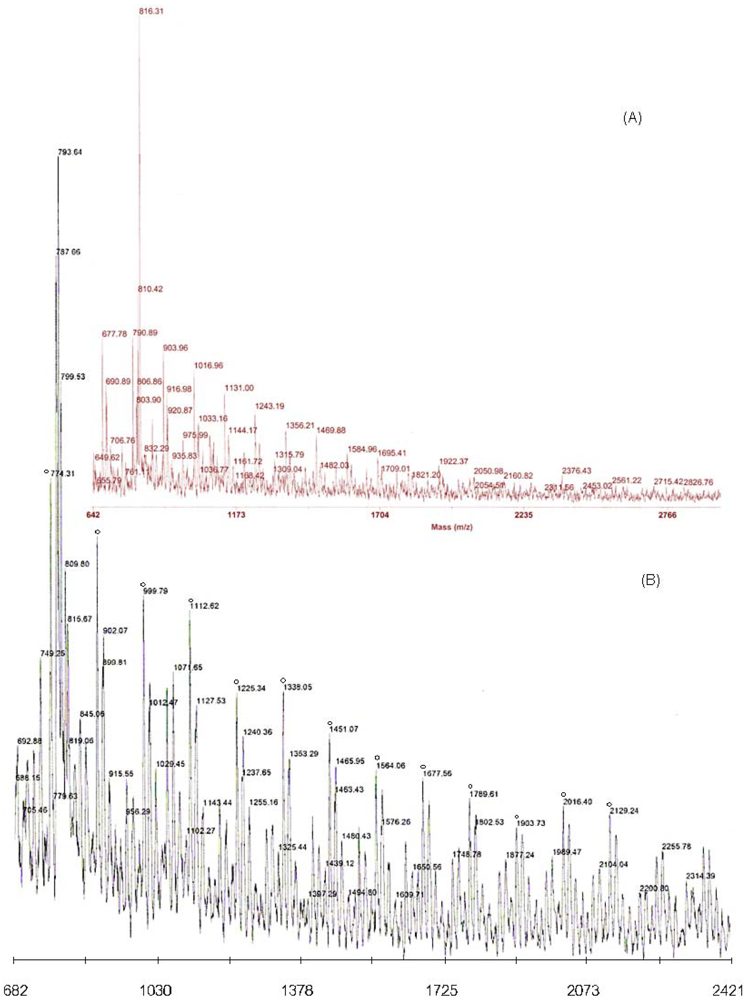
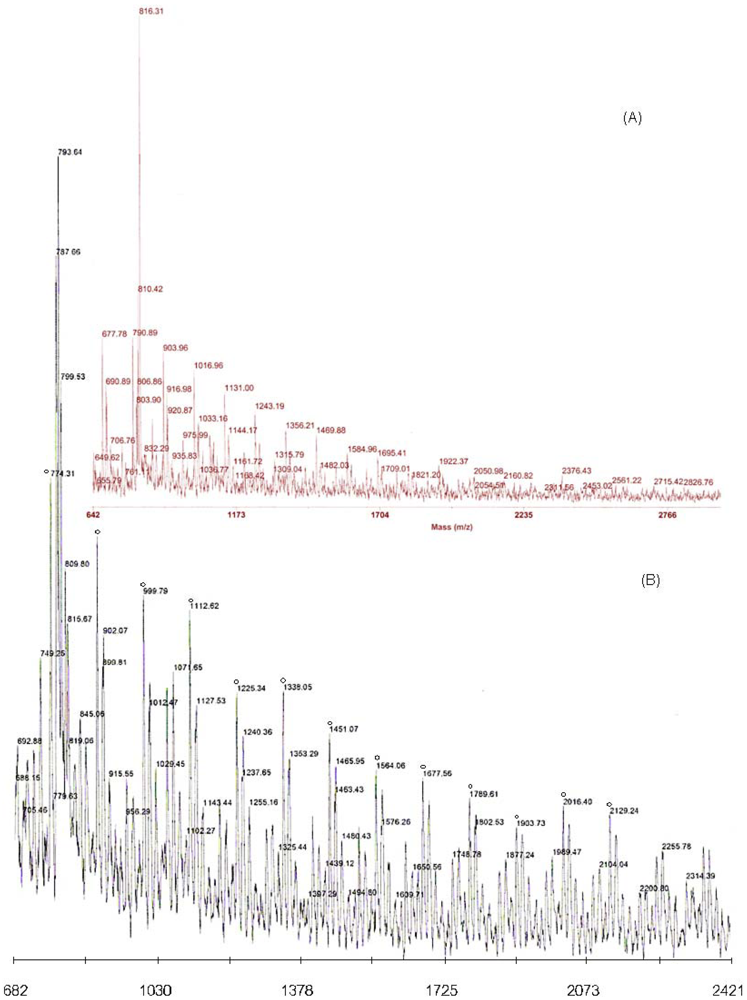
Matrix-Assisted Laser Desorption and Ionization Time-of-Flight Mass Spectrometry (MALDI-TOF-MS)
Electrospray Ionization Time-of-Flight Mass Spectrometry (ESI-TOF MS)
Gel Permeation Chromatography (GPC)
Differential Scanning Calorimetry (DSC)
UV-Vis
In vitro degradation
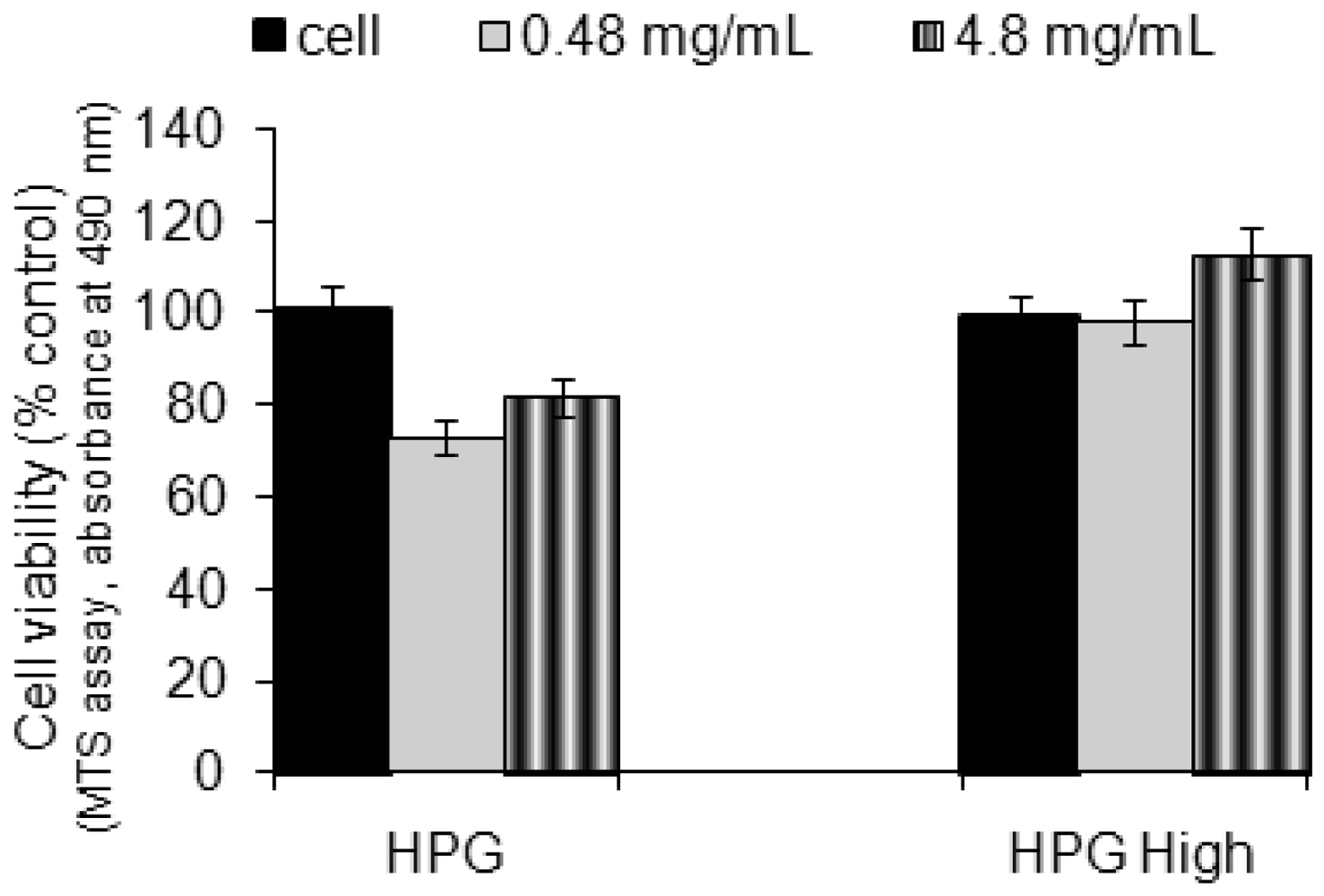
2.4. Cytotoxicity Assay
3. Results and Discussion
3.1. Synthesis of HPG-MA


3.2. Synthesis of HEMAPLA

3.3. Synthesis of Copolymers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (NIPAAm∣HEMAPLA∣AAc∣HPG-MA) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GPC | LCST (°C)a | |||||||||||
| Sample ID | Yield | Feed Ratio | 1H NMR (Mn) | Mn | ||||||||
| HPG High | 88% | (80∣ | 10∣ | 1∣ | 9) | (79∣ | 1∣ | 3.3∣ | 17) | 3689 | 1.7 | 28 ± 0.1 |
| HPG Med | 83% | (86∣ | 7∣ | 1∣ | 6) | (85∣ | 8∣ | 0.8∣ | 6) | 3455 | 1.7 | 24 ± 0.2 |
| HPG Low | 87% | (85∣ | 10∣ | 1∣ | 4) | (85∣ | 11∣ | 0.2∣ | 4) | 4465 | 1.5 | 22 ± 0.1 |
| Control | 90% | (87∣ | 10∣ | 3∣ | 0) | (86∣ | 12∣ | 1.8∣ | 0) | 1253 | 1.5 | 20 ± 0.4 |


3.4. The LCST of Copolymers


3.5. Cytotocity

3.6. Degradation Studies
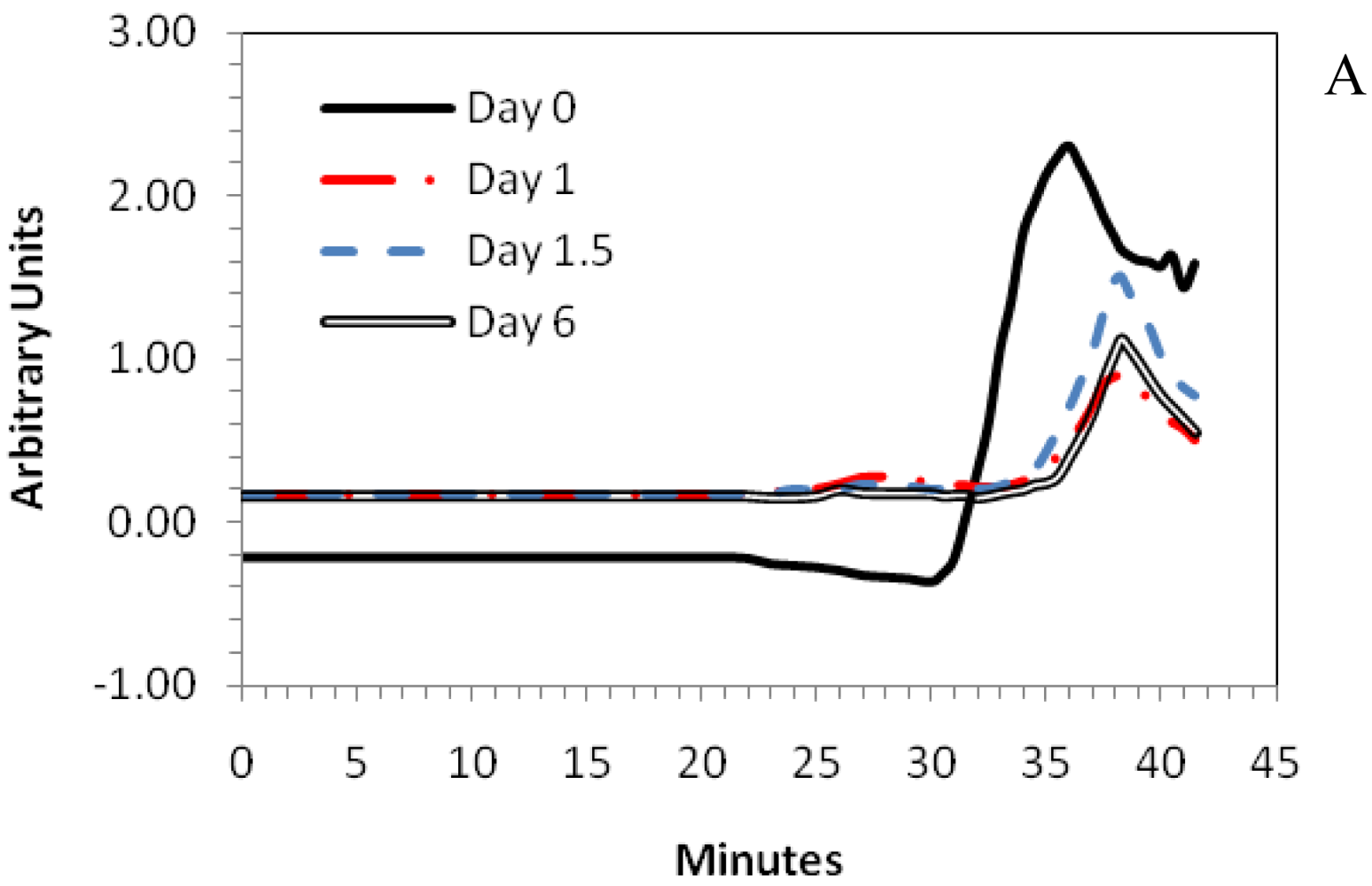


4. Conclusions
Acknowledgments
Appendix


| Region | Shift (ppm) | Relative Integral | ||
|---|---|---|---|---|
| HPG-1 | HPG-2 | HPG-3 | ||
| L13 | 81.0–82.0 | 1.00 | 1.00 | 1.00 |
| D | 79.5–80.5 | 2.10 | 2.15 | 1.90 |
| 2L14 | 73.5–74.5 | 6.94 | 6.69 | 2.58 |
| 2D,2T | 72.0–73.5 | 11.18 | 11.17 | 8.18 |
| L13, L14 | 70.5–72.0 | 3.88 | 3.96 | 2.25 |
| T | 64.0–65.0 | 4.05 | 3.86 | 0.59 |
| L13 | 62.0–63.5 | 1.07 | 1.11 | -* |
| Structural Unit | Relative Abundance (%) | ||
|---|---|---|---|
| T | 38 | 37 | 29 |
| L13 | 9.4 | 10 | 4 |
| L14 | 33 | 32 | 27 |
| D | 20 | 21 | 40 |
| Sample | 13C NMR | MALDI-TOF | |||
|---|---|---|---|---|---|
| DB | DPn | Mn | Mn | Mw/Mn | |
| HPG-1 | 0.48 | 16.3 | 1341 | 1367 | 1.15 |
| HPG-2 | 0.50 | 18.1 | 1473 | 1257 | 1.18 |
| HPG-3+ | 0.57 | 10.9 | 672 | 890 | 1.23 |
References
- Huang, X.; Misra, G.P.; Vaish, A.; Flanagan, J.M.; Sutermaster, B.; Lowe, T.L. Novel nanogels with both thermoreponsive and hydrolytically degradable properties. Macromolecules 2008, 41, 8339–8345. [Google Scholar]
- Rubio-Retama, J.; Zafeiropoulos, N.E.; Serafinelli, C.; Rojas-Reyna, R.; Voit, B.; Cabarcos, E.L.; Stamm, M. Synthesis and characterization of thermosensitive PNIPAM microgels covered with superparamagnetic gamma-Fe2O3 nanoparticles. Langmuir 2007, 23, 10280–10285. [Google Scholar]
- Ruel-Gariepy, E.; Leroux, J.C. In situ-forming hydrogels-review of temperature-sensitive systems. Eur. J. Pharm. Biopharm. 2004, 58, 409–426. [Google Scholar]
- Al-Tahami, K.; Singh, J. Smart polymer based delivery systems for peptides and proteins. Recent Patents Drug Deliv. Formulation 2007, 1, 65–71. [Google Scholar]
- Geever, L.M.; Devine, D.M.; Nugent, M.J.D.; Kennedy, J.E.; Lyons, J.G.; Higginbotham, C.L. The synthesis, characterisation, phase behaviour and swelling of temperature sensitive physically crosslinked poly(1-vinyl-2-pyrrolidinone)/poly(N-isopropylacrylamide) hydrogels. Eur. Polym. J. 2006, 42, 69–80. [Google Scholar]
- He, C.; Kim, S.W.; Lee, D.S. In situ gelling stimuli-sensitive block copolymer hydrogels for drug delivery. J. Control Release 2008, 127, 189–207. [Google Scholar]
- Hacker, M.C.; Klouda, L.; Ma, B.B.; Kretlow, J.D.; Mikos, A.G. Synthesis and characterization of injectable, thermally and chemically gelable, amphiphilic poly(N-isopropylacrylamide)-based macromers. Biomacromolecules 2008, 9, 1558–1570. [Google Scholar]
- Klouda, L.; Mikos, A.G. Thermoresponsive hydrogels in biomedical applications. Eur. J. Pharm. Biopharm. 2008, 68, 34–45. [Google Scholar]
- Fujimoto, K.L.; Ma, Z.; Nelson, D.M.; Hashizume, R.; Guan, J.; Tobita, K.; Wagner, W.R. Synthesis, characterization and therapeutic efficacy of a biodegradable, thermoresponsive hydrogel designed for application in chronic infarcted myocardium. Biomaterials 2009, 30, 4357–4368. [Google Scholar]
- Rzaev, Z.M.O.; Dinçer, S.; Piskin, E. Functional copolymers of N-isopropylacrylamide for bioengineering applications. Prog. Polym. Sci. 2007, 32, 534–595. [Google Scholar]
- Li, Z.; Wang, F.; Roy, S.; Sen, C.K.; Guan, J. Injectable, highly flexible, and thermosensitive hydrogels capable of delivering superoxide dismutase. Biomacromolecules 2009, 10, 3306–3316. [Google Scholar]
- Metters, A.T.; Anseth, K.S.; Bowman, C.N. Fundamental studies of a novel, biodegradable PEG-b-PLA hydrogel. Polymer 2000, 41, 3993–4004. [Google Scholar]
- Ma, Z.; Nelson, D.M.; Hong, Y.; Wagner, W.R. Thermally responsive injectable hydrogel incorporating methacrylate-polylactide for hydrolytic lability. Biomacromolecules 2010, 11, 1873–1881. [Google Scholar]
- Kainthan, R.K.; Janzen, J.; Levin, E.; Devine, D.V.; Brooks, D.E. Biocompatibility testing of branched and linear polyglycidol. Biomacromolecules 2006, 7, 703–709. [Google Scholar]
- Kainthan, R.K.; Muliawan, E.B.; Hatzikiriakos, S.G.; Brooks, D.E. Synthesis, charaterization, and viscoelastic properties of high molecular weight hyperbranched polyglycerols. Macromolecules 2006, 39, 7708–7717. [Google Scholar]
- Calderon, M.; Quadir, M.A.; Sharma, S.K.; Haag, R. Dendritic polyglycerols for biomedical applications. Adv. Mater. 2010, 22, 190–218. [Google Scholar]
- Calderon, M.; Quadir, M.A.; Strumia, M.; Haag, R. Functional dendritic polymer architectures as stimuli-responsive nanocarriers. Biochimie 2010, 92, 1242–1251. [Google Scholar]
- Frey, H.; Haag, R. Dendritic polyglycerol: A new versatile biocompatible material. Rev. Mol. Biotechnol. 2002, 90, 257–267. [Google Scholar]
- Papp, I.; Dernedde, J.; Enders, S.; Haag, R. Modular synthesis of multivalent glycoarchitectures and their unique selection binding behavior. Chem. Commun. (Camb.) 2008, 44, 5851–5853. [Google Scholar]
- Quadir, M.A.; Radowski, M.R.; Kratz, F.; Licha, K.; Hauff, P.; Haag, R. Dendritic multishell architectures for drug and dye transport. J. Control Release 2008, 132, 289–294. [Google Scholar]
- Sunder, A.; Hanselmann, R.; Frey, H.; Mulhaupt, R. Controlled synthesis of hyperbranched polyglycerols by ring-opening multibranching polymerization. Macromolecules 1999, 32, 4240–4246. [Google Scholar]
- André, P.; Lacroix-Desmazes, P.; Taylor, D.K.; Boutevin, B. Solubility of fluorinated homopolymer and block copolymer in compressed CO2. J. Supercritical Fluids 2006, 37, 263–270. [Google Scholar]
- Wells, S.L.; Taylor, D.; Adam, M.; DeSimone, J.M.; Farago, B. Study of the association of a diblock copolymer and absorption of an insoluble homopolymer in CO2. Macromolecules 2001, 34, 6161–6163. [Google Scholar]
- Duncan, R.; Izzo, L. Dendrimer biocompatibility and toxicity. Advan. Drug Delivery Rev. 2005, 57, 2215–2237. [Google Scholar]
- Kojima, C.; Yoshimura, K.; Harada, A.; Sakanishi, Y.; Kono, K. Temperature-sensitive hyperbranched poly(glycidol)s with oligo(ethylene glycol) monoethers. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 4047–4054. [Google Scholar]
- Oudshoorn, M.H.; Rissmann, R.; Bouwstra, J.A.; Hennink, W.E. Synthesis and characterization of hyperbranched polyglycerol hydrogels. Biomaterials 2006, 27, 5471–5479. [Google Scholar]
- Feil, H.; Bae, Y.H.; Feijen, J.; Kim, S.W. Effect of comonomer hydrophilicity and ionization on the lower critical solution temperature of N-isopropylacrylamide copolymers. Macromolecules 1993, 26, 2496–2500. [Google Scholar]
- Schick, M.J.; Fowkes, F.M. Surface Science Series; Marcel Dekker: New York, NY, USA, 1990. [Google Scholar]
- Wellons, S.L.; Carey, M.A.; Elder, D.K. Determination of hydroxyl content of polyurethane polyols and other alcohols. Anal. Chem. 1980, 52, 7376–7377. [Google Scholar]
- Tian, J.; Seery, T.A.P.; Weiss, R.A. Physically cross-linked alkylacrylamide hydrogels: Phase behavior and microstructure. Macromolecules 2004, 37, 9994–10000. [Google Scholar]
- Takizawa, K.; Nulwala, H.; Hu, J.; Yoshinaga, K.; Hawker, C.J. Molecularly defined (L)-lactic acid oligomers and polymers: Synthesis and characterization. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 5977–5990. [Google Scholar]
- Weir, N.A.; Buchanan, F.J.; Orr, J.F.; Dickson, G.R. Degradation of poly-L-lactide. Part 1: In vitro and in vivo physiological temperature degradation. J. Eng. Med. 2004, 218, 307–319. [Google Scholar]
- Törmälä, P.; Pohjonen, T.; Rokkanen, P. Bioabsorbable Polymers: Materials technology and surgical applications. J. Eng. Med. 1998, 212, 101–112. [Google Scholar]
- Lee, B.H.; Vernon, B. In situ-gelling, erodible N-Isopropylacrylamide copolymers. Macromol. Biosci. 2005, 5, 629–635. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Taylor, D.K.; Jayes, F.L.; House, A.J.; Ochieng, M.A. Temperature-Responsive Biocompatible Copolymers Incorporating Hyperbranched Polyglycerols for Adjustable Functionality. J. Funct. Biomater. 2011, 2, 173-194. https://doi.org/10.3390/jfb2030173
Taylor DK, Jayes FL, House AJ, Ochieng MA. Temperature-Responsive Biocompatible Copolymers Incorporating Hyperbranched Polyglycerols for Adjustable Functionality. Journal of Functional Biomaterials. 2011; 2(3):173-194. https://doi.org/10.3390/jfb2030173
Chicago/Turabian StyleTaylor, Darlene K., Friederike L. Jayes, Alan J. House, and Melony A. Ochieng. 2011. "Temperature-Responsive Biocompatible Copolymers Incorporating Hyperbranched Polyglycerols for Adjustable Functionality" Journal of Functional Biomaterials 2, no. 3: 173-194. https://doi.org/10.3390/jfb2030173
APA StyleTaylor, D. K., Jayes, F. L., House, A. J., & Ochieng, M. A. (2011). Temperature-Responsive Biocompatible Copolymers Incorporating Hyperbranched Polyglycerols for Adjustable Functionality. Journal of Functional Biomaterials, 2(3), 173-194. https://doi.org/10.3390/jfb2030173