Percolation Diffusion into Self-Assembled Mesoporous Silica Microfibres

 ,
,
Abstract
:1. Introduction

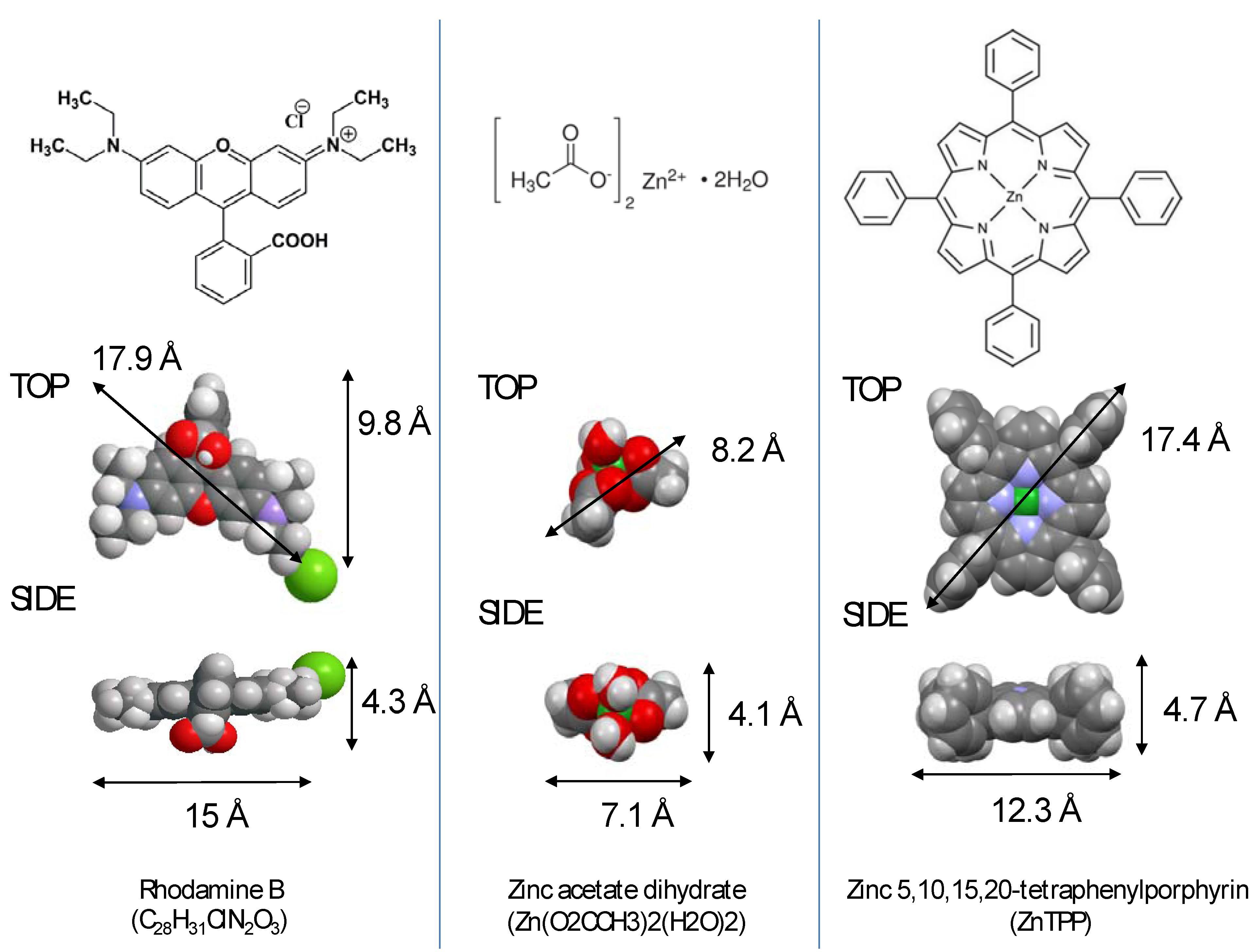
2. Results and Analysis
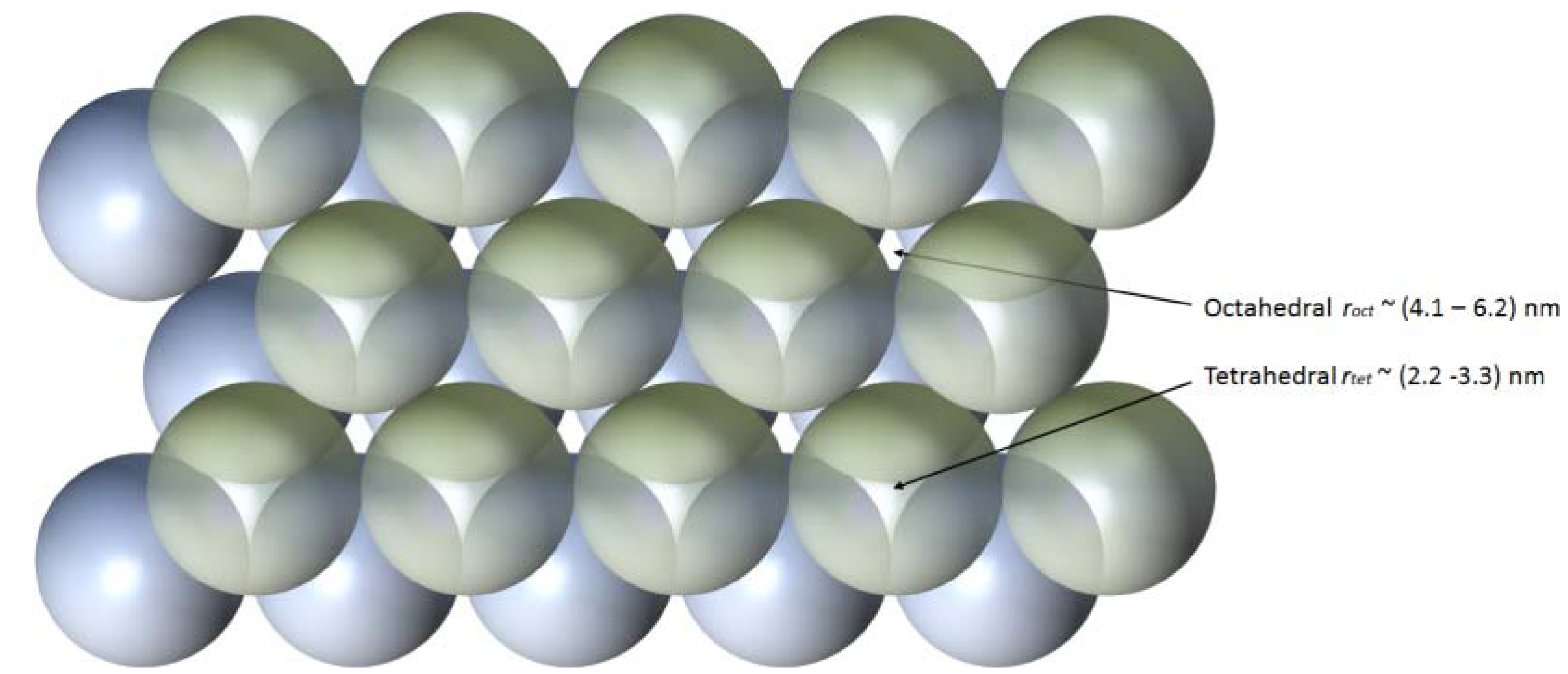
2.1. Fabricating Self-Assembled, Long Silica Microfibres

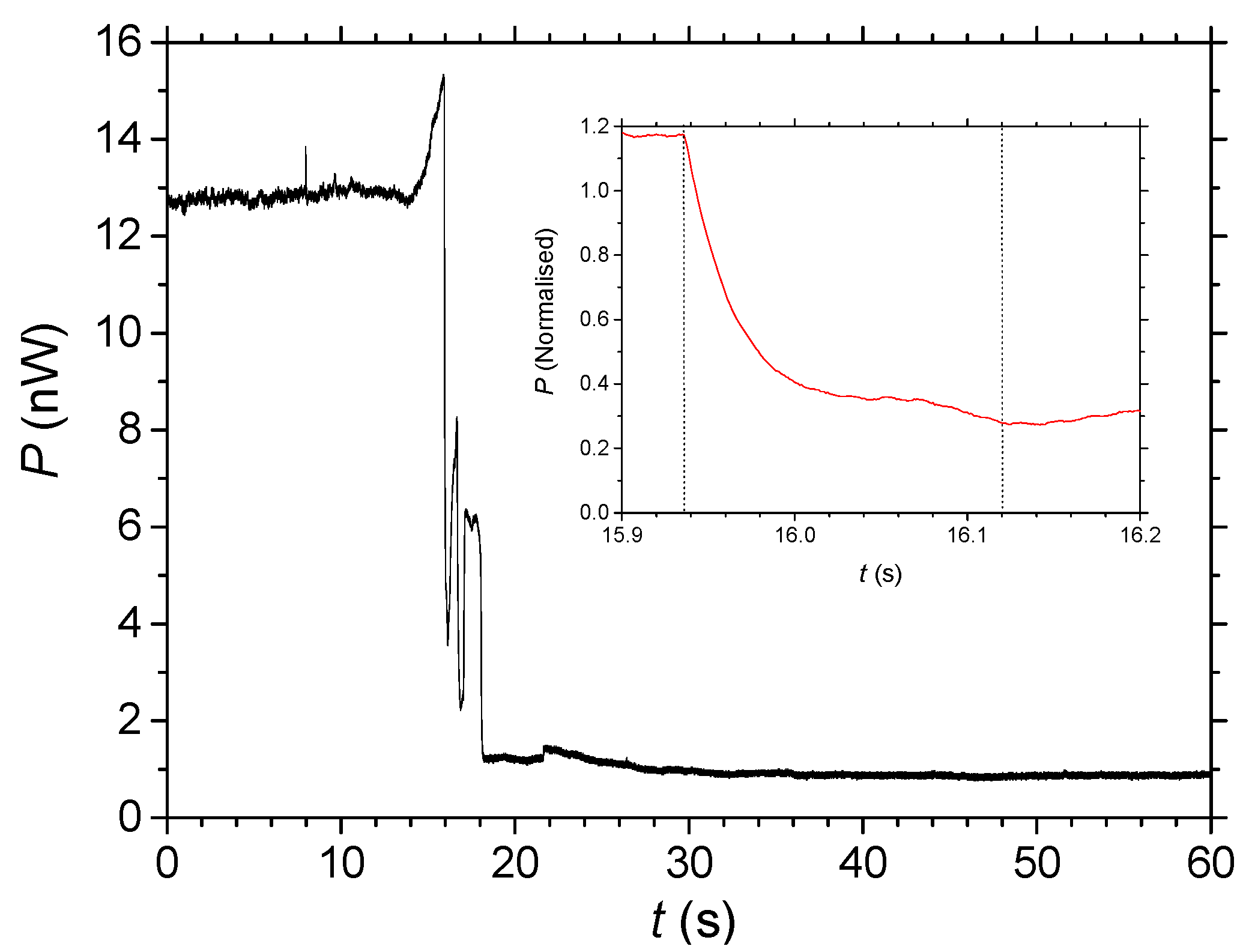
2.2. Diffusion Measured by Optical Transmission

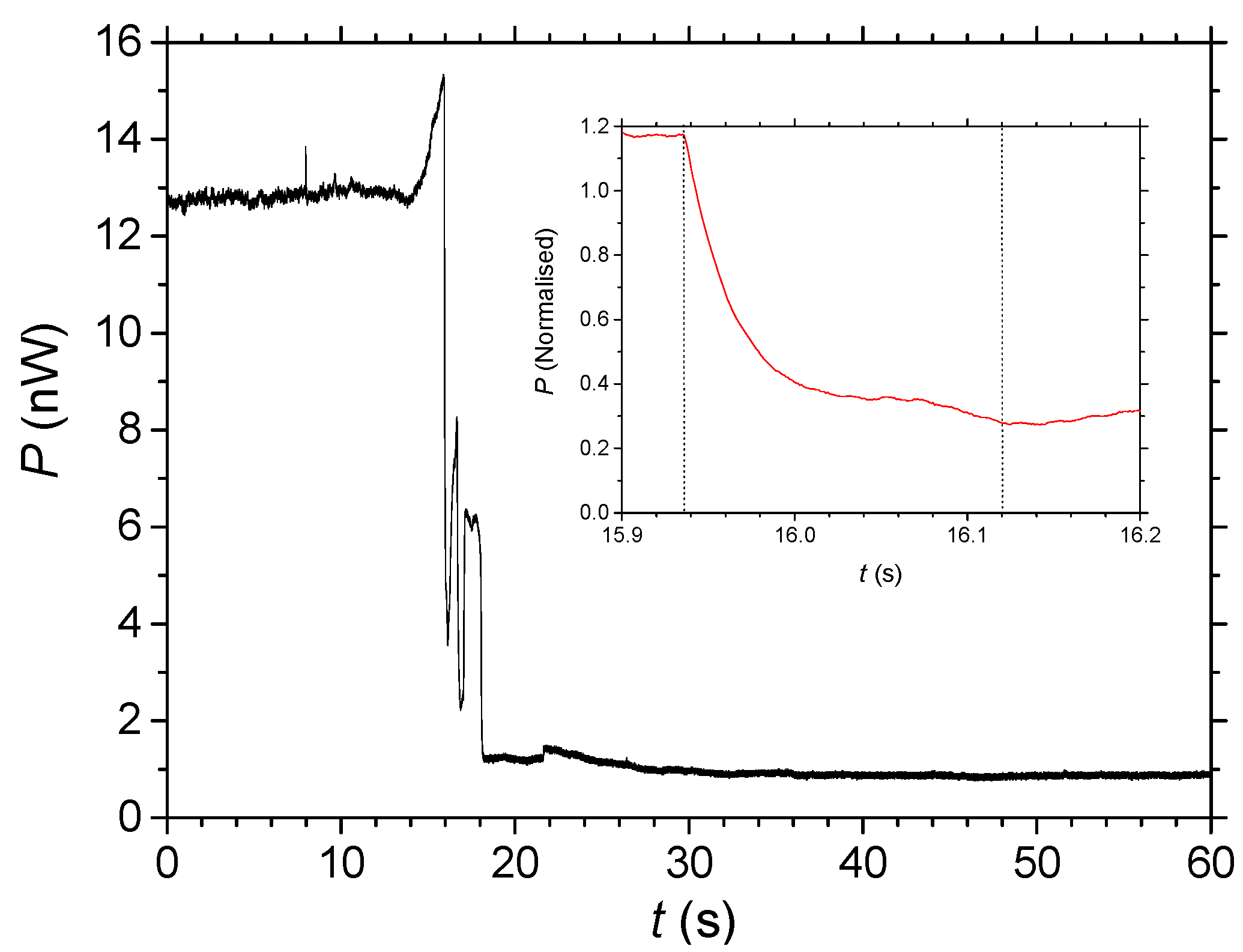
2.3. Measuring Diffusion through Laser Ablation of Self-Assembled Wires


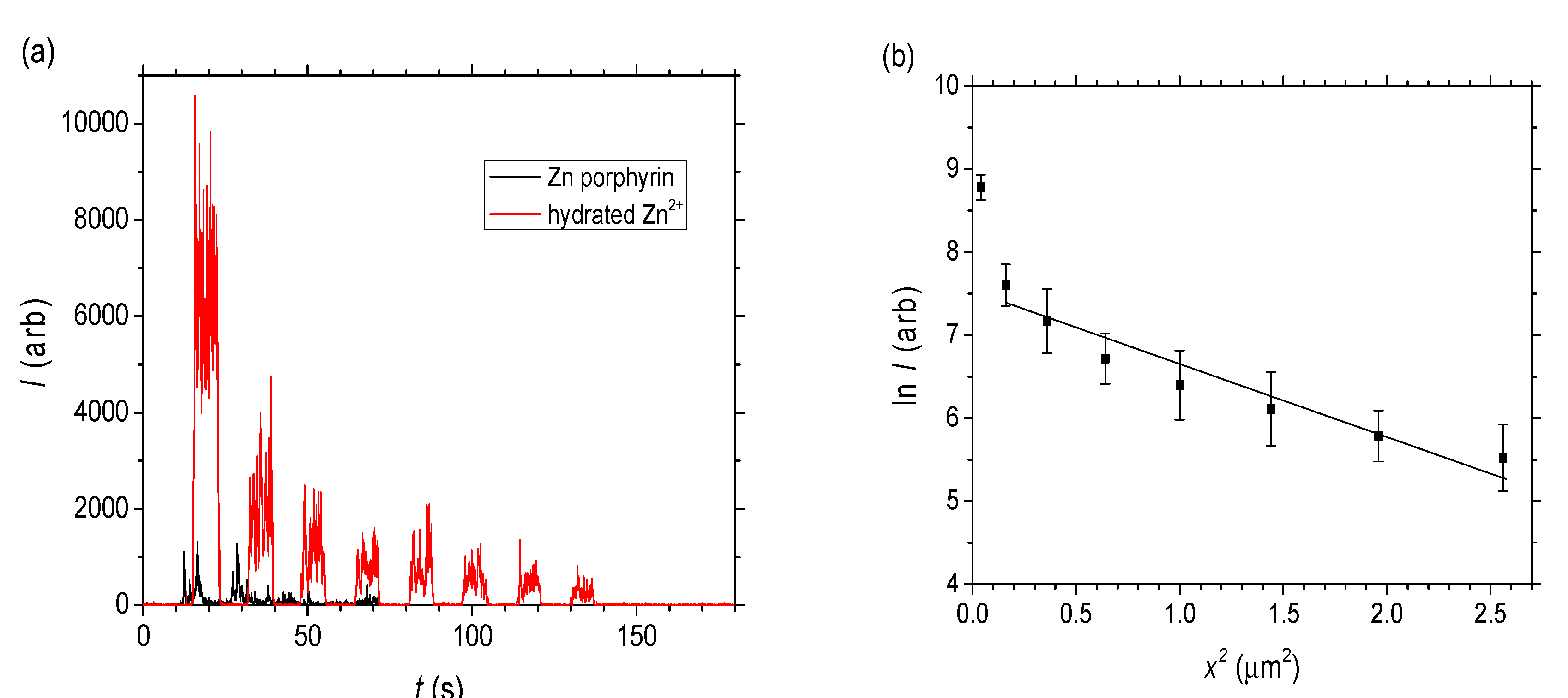
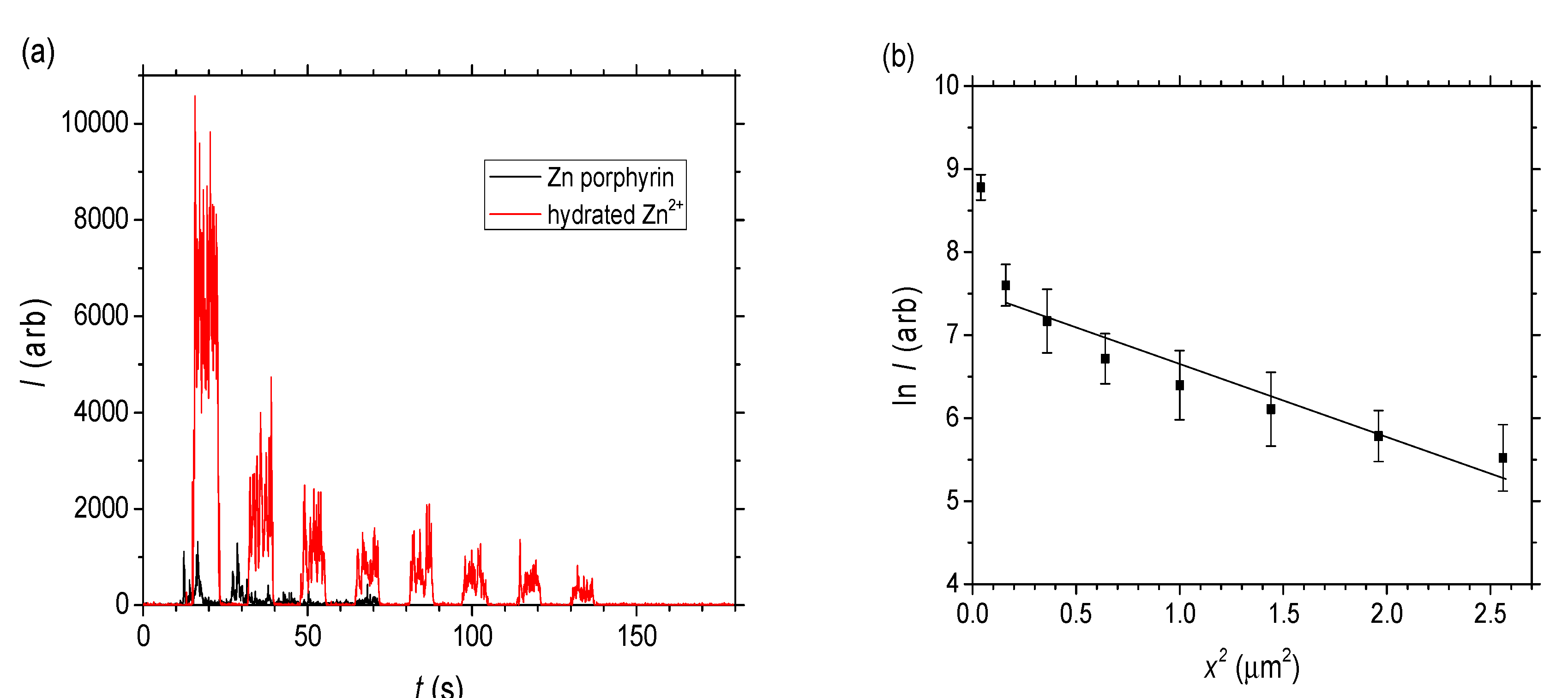
 C = c∙NA where NA is Avogadro’s number, the diffusion coefficient for the hydrated Zn2+ can also be extracted from the slope of ln C vs. x2 by converting to C noting I0 obtained from extrapolating to x2 = 0 in Figure 6b is assumed to equal 0.1 M × 6.022 × 1023. From this, the effective molecular diffusion coefficient is estimated to be a little lower: D~3 × 10−4 nm2 s−1. Overall it is clear that even for the hydrated Zn2+ which are small compared to the pore radius, liquid diffusion or percolation is very low compared to the Rhodamine B (and indeed compared to previous work with Rhodamine 6G in smaller channels [26]).
C = c∙NA where NA is Avogadro’s number, the diffusion coefficient for the hydrated Zn2+ can also be extracted from the slope of ln C vs. x2 by converting to C noting I0 obtained from extrapolating to x2 = 0 in Figure 6b is assumed to equal 0.1 M × 6.022 × 1023. From this, the effective molecular diffusion coefficient is estimated to be a little lower: D~3 × 10−4 nm2 s−1. Overall it is clear that even for the hydrated Zn2+ which are small compared to the pore radius, liquid diffusion or percolation is very low compared to the Rhodamine B (and indeed compared to previous work with Rhodamine 6G in smaller channels [26]).3. Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Rhodamine B | Zinc acetate dihydrate | ZnTPP |
|---|---|---|---|
| D (m2 s−1) | ~(80–2000) × 10−12 m2 s−1 | ~(3–4.8) × 10−22 m2 s−1 | < 10−22 m2 s−1 |
4. Experimental Section
4.1. Microfibre Fabrication
4.2. Optical Diffusion Measurement
4.3. Laser Ablation - Inductively Coupled Plasma - Mass Spectrometry (LA-ICP-MS)
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Naqshbandi, M.; Canning, J.; Gibson, B.C.; Nash, M.; Crossley, M.J. Room temperature self-assembly of mixed nanoparticles into photonic structures. Nat. Comm. 2012, 3, 1188. [Google Scholar] [CrossRef]
- Canning, J.; Weil, H.; Naqshbandi, M.; Cook, K.; Lancry, M. Laser tailoring surface interactions, contact angles, drop topologies and the self-assembly of optical microwires. Opt. Mat. Express 2013, 3, 284–294. [Google Scholar] [CrossRef]
- Canning, J.; Ma, M.; Gibson, B.C.; Shi, J.; Cook, K.; Crossley, M.J. Highly ordered mesoporous silica microfibres produced by evaporative self-assembly and fracturing. Opt. Mat. Express 2013, 3, 2028–2036. [Google Scholar] [CrossRef]
- De Angelis, R.; Venditti, I.; Fratoddi, I.; De Matteis, F.; Prosposito, P.; Cacciotti, I.; D’Amico, L.; Nanni, F.; Yadav, A.; Casalboni, M.; et al. From nanospheres to microribbons: Self-assembled Eosin Y doped PMMA nanoparticles as photonic crystals. J. Colloid Interface Sci. 2014, 414, 24–32. [Google Scholar] [CrossRef]
- Vos, W.L.; Sprik, R.; van Blaaderen, A.; Imhof, A.; Lagendijk, A.; Wegdam, G.H. Strong effects of photonic band structures on the diffraction of colloidal crystals. Phys. Rev. B 1996, 53, 16231–16235. [Google Scholar] [CrossRef]
- Schroden, R.C.; Al-Daous, M.; Blanford, C.F.; Stein, A. Optical properties of inverse opal photonic crystals. Chem. Mater. 2002, 14, 3305–3315. [Google Scholar] [CrossRef]
- Cai, Z.; Teng, J.; Xiong, Z.; Li, Y.; Li, Q.; Lu, X.; Zhao, X.S. Fabrication of TiO2 binary inverse opals without overlayers via the sandwich-vacuum infiltration of precursor. Langmuir 2011, 27, 5157–5164. [Google Scholar] [CrossRef]
- Cai, Z.; Liu, Y. J.; Teng, J.; Lu, X. Fabrication of large domain crack-free colloidal crystal heterostructures with superposition bandgaps using hydrophobic polystyrene spheres. ACS App. Mater. Interfaces 2012, 4, 5562–5569. [Google Scholar] [CrossRef]
- Cademartiri, L.; Bishop, K.J.M.; Snyder, P.W.; Ozin, G.A. Using shape for self-assembly. Phil. Tran. R. Soc. A 2012, 28, 2824–2847. [Google Scholar] [CrossRef]
- Adams, D.M. Inorganic Solids; John Wiley & Sons: Chichester, UK, 1974. [Google Scholar]
- Hulsken, B.; van Hameren, R.; Gerritsen, J.W.; Khoury, T.; Thordarson, P.; Crossley, M.J.; Rowan, A.E.; Nolte, R.J.M.; Elemens, A.A.W.; Speller, S. Real-time single-molecule imaging of oxidation catalysis at a liquid-solid interface. Nat. Nanotech. 2007, 2, 285–289. [Google Scholar] [CrossRef]
- Canning, J.; Khoury, T.; Jensen, P.; Huyang, G.; Sum, T.; Chin, Y.; Reimers, J.; Crossley, M.J. Alkyl Chain Control of The Formation of Self-Assembled Monolayers, Iridescent Microrods and Crystals of Alkylporphyrins of Potential Use in Photonics. In Proceedings of the Australian Conference on Optics, Lasers and Spectroscopy (ACOLS), Adelaide, Australia, 23–27 November 2009.
- Tong, L.; Gattass, R.R.; Ashcom, J.B.; He, S.; Lou, J.; Shen, M.; Maxwell, I.; Mazur, E. Subwavelength-diameter silica wires for low-loss optical wave guiding. Nature 2003, 426, 816–819. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Arai, D.; Taneda, N.; Mukaaiyama, T.; Ikemura, M. Continuous silica glass fiber produced by sol-gel process. J. Non-Cryst. Sol. 1989, 112, 437–441. [Google Scholar]
- Huo, Q.; Zhao, D.; Fang, J.; Weston, K.; Buratto, S.K.; Stucky, G.D.; Schacht, S.; Schuth, F. Room temperature growth of mesoporous silica fibers: A new high-surface-area optical waveguide. Adv. Mater. 1997, 9, 974–978. [Google Scholar]
- Levitz, P.; Ehret, G.; Sinha, S.K.; Drake, J.M. Porous vycor glass: The microstructure as probed by electron microscopy, direct energy transfer, small angle scattering, and molecular adsorption. J. Chem. Phys. 1991, 95, 6151–6161. [Google Scholar] [CrossRef]
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C.; Beck, J.S. Ordered mesoporous molecular sieves synthesized by a liquid-crystal template mechanism. Nature 1992, 359, 710–712. [Google Scholar] [CrossRef]
- Moller, K.; Bein, T. Inclusion chemistry in mesoporous materials. Chem. Mater. 1998, 10, 2950–2963. [Google Scholar] [CrossRef]
- Günther, D.; Jackson, S.E.; Longerichm, H.P. Laser ablation and arc/spark solid sample introduction into inductively coupled plasma mass spectrometers. Spectrochim.Acta B 1999, 54, 381–409. [Google Scholar] [CrossRef]
- Russo, R.E.; Mao, X.; Liu, H.; Gonzalez, J.; Mao, S.S. Laser ablation in analytical chemistry—A review. Talanta 2002, 57, 425–451. [Google Scholar] [CrossRef]
- Becker, J.S. Imaging of metals in biological tissue by laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS): State of the art and future developments. J. Mass Spectrom. 2013, 48, 255–268. [Google Scholar] [CrossRef]
- Snyder, A.W.; Love, J.D. Optical Waveguide Theory; Chapman & Hall: New York, NY, USA, 1983. [Google Scholar]
- Aslund, M.; Canning, J. Air-clad fibres for astronomical instrumentation: Focal-ratio degradation. Exp. Astron. 2009, 24, 1–7. [Google Scholar] [CrossRef]
- Blahut, M.; Opilski, A. Multimode interference structures—New way of passive element technology for photonics. Opto-Electron. Rev. 2000, 9, 293–300. [Google Scholar]
- Hoyst, R.; Bielejewska, A.; Szymanski, J.; Wilk, A.; Patkowski, A.; Gapinski, J.; Zywocinski, A.; Kalwarczyk, T.; Kalwarcyzk, E.; Tabaka, M.; et al. Scaling form of viscosity at all length scales in poly(ethylene glycol) solutions studied by fluorescence correlation spectroscopy and capillary electrophoresis. Phys. Chem. Chem. Phys. 2009, 11, 9025–9032. [Google Scholar] [CrossRef]
- Kievsky, Y.Y.; Carey, B.; Naik, S.; Mangan, N.; Ben-Avraham, D.; Sokolov, I. Dynamics of molecular diffusion of Rhodamine 6G in silica nanochannels. J. Chem. Phys. 2002, 128, 151102. [Google Scholar]
- Austin, C.; Hare, D.; Rawling, T.; McDonagh, A.M.; Doble, P.J. Quantification method for elemental bio-imaging by LA-ICP-MS using metal spiked PMMA films. J. Anal. Atom. Spect. 2010, 25, 722–725. [Google Scholar] [CrossRef]
- Huyang, G.; Canning, J.; Petermann, I.; Bishop, D.; McDonagh, A.M.; Crossley, M.J. Room temperature sol-gel fabrication and functionalization for sensor applications. Photon. Sensors 2013, 3, 168–177. [Google Scholar] [CrossRef]
- Becker, J.S.; Zoriy, M.; Dobrowolska, J.; Matusch, A. Laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MZ) in elemental imaging of biological tissues and in proteomics. J. Anal. Atom. Spect. 2007, 22, 736–744. [Google Scholar] [CrossRef]
- Sholl, D.S.; Fichthorn, K.A. Normal, single-file, and dual-mode diffusion of binary adsorbate mixtures in AlPO4-5. J. Chem. Phys. 1997, 107, 4384–4389. [Google Scholar] [CrossRef]
- Atkins, P.W. Physical Chemistry, 3rd ed; Oxford University Press: Oxford, UK, 1986. [Google Scholar]
- Gitterman, M. Mean first passage time for anomalous diffusion. Phys. Rev. E 2000, 62, 6065–6070. [Google Scholar] [CrossRef]
- Richardson, L.F. Atmospheric diffusion shown on a distance-neighbour graph. Proc. R. Soc. London 1926, 110, 709–737. [Google Scholar] [CrossRef]
- Wang, B.S.; Teoh, S.-H.; Kang, L. Polycaprolactone scaffold as targeted drug delivery system and cell attachment scaffold for postsurgical care of limb salvage. Drug Deliv. Transl. Res. 2012, 2, 272–283. [Google Scholar] [CrossRef]
- Pollack, G.H. Cells, Gels and the Engines of Life: A New Unifying Approach to Cell Function; Ebner & Sons: Seattle, Washington, USA, 2001. [Google Scholar]
- MGeoch, J.E.M.; McGeoch, M.W. Entrapment of water by subunit c of ATP synthase. J. Royal Soc. Interface 2008, 5, 311–318. [Google Scholar] [CrossRef]
- Henniker, J.C. The depth of the surface zone of a liquid. Rev. Mod. Phys. 1949, 21, 322–341. [Google Scholar] [CrossRef]
- Zheng, J.-M.; Chin, W.-C.; Khijniak, E.; Pollack, G.H. Surfaces and interfacial water: Evidence that hydrophilic surfaces have long-range impact. Adv. Colloid Interface Sci. 2006, 127, 19–27. [Google Scholar] [CrossRef]
- Gray-Weale, A.; Beattie, J.K. An explanation for the charge on water’s surface. Phys. Chem. Chem. Phys. 2009, 11, 10994–11005. [Google Scholar] [CrossRef]
- Ishikawa, T.; Matsuda, M.; Yasukawa, A.; Kandori, K.; Inagaki, S.; Fukushima, T.; Kondo, S. Surface silanol groups of mesoporous silica FSM-16. J. Chem. Soc. Faraday Trans. 1996, 92, 1985–1989. [Google Scholar] [CrossRef]
- Duval, Y.; Mielczarski, J.A.; Pokrovsky, O.S.; Mielczarski, E.; Ehrhardt, J.J. Evidence of the existence of three types of species at the quartz-aqueous solution interface at pH 0–10: XPS surface group quantification and surface complexation modeling. J. Phys. Chem.B 2002, 106, 2937–2945. [Google Scholar] [CrossRef]
- Ostroverkhov, V.; Waychunas, G.A.; Shen, Y.R. New information on water interfacial structure revealed by phase-sensitive surface spectroscopy. Phys. Rev. Lett. 2005, 94, 046102. [Google Scholar] [CrossRef]
- Kasuya, M.; Hino, M.; Yamada, H.; Mizukami, M.; Mori, H.; Kajita, S.; Ohmori, T.; Suzuki, A.; Kurihara, K. Characterisation of water confined between silica surfaces using the resonance shear method. J. Phys. Chem. C 2013, 117, 13540–13546. [Google Scholar] [CrossRef]
- Zheng, J.-M.; Pollack, G.H. Long range forces extending from polymer-gel surfaces. Phys. Rev. E 2003, 68, 031408. [Google Scholar] [CrossRef]
- Tada, T.; Kanekoa, D.; Gong, J.-P.; Kanekoa, T.; Osada, Y. Surface friction of poly(dimethyl siloxane) gel and its transition phenomena. Tribol. Lett. 2004, 17, 505–511. [Google Scholar] [CrossRef]
- Martelli, C.; Canning, J.; Kristensen, M.; Groothoff, N. Impact of water and ice 1 h formation in a photonic crystal fibre grating. Opt. Lett. 2006, 31, 706–708. [Google Scholar] [CrossRef]
- Derjaguin, B.V.; Churaev, N.V.; Kiseleva, A.; Sobolev, V.D. Evaluation of the thickness of non-freezing water films from the measurement of thermocrystallization and thermocapillary flows. Langmuir 1987, 3, 631–634. [Google Scholar] [CrossRef]
- Nogami, M.; Abe, Y. Evidence of water-cooperative proton conduction in silica glasses. Phys. Rev. B 1997, 55, 12108–12112. [Google Scholar] [CrossRef]
- Uma, T.; Izuhura, S.; Nogami, M. Structural and proton conductivity study of P2O5-TiO2-SiO2 glasses. J. Eur. Ceram. Soc. 2006, 26, 2365–2372. [Google Scholar] [CrossRef]
- Colomer, M.T.; Rubio, F.; Jurado, J.R. Transport properties of fast proton conducting mesoporous silica xerogels. J. Power Sources 2007, 167, 53–57. [Google Scholar] [CrossRef]
- Yang, Y.; Lu, F.; Kostiuk, L.W.; Kwok, D.Y. Electrokinetic microchannel battery by means of electrokinetic and microfluidic phenomena. J. Micromech. Microeng. 2013, 13, 963–970. [Google Scholar]
- Canning, J.; Buckley, E.; Huntington, S.; Lyytikäinen, K. Using multi-microchannel capillaries for determination of the zeta potential of a microfluidic channel. Electrochim.Acta 2004, 49, 3581–3586. [Google Scholar] [CrossRef]
- Huyang, G.; Canning, J.; Gibson, B.; Khoury, T.; Neto, C.; Crossley, M.J. Focused ion beam processing and engineering of devices in self-assembled supramolecular structures. Nanotechnology 2009, 20. [Google Scholar] [CrossRef]
- Martelli, C.; Canning, J.; Reimers, J.R.; Sintic, M.; Stocks, D.; Khoury, T.; Crossley, M.J. Evanescent-field spectroscopy using structured optical fibers: Detection of charge-transfer at the porphyrin-silica interface. J. Am. Chem. Soc. 2009, 131, 2925–2933. [Google Scholar]
- Lu, S.; Song, Z.; He, J. Diffusion-controlled protein adsorption in mesoporous silica. J. Phys. Chem. B 2011, 115, 7744–7750. [Google Scholar]
- Cavallaro, G.; Pierro, P.; Palumbo, F.S.; Tesat, F.; Pasqua, L.; Aiello, R. Drug delivery devices based on mesoporous silicate. Drug Deliv. 2004, 11, 41–46. [Google Scholar] [CrossRef]
- Slowing, I.I.; Vivero-Escoto, J.L.; Wu, C.-W.; Lin, V.S.-Y. Mesoporous silica nanoparticles as controlled release drug delivery and gene transfection carriers. Adv. Drug Deliv.Rev. 2008, 60, 1278–1288. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Canning, J.; Huyang, G.; Ma, M.; Beavis, A.; Bishop, D.; Cook, K.; McDonagh, A.; Shi, D.; Peng, G.-D.; Crossley, M.J. Percolation Diffusion into Self-Assembled Mesoporous Silica Microfibres. Nanomaterials 2014, 4, 157-174. https://doi.org/10.3390/nano4010157
Canning J, Huyang G, Ma M, Beavis A, Bishop D, Cook K, McDonagh A, Shi D, Peng G-D, Crossley MJ. Percolation Diffusion into Self-Assembled Mesoporous Silica Microfibres. Nanomaterials. 2014; 4(1):157-174. https://doi.org/10.3390/nano4010157
Chicago/Turabian StyleCanning, John, George Huyang, Miles Ma, Alison Beavis, David Bishop, Kevin Cook, Andrew McDonagh, Dongqi Shi, Gang-Ding Peng, and Maxwell J. Crossley. 2014. "Percolation Diffusion into Self-Assembled Mesoporous Silica Microfibres" Nanomaterials 4, no. 1: 157-174. https://doi.org/10.3390/nano4010157