A CRISPR/Cas12a-Based System for Sensitive Detection of Antimicrobial-Resistant Genes in Carbapenem-Resistant Enterobacterales


Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Bacterial Culturing and Genomic-DNA Extraction
2.3. Preparation of gRNA
2.4. Antimicrobial Gene (blaNDM-targeted) Preparation
2.5. CRISPR/Cas12a Fluorescence Assay
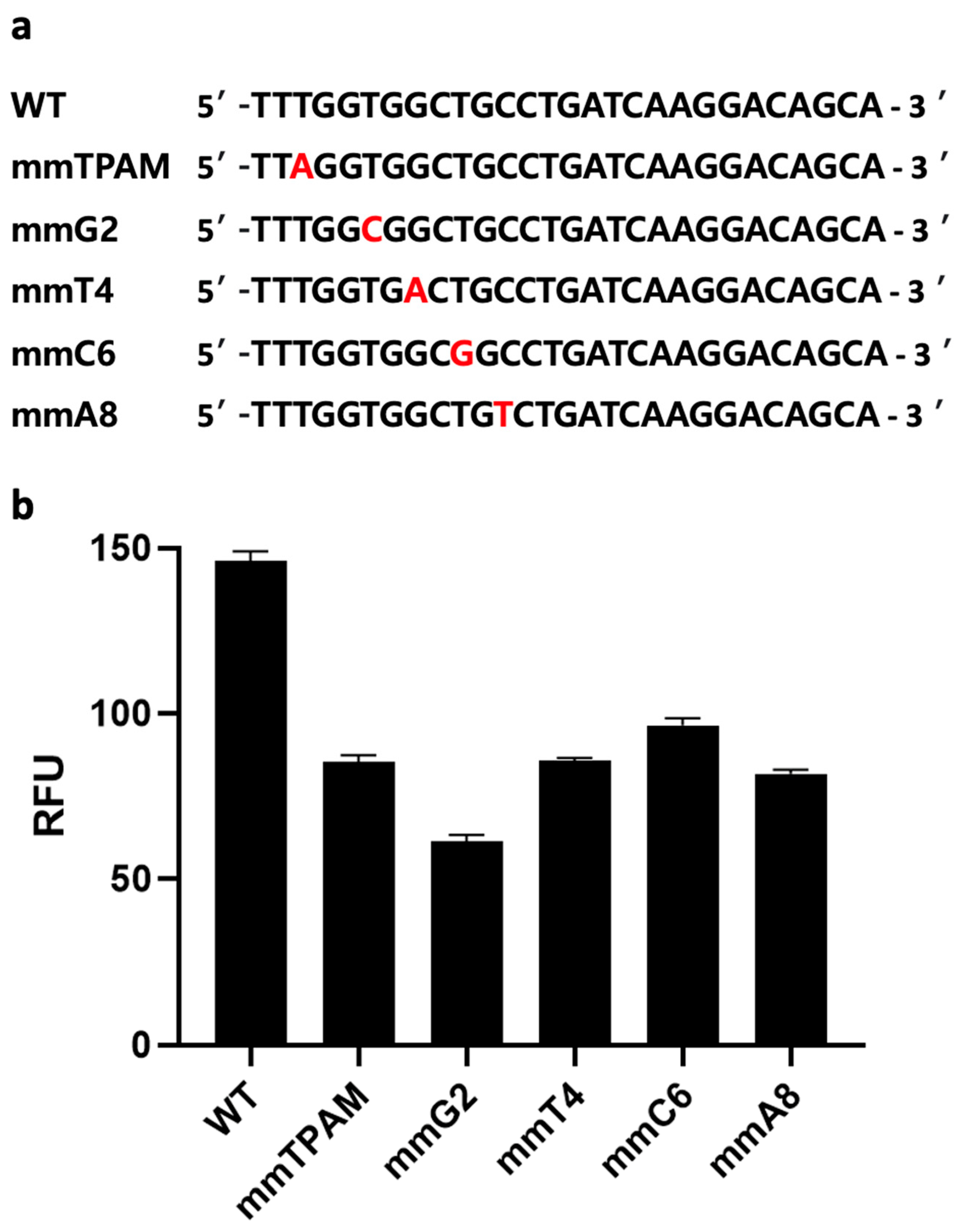
2.6. Mismatch Analysis
2.7. Detection of the Target AMR Gene in Real Food Samples
2.8. Data Collection and Processing
3. Results and Discussion
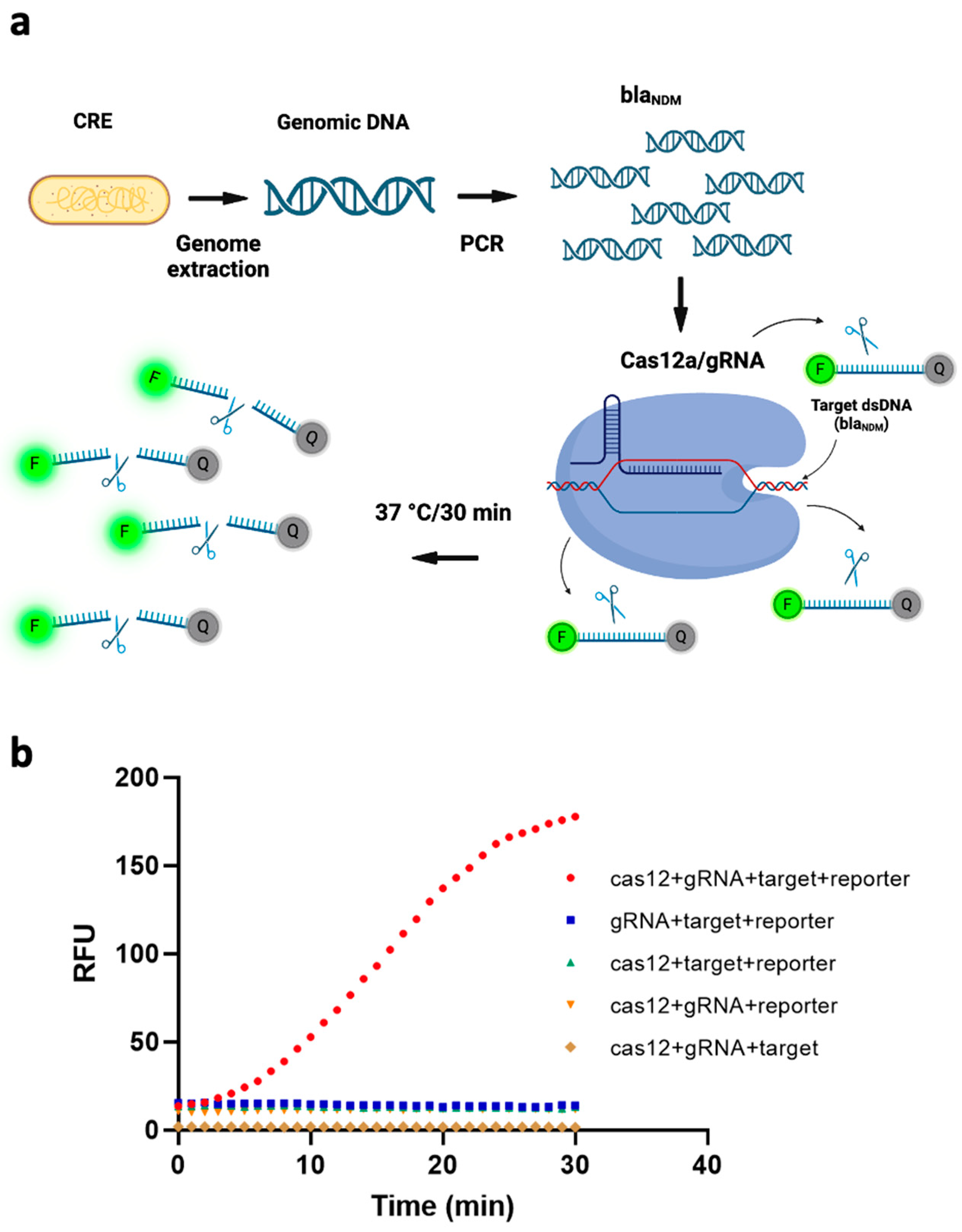
3.1. Design of CRISPR/Cas12a Fluorescence Assays
3.2. Assay Optimization
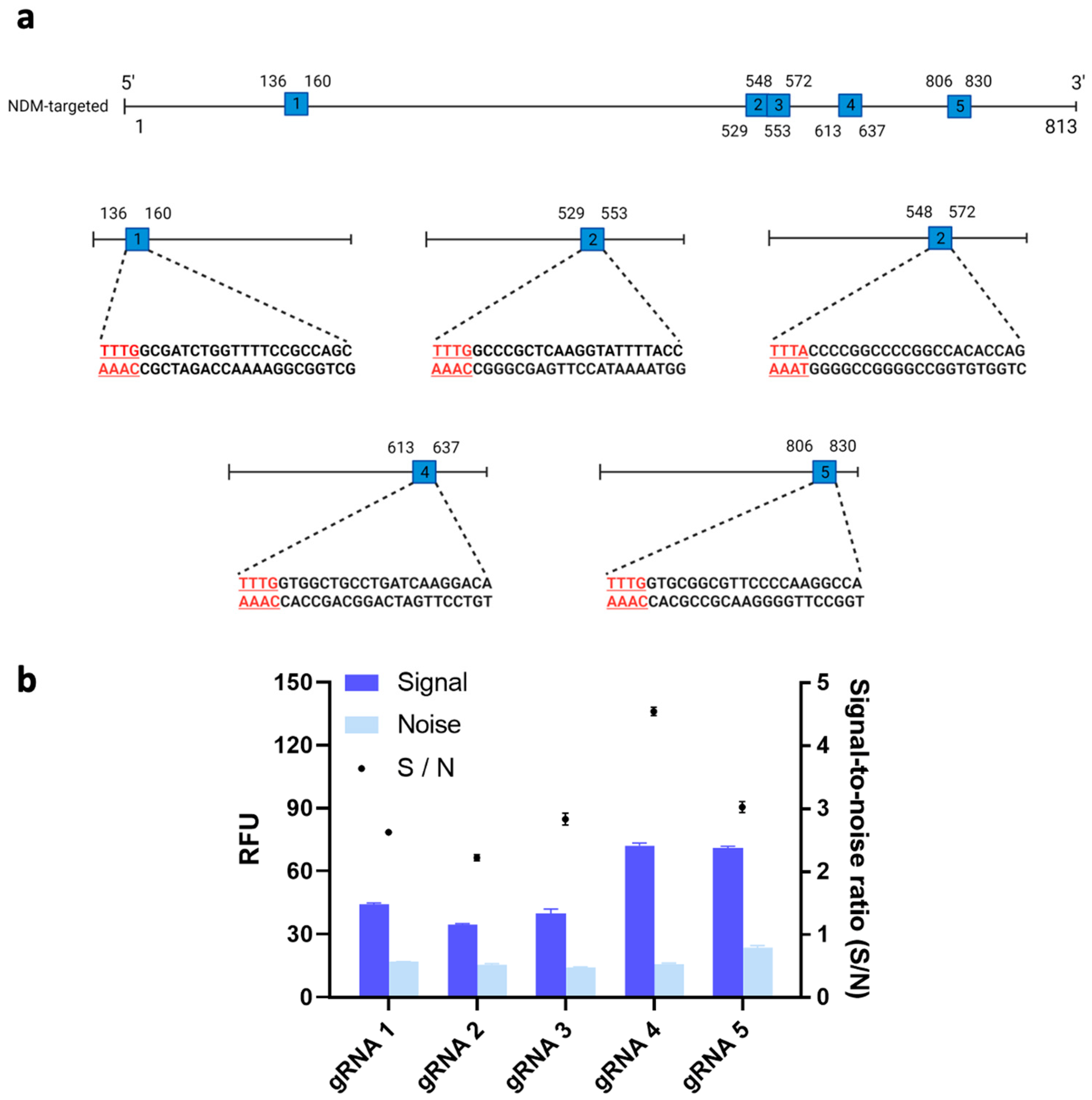
3.2.1. gRNA Selection
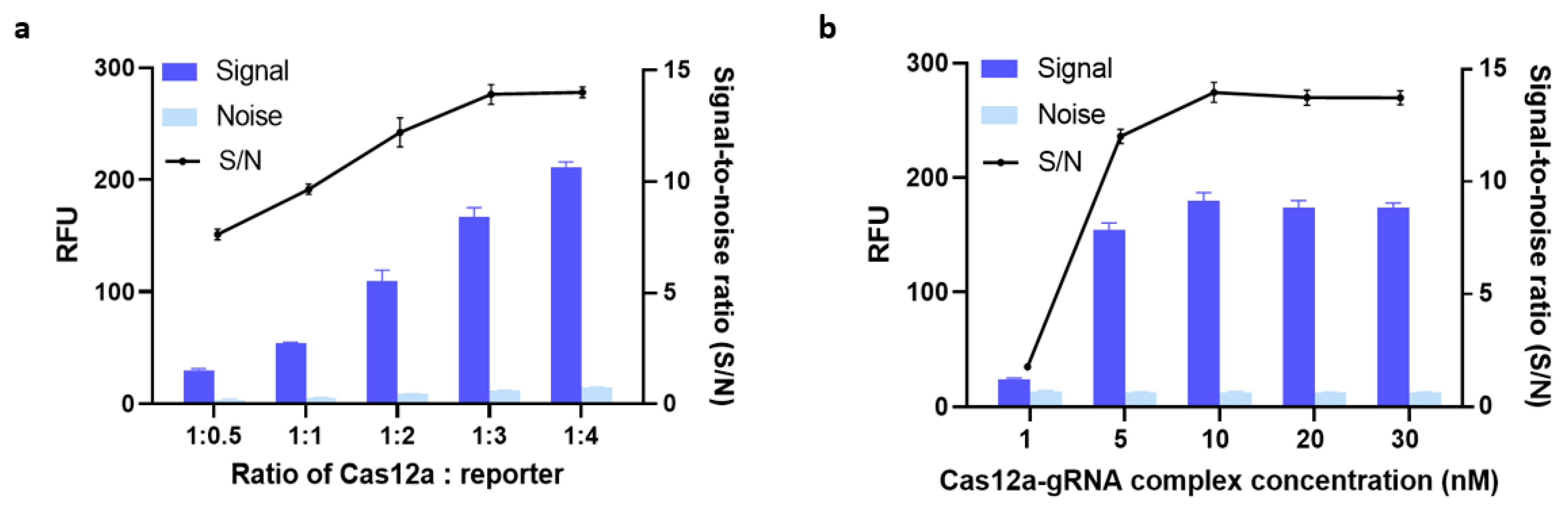
3.2.2. Optimization of the Amounts of Cas12a, gRNA, and ssDNA FQ-Reporter
3.3. Performance Analysis of the CRISPR/Cas12a Fluorescence Assay
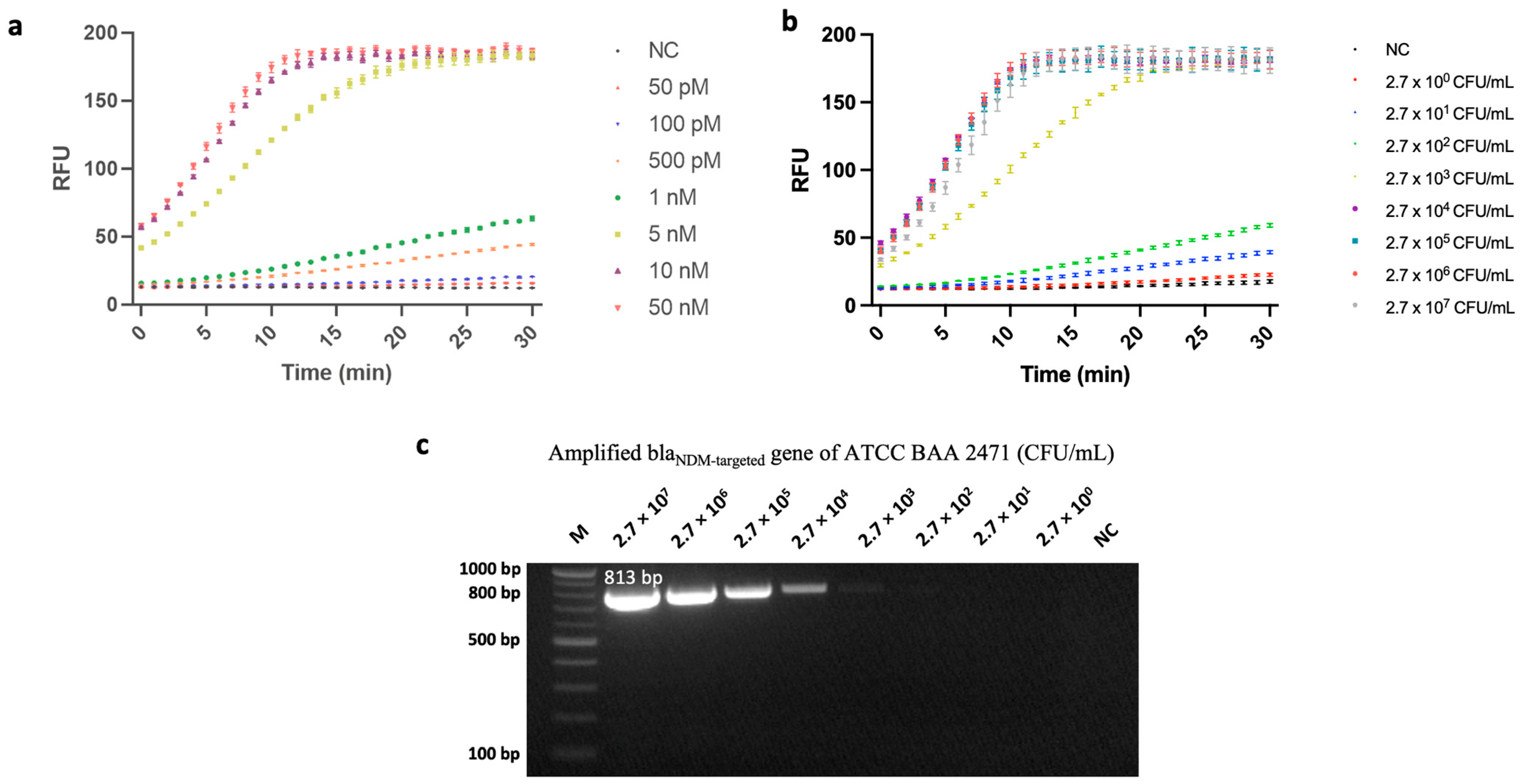
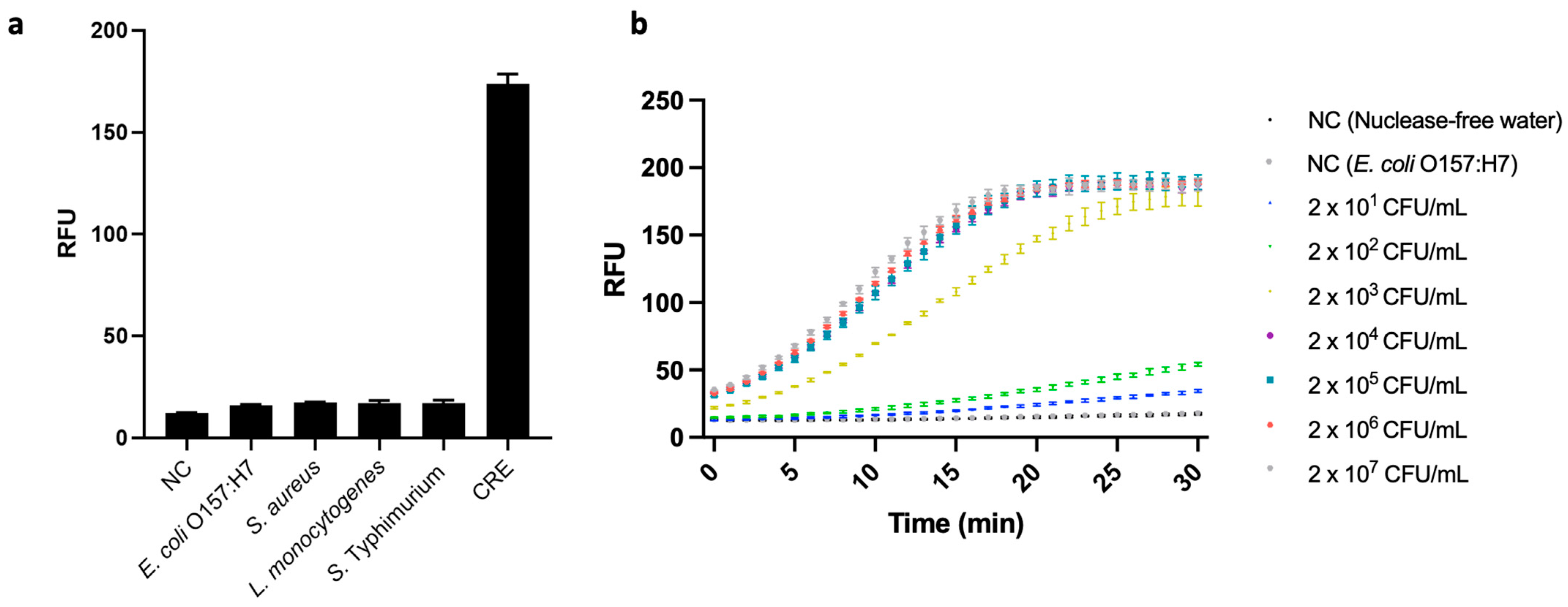
3.3.1. Sensitivity
3.3.2. Specificity of the CRISPR/Cas12a Fluorescence Assay
3.3.3. Detecting blaNDM in a Sample of Real Food
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dadgostar, P. Antimicrobial resistance: Implications and costs. Infect. Drug Resist. 2019, 12, 3903–3910. [Google Scholar] [CrossRef] [PubMed]
- CDC. About Antibiotic Resistance|CDC. 5 October 2022. Available online: https://www.cdc.gov/drugresistance/about.html/ (accessed on 27 December 2022).
- World Health Organization (WHO). Top Ten Threats to Global Health in 2021. 2021. Available online: https://www.who.int/news-room/spotlight/10-global-health-issues-to-track-in-2021/ (accessed on 25 October 2022).
- Codjoe, F.S.; Donkor, E.S. Carbapenem Resistance: A Review. Med. Sci. 2017, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Papp-Wallace, K.M.; Endimiani, A.; Taracila, M.A.; Bonomo, R.A. Carbapenems: Past, present, and future. Antimicrob. Agents Chemother. 2011, 55, 4943–4960. [Google Scholar] [CrossRef] [PubMed]
- CDC. Antibiotic Resistance Threats in the United States; CDC: Atlanta, GA, USA, 2019. [CrossRef]
- Wang, Y.; Wu, C.; Zhang, Q.; Qi, J.; Liu, H.; Wang, Y.; He, T.; Ma, L.; Lai, J.; Shen, Z.; et al. Identification of New Delhi metallo-β-lactamase 1 in Acinetobacter lwoffii of food animal origin. PLoS ONE 2012, 7, e37152. [Google Scholar] [CrossRef] [PubMed]
- Köck, R.; Daniels-Haardt, I.; Becker, K.; Mellmann, A.; Friedrich, A.W.; Mevius, D.; Schwarz, S.; Jurke, A. Carbapenem-resistant Enterobacteriaceae in wildlife, food-producing, and companion animals: A systematic review. Clin. Microbiol. Infect. 2018, 24, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Kluytmans, J. Ban resistant strains from food chain. Nature 2013, 501, 316. [Google Scholar] [CrossRef] [PubMed]
- Schrijver, R.; Stijntjes, M.; Rodríguez-Baño, J.; Tacconelli, E.; Babu Rajendran, N.; Voss, A. Review of antimicrobial resistance surveillance programmes in livestock and meat in EU with focus on humans. Clin. Microbiol. Infect. 2018, 24, 577–590. [Google Scholar] [CrossRef]
- Rabaan, A.A.; Eljaaly, K.; Alhumaid, S.; Albayat, H.; Al-Adsani, W.; Sabour, A.A.; Alshiekheid, M.A.; Al-Jishi, J.M.; Khamis, F.; Alwarthan, S.; et al. An Overview on Phenotypic and Genotypic Characterisation of Carbapenem-Resistant Enterobacterales. Medicina 2022, 58, 1675. [Google Scholar] [CrossRef]
- Nordmann, P.; Poirel, L.; Carrër, A.; Toleman, M.A.; Walsh, T.R. How to detect NDM-1 producers. J. Clin. Microbiol. 2011, 49, 718–721. [Google Scholar] [CrossRef]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Shin, J.; Miller, M.; Wang, Y.-C. Recent advances in CRISPR-based systems for the detection of foodborne pathogens. Compr. Rev. Food Sci. Food Saf. 2022, 21, 3010–3029. [Google Scholar] [CrossRef]
- Chen, J.S.; Ma, E.; Harrington, L.B.; da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Song, M.; Lee, J.; Menon, V.; Jung, S.; Kang, Y.-M.; Choi, J.W.; Woo, E.; Koh, H.C.; Nam, J.-W.; et al. In vivo high-throughput profiling of crisPr-cpf1 activity. Nat. Methods 2017, 14, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Tao, D.; Liu, J.; Nie, X.; Xu, B.; Tran-Thi, T.N.; Niu, L.; Liu, X.; Ruan, J.; Lan, X.; Peng, G.; et al. Application of CRISPR-Cas12a Enhanced Fluorescence Assay Coupled with Nucleic Acid Amplification for the Sensitive Detection of African Swine Fever Virus. ACS Synth. Biol. 2020, 9, 2339–2350. [Google Scholar] [CrossRef]
- Li, Y.; Shi, Z.; Hu, A.; Cui, J.; Yang, K.; Liu, Y.; Deng, G.; Zhu, C.; Zhu, L. Rapid one-tube RPA-CRISPR/Cas12 detection platform for methicillin-resistant Staphylococcus aureus. Diagnostics 2022, 12, 829. [Google Scholar] [CrossRef]
- Khan, A.U.; Maryam, L.; Zarrilli, R. Structure, Genetics and Worldwide Spread of New Delhi Metallo-β-lactamase (NDM): A threat to public health. BMC Microbiol. 2017, 17, 101. [Google Scholar] [CrossRef]
- Hong, J.; Woo, K.J.; Jun, M.K.; So, H.K.; Hye, C.K.; Ser, J.; Yong, H.P. Quantification and differentiation of Campylobacter jejuni and Campylobacter coli in raw chicken meats using a real-time PCR method. J. Food Prot. 2007, 70, 2015–2022. [Google Scholar] [CrossRef]
- Wang, L.; Mustapha, A. EMA-real-time PCR as a reliable method for detection of viable Salmonella in chicken and eggs. J. Food Sci. 2010, 75, M134–M139. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Dong, Y.; Shi, Y.; Yang, H.; Zhang, J.; Rizwan Khan, M.; Deng, S.; He, G.; He, Q.; Lv, Y.; et al. CRISPR-Cas12-Based Rapid Authentication of Halal Food. J. Agric. Food Chem. 2021, 69, 10321–10328. [Google Scholar] [CrossRef]
- Niu, C.; Wang, C.; Li, F.; Zheng, X.; Xing, X.; Zhang, C. Aptamer assisted CRISPR-Cas12a strategy for small molecule diagnostics. Biosens. Bioelectron. 2021, 183, 113196. [Google Scholar] [CrossRef]
- Jeon, Y.; Choi, Y.H.; Jang, Y.; Yu, J.; Goo, J.; Lee, G.; Jeong, Y.K.; Lee, S.H.; Kim, I.S.; Kim, J.S.; et al. Direct observation of DNA target searching and cleavage by CRISPR-Cas12a. Nat. Commun. 2018, 9, 2777. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.M.; Nielsen, K.M. Mechanisms of, and Barriers to, Horizontal Gene Transfer between Bacteria. Nat. Rev. Microbiol. 2005, 3, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ye, Q.; Chen, M.; Xiang, X.; Zhang, J.; Pang, R.; Xue, L.; Wang, J.; Gu, Q.; Lei, T.; et al. Cas12aFDet: A CRISPR/Cas12a-based fluorescence platform for sensitive and specific detection of Listeria monocytogenes serotype 4c. Anal. Chim. Acta 2021, 1151, 338248. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, R.; Wang, D.; Wu, J.; Li, J.; Wang, J.; Liu, H.; Wang, Y. Cas12aVDet: A CRISPR/Cas12a-Based Platform for Rapid and Visual Nucleic Acid Detection. Anal. Chem. 2019, 91, 12156–12161. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, S.; Wu, H.; Cheng, P.; Wang, X.; Qian, S.; Zhang, M.; Xu, J.; Ji, F.; Wu, J. CRISPR/Cas12a-Based Versatile Method for Checking Quantitative Polymerase Chain Reaction Samples with Cycles of Threshold Values in the Gray Zone. ACS Sens. 2021, 6, 1963–1970. [Google Scholar] [CrossRef]
- Mikutis, G.; Schmid, L.; Stark, W.J.; Grass, R.N. Length-dependent DNA degradation kinetic model: Decay compensation in DNA tracer concentration measurements. AIChE J. 2019, 65, 40–48. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, C.; Shi, Y.; Wu, J.; Wu, J.; Chen, H. Selective endpoint visualized detection of Vibrio parahaemolyticus with CRISPR/Cas12a assisted PCR using thermal cycler for on-site application. Talanta 2020, 214, 120818. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Tang, H.; Li, R.; Xia, Z.; Yang, W.; Zhu, Y.; Liu, Z.; Lu, G.; Ni, S.; Shen, J. A new method based on LAMP-CRISPR–Cas12a-lateral flow immunochromatographic strip for detection. Infect. Drug Resist. 2022, 15, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef]
- Shi, J.; Yan, Y.; Links, M.G.; Li, L.; Dillon, J.-A.R.; Horsch, M.; Kusalik, A. Antimicrobial resistance genetic factor identification from whole-genome sequence data using deep feature selection. BMC Bioinform. 2019, 20, 535. [Google Scholar] [CrossRef]
- Liu, B.T.; Song, F.J.; Zou, M.; Zhang, Q.D.; Shan, H. High Incidence of Escherichia coli Strains Coharboring mcr-1 and blaNDM from Chickens. Antimicrob. Agents Chemother. 2017, 61, e02347-16. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Hu, F.; Wang, Y.; Yin, D.; Yang, L.; Chen, Y.; Xu, C.; Li, J.; Jiang, J.; Wang, X.; et al. Transmission of Carbapenem Resistance Between Human and Animal NDM-Positive Escherichia coli Strains. Engineering 2022, 15, 24–33. [Google Scholar] [CrossRef]
- Koutsoumanis, K.; Allende, A.; Álvarez-Ordóñez, A.; Bolton, D.; Bover-Cid, S.; Chemaly, M.; Davies, R.; de Cesare, A.; Herman, L.; Hilbert, F.; et al. Role played by the environment in the emergence and spread of antimicrobial resistance (AMR) through the food chain. EFSA J. 2021, 19, e06651. [Google Scholar] [CrossRef] [PubMed]
- Ong DC, T.; Koh, T.H.; Syahidah, N.; Krishnan, P.; Tan, T.Y. Rapid detection of the blaNDM-1 gene by real-time PCR. J. Antimicrob. Chemother. 2011, 66, 1647–1649. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Oligonucleotide Sequence |
|---|---|
| NDM-targeted FP | 5′ ATGGAATTGCCCAATATTATGCACCCGG 3′ |
| NDM-targeted RP | 5′ TCAGCGCAGCTTGTCGGCCATG 3′ |
| gRNA synthesis FP | 5′ GAAATTAATACGACTCACTATAGTAATTTCTACTAAGTGTAGAT 3′ |
| gRNA 1 synthesis RP | 5′ GCTGGCGGAAAACCAGATCGCATCTACACTTAGTAGAAATTA 3′ |
| gRNA 2 synthesis RP | 5′ GGTAAAATACCTTGAGCGGGCATCTACACTTAGTAGAAATTA 3′ |
| gRNA 3 synthesis RP | 5′ CTGGTGTGGCCGGGGCCGGGGATCTACACTTAGTAGAAATTA 3′ |
| gRNA 4 synthesis RP | 5′ TGTCCTTGATCAGGCAGCCACATCTACACTTAGTAGAAATTA 3′ |
| gRNA 5 synthesis RP | 5′ TGGCCTTGGGGAACGCCGCACATCTACACTTAGTAGAAATTA 3′ |
| gRNA 1 | 5′ UAAUUUCUACUAAGUGUAGAUGCGAUCUGGUUUUCCGCCAGC 3′ |
| gRNA 2 | 5′ UAAUUUCUACUAAGUGUAGAUGCCCGCUCAAGGUAUUUUACC 3′ |
| gRNA 3 | 5′ UAAUUUCUACUAAGUGUAGAUCCCCGGCCCCGGCCACACCAG 3′ |
| gRNA 4 | 5′ UAAUUUCUACUAAGUGUAGAUGUGGCUGCCUGAUCAAGGACA 3′ |
| gRNA 5 | 5′ UAAUUUCUACUAAGUGUAGAUGUGCGGCGUUCCCCAAGGCCA 3′ |
| ssDNA-FQ (HEX labeled) | 5′ HEX-TTTTTTTTTT-IABkFQ 3′ |
| WT (gRNA4) * | 5′ TTTGGTGGCTGCCTGATCAAGGACAGCA 3′ |
| mmTPAM * | 5′ TTAGGTGGCTGCCTGATCAAGGACAGCA 3′ |
| mmG2 * | 5′ TTTGGCGGCTGCCTGATCAAGGACAGCA 3′ |
| mmT4 * | 5′ TTTGGTGACTGCCTGATCAAGGACAGCA 3′ |
| mmC6 * | 5′ TTTGGTGGCGGCCTGATCAAGGACAGCA 3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, J.; Kim, S.R.; Xie, Z.; Jin, Y.-S.; Wang, Y.-C. A CRISPR/Cas12a-Based System for Sensitive Detection of Antimicrobial-Resistant Genes in Carbapenem-Resistant Enterobacterales. Biosensors 2024, 14, 194. https://doi.org/10.3390/bios14040194
Shin J, Kim SR, Xie Z, Jin Y-S, Wang Y-C. A CRISPR/Cas12a-Based System for Sensitive Detection of Antimicrobial-Resistant Genes in Carbapenem-Resistant Enterobacterales. Biosensors. 2024; 14(4):194. https://doi.org/10.3390/bios14040194
Chicago/Turabian StyleShin, Jiyong, Sei Rim Kim, Zifan Xie, Yong-Su Jin, and Yi-Cheng Wang. 2024. "A CRISPR/Cas12a-Based System for Sensitive Detection of Antimicrobial-Resistant Genes in Carbapenem-Resistant Enterobacterales" Biosensors 14, no. 4: 194. https://doi.org/10.3390/bios14040194