
A Cytochrome P450 3A4 Biosensor Based on Generation 4.0 PAMAM Dendrimers for the Detection of Caffeine

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Materials
2.2. Methods
2.2.1. Preparation of PAMAM G4.0-Au Nanocomposites
2.2.2. Layer-by-Layer Assembly of the Au/MPS/G4.0-Au/CYP3A4 Biosensor
2.2.3. Electrochemical Experiments using Au/MPS/PAMAM G4.0-Au/CYP3A4 Biosensor
3. Results and Discussion
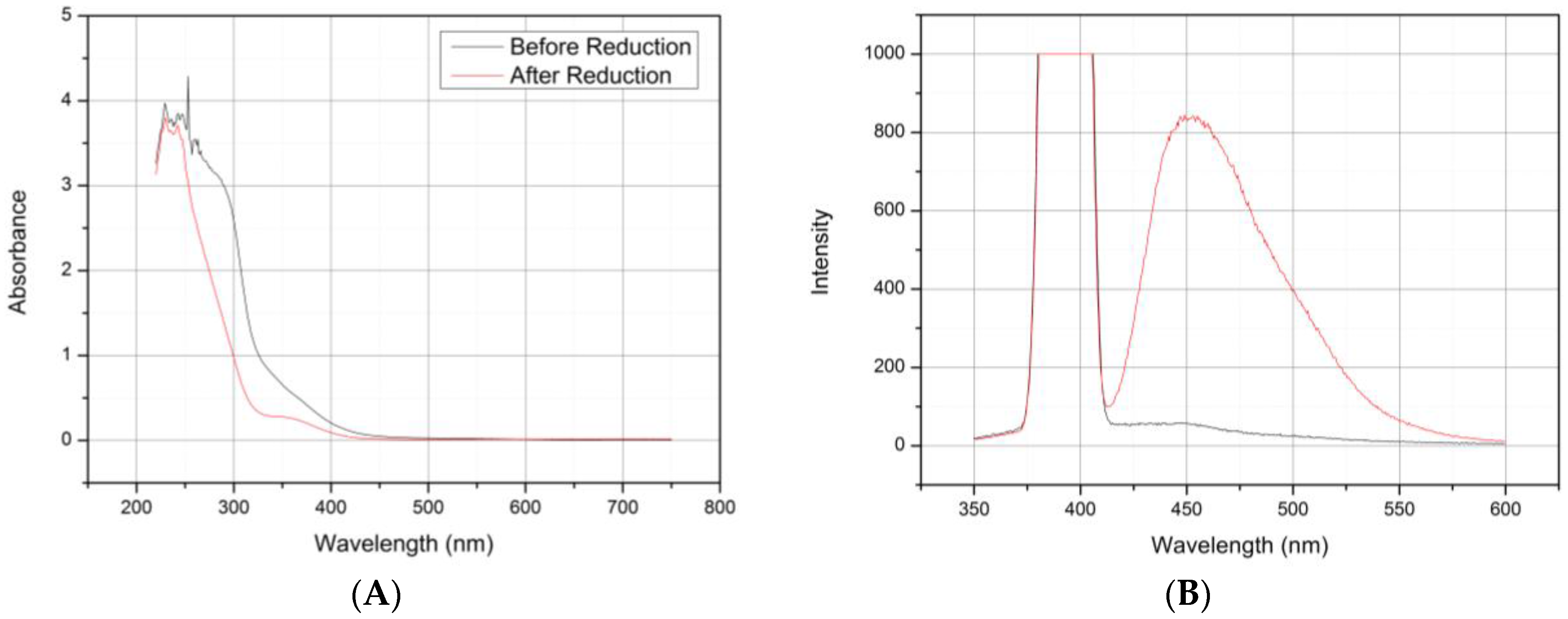
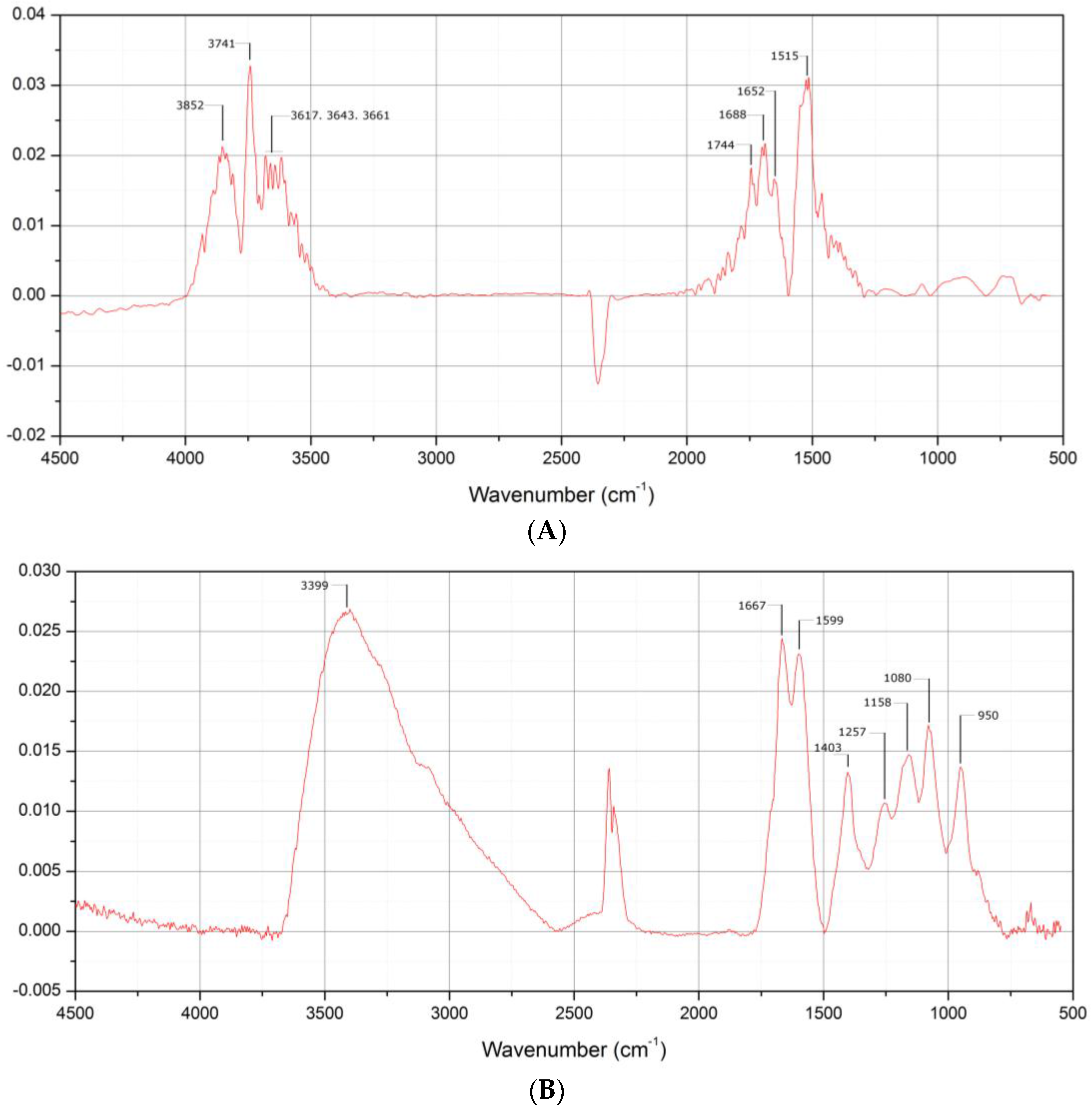
3.1. Preparation of PAMAM G4.0-Au Nanocomposites
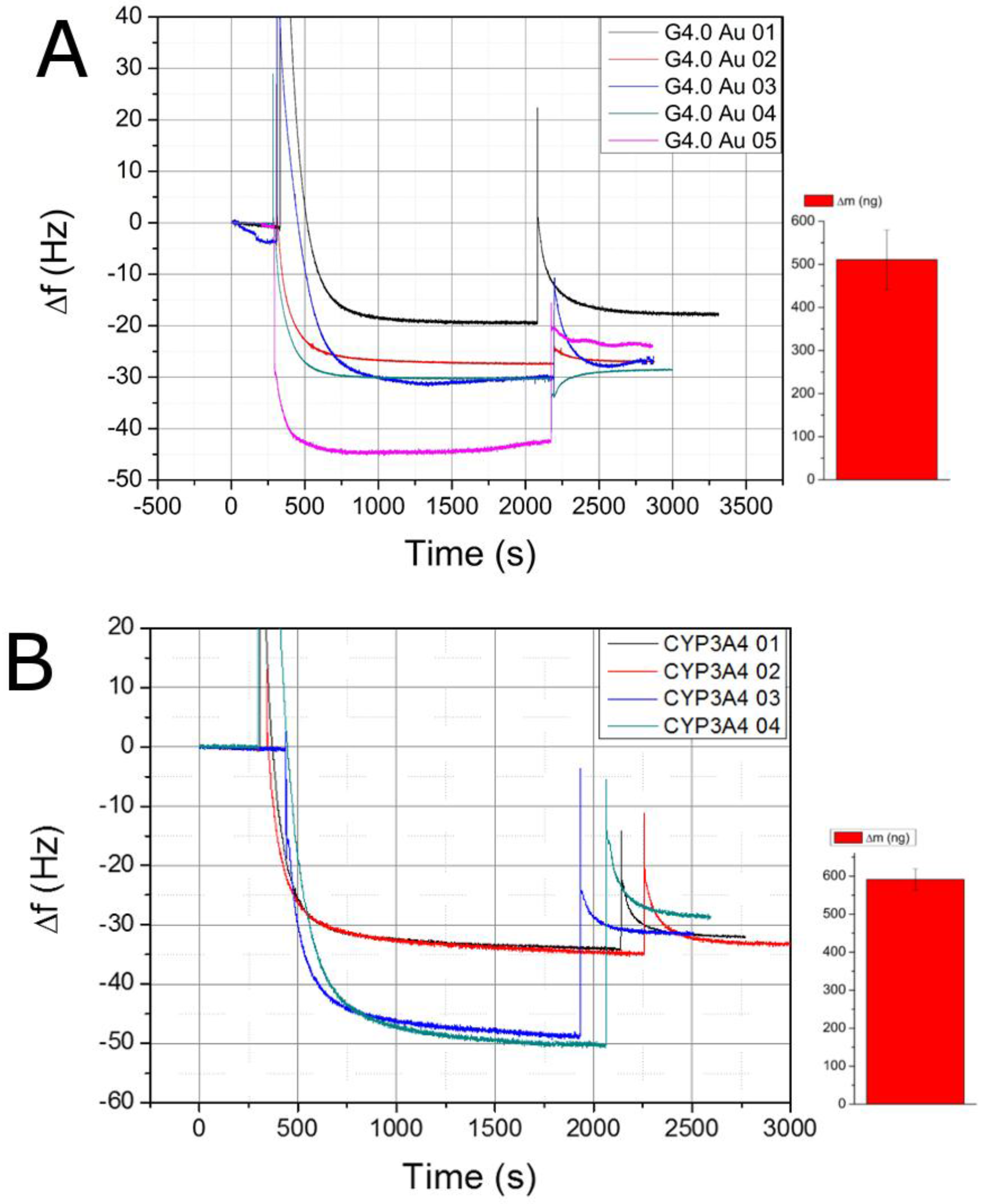
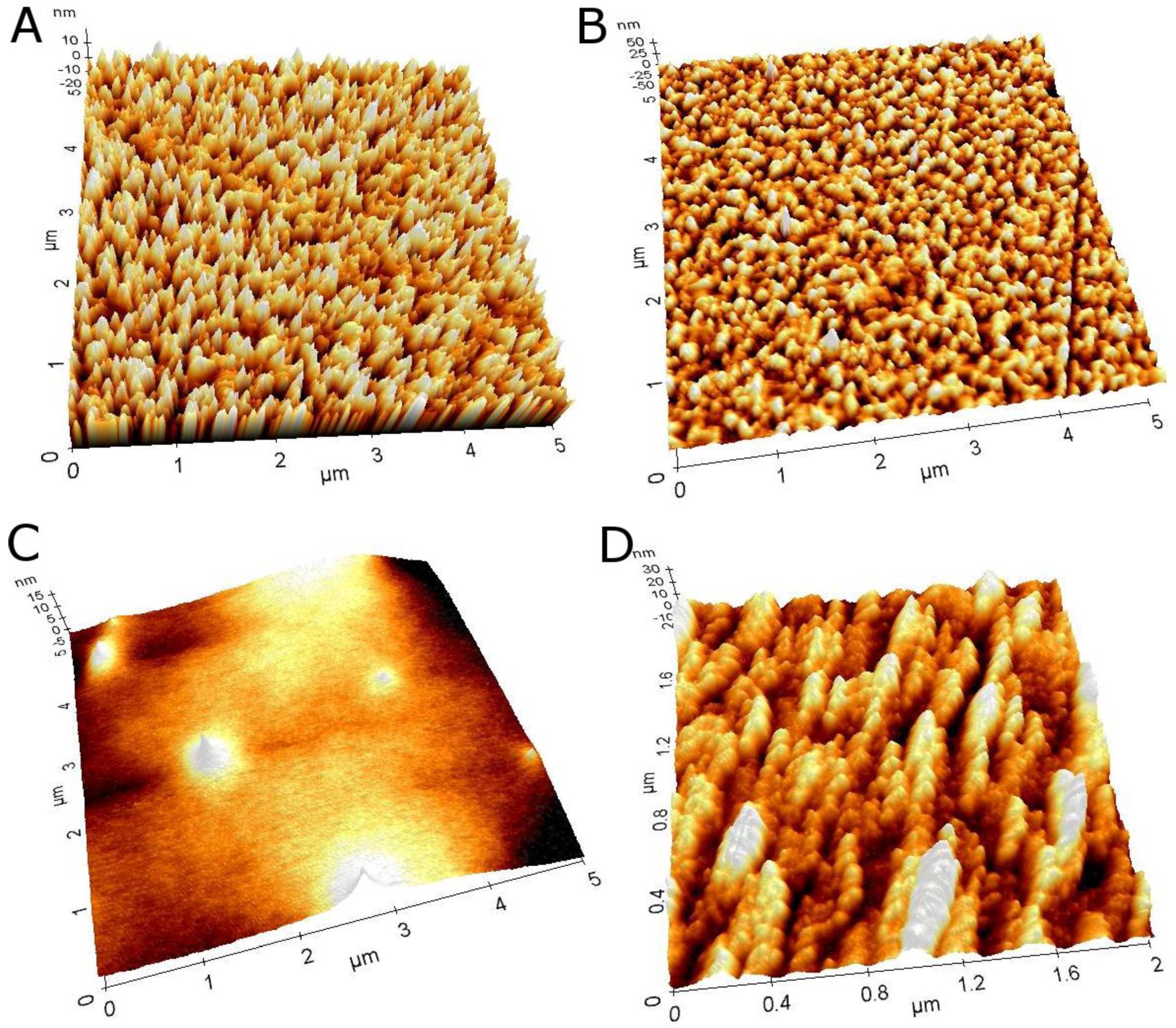
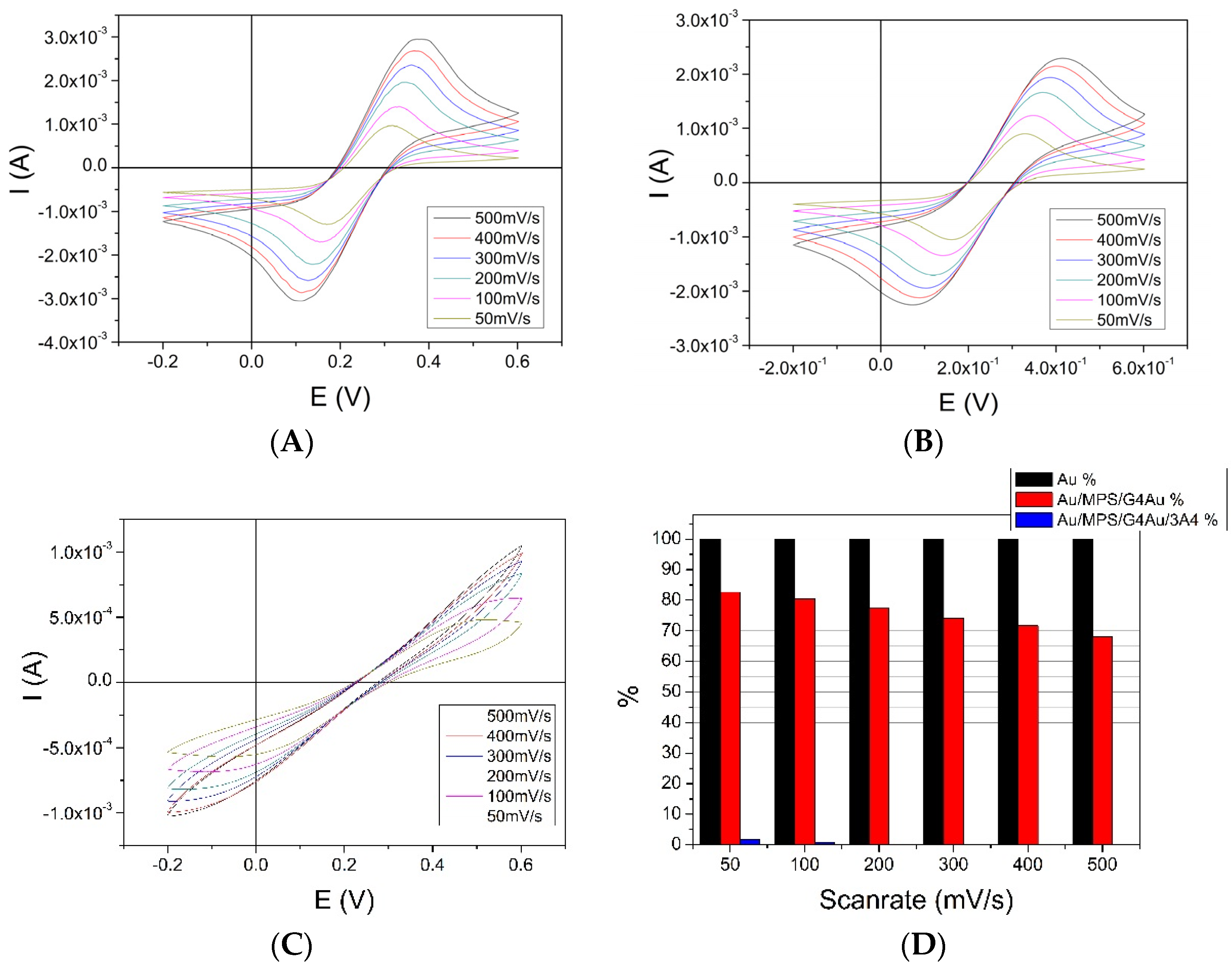
3.2. Layer-by-Layer Assembly of the Au/MPS/G4.0-Au/CYP3A4 Biosensor
3.3. Au/MPS/PAMAM G4.0-Au/CYP3A4 Electrochemistry Using Potassium-Ferricyanide
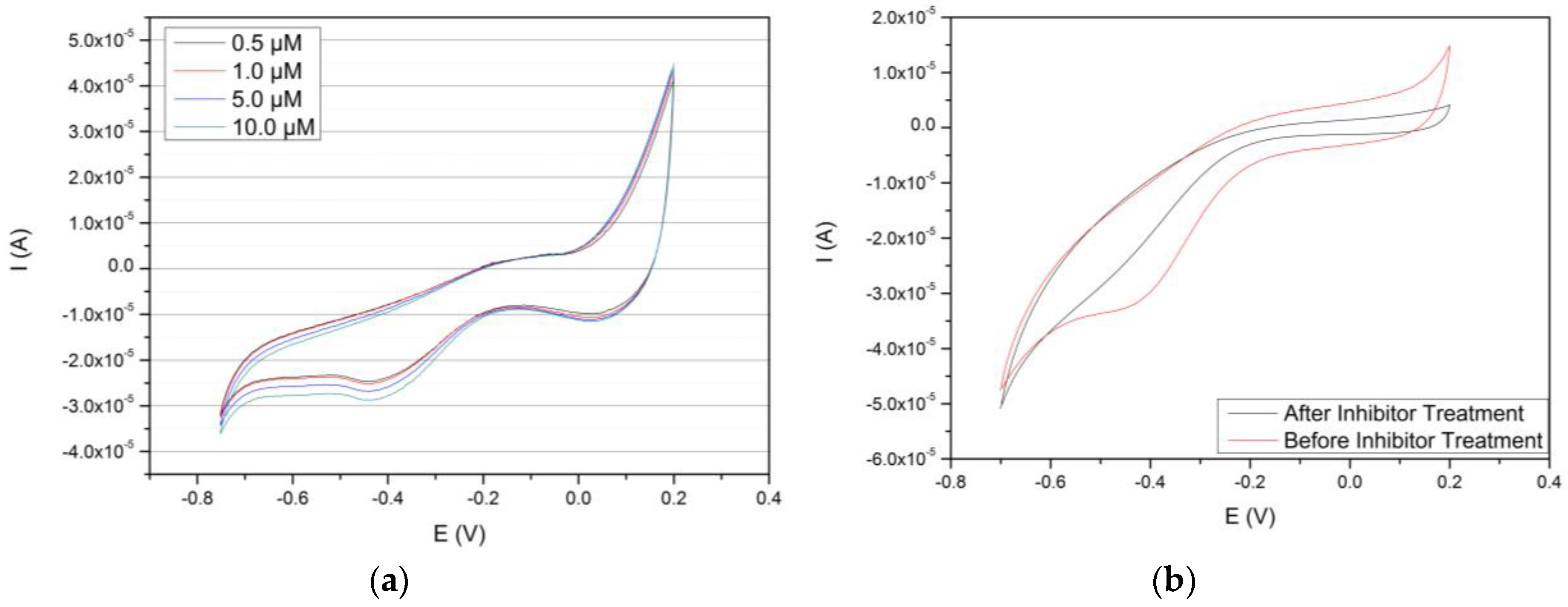
3.4. Concentration-Based Sensing of the Au/MPS/PAMAM G4.0-Au/CYP3A4 Biosensor of Caffeine/Cyclic Voltammetry
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Urlacher, V.B.; Lutz-Wahl, S.; Schmid, R.D. Microbial p450 enzymes in biotechnology. Appl. Microbiol. Biotechnol. 2004, 64, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Shafiee, A.; Hutchinson, C.R. Purification and reconstitution of the electron transport components for 6-deoxyerythronolide b hydroxylase, a cytochrome p-450 enzyme of macrolide antibiotic (erythromycin) biosynthesis. J. Bacteriol. 1988, 170, 1548–1553. [Google Scholar] [PubMed]
- Anzenbacher, P.; Anzenbacherová, E. Cytochromes p450 and metabolism of xenobiotics. CMLS Cell. Mol. Life Sci. 2001, 58, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Coon, M.J. Cytochrome p450: Nature’s most versatile biological catalyst. Ann. Rev. Pharmacol. Toxicol. 2005, 45, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Hannemann, F.; Bichet, A.; Ewen, K.M.; Bernhardt, R. Cytochrome p450 systems—Biological variations of electron transport chains. Biochim. Biophys. Acta 2007, 1770, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Makar, A.B.; McMartin, K.E.; Palese, M.; Tephly, T.R. Formate assay in body fluids: Application in methanol poisoning. Biochem. Med. 1975, 13, 117–126. [Google Scholar] [CrossRef]
- Whirl-Carrillo, M.; McDonagh, E.M.; Hebert, J.M.; Gong, L.; Sangkuhl, K.; Thorn, C.F.; Altman, R.B.; Klein, T.E. Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 2012, 92, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Boetsch, C.; Parrott, N.; Fowler, S.; Poirier, A.; Hainzl, D.; Banken, L.; Martin-Facklam, M.; Hofmann, C. Effects of cytochrome p450 3a4 inhibitors—Ketoconazole and erythromycin—On bitopertin pharmacokinetics and comparison with physiologically based modelling predictions. Clin. Pharmacokinet. 2016, 55, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Shang, W.; Liu, J.; Chen, R.; Ning, R.; Xiong, J.; Liu, W.; Mao, Z.; Hu, G.; Yang, J. Fluoxetine reduces ces1, ces2, and cyp3a4 expression through decreasing pxr and increasing dec1 in hepg2 cells. Xenobiotica 2016, 46, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Cytochrome p450 enzymes in the generation of commercial products. Nat. Rev. Drug Discov. 2002, 1, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Rendic, S.; Carlo, F.J.D. Human cytochrome p450 enzymes: A status report summarizing their reactions, substrates, inducers, and inhibitors. Drug Metab. Rev. 1997, 29, 413–580. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Iwatsubo, T.; Kanamitsu, S.; Nakajima, Y.; Sugiyama, Y. Quantitative prediction of in vivo drug clearance and drug interactions from in vitro data on metabolism, together with binding and transport. Ann. Rev. Pharmacol. Toxicol. 1998, 38, 461–499. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Dannert, C.; Umeno, D.; Arnold, F.H. Molecular breeding of carotenoid biosynthetic pathways. Nat. Biotech. 2000, 18, 750–753. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.R.; Reddy, Y.K.; Haines, D.C.; Reddy, K.M.; Krishna, U.M.; Graham, S.; Murry, B.; Peterson, J.A. Practical, enantiospecific syntheses of 14,15-eet and leukotoxin b (vernolic acid). Tetrahedron Lett. 2001, 42, 4131–4133. [Google Scholar] [CrossRef]
- Jennewein, S.; Rithner, C.D.; Williams, R.M.; Croteau, R.B. Taxol biosynthesis: Taxane 13α-hydroxylase is a cytochrome p450-dependent monooxygenase. Proc. Natl. Acad. Sci. USA 2001, 98, 13595–13600. [Google Scholar] [CrossRef] [PubMed]
- Chumsri, S.; Howes, T.; Bao, T.; Sabnis, G.; Brodie, A. Aromatase, aromatase inhibitors, and breast cancer. J. Steroid Biochem. Mol. Biol. 2011, 125, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Scheller, F.; Renneberg, R.; Strnad, G.; Pommerening, K.; Mohr, P. Electrochemical aspects of cytochrome p-450 system from liver microsomes. Bioelectrochem. Bioenerg. 1977, 4, 500–507. [Google Scholar] [CrossRef]
- Renneberg, R.; Scheller, F.; Ruckpaul, K.; Pirrwitz, J.; Mohr, P. Nadph and h2o2-dependent reactions of cytochrome p-450lm compared with peroxidase catalysis. FEBS Lett. 1978, 96, 349–353. [Google Scholar] [CrossRef]
- Sadeghi, S.J.; Fantuzzi, A.; Gilardi, G. Breakthrough in p450 bioelectrochemistry and future perspectives. Biochim. Biophys. Acta 2011, 1814, 237–248. [Google Scholar] [PubMed]
- Kazlauskaite, J.; Westlake, A.C.G.; Wong, L.-L.; Hill, H.A.O. Direct electrochemistry of cytochrome p450cam. Chem. Commun. 1996, 2189–2190. [Google Scholar] [CrossRef]
- Fantuzzi, A.; Fairhead, M.; Gilardi, G. Direct electrochemistry of immobilized human cytochrome p450 2e1. J. Am. Chem. Soc. 2004, 126, 5040–5041. [Google Scholar] [CrossRef] [PubMed]
- Putzbach, W.; Ronkainen, N. Immobilization techniques in the fabrication of nanomaterial-based electrochemical biosensors: A review. Sensors 2013, 13, 4811. [Google Scholar] [CrossRef] [PubMed]
- Holzinger, M.; Le Goff, A.; Cosnier, S. Nanomaterials for biosensing applications: A review. Front. Chem. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Gillam, E.M.; Mitchell, D.J.; Bernhardt, P.V. Direct electrochemistry of enzymes from the cytochrome p450 2c family. Electrochem. Commun. 2005, 7, 437–442. [Google Scholar] [CrossRef]
- Alonso-Lomillo, M.A.; Gonzalo-Ruiz, J.; Domínguez-Renedo, O.; Muñoz, F.J.; Arcos-Martínez, M.J. Cyp450 biosensors based on gold chips for antiepileptic drugs determination. Biosens. Bioelectron. 2008, 23, 1733–1737. [Google Scholar] [CrossRef] [PubMed]
- Udit, A.K.; Hagen, K.D.; Goldman, P.J.; Star, A.; Gillan, J.M.; Gray, H.B.; Hill, M.G. Spectroscopy and electrochemistry of cytochrome p450 bm3-surfactant film assemblies. J. Am. Chem. Soc. 2006, 128, 10320–10325. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Lomillo, M.A.; Yardimci, C.; Domínguez-Renedo, O.; Arcos-Martínez, M.J. Cyp450 2b4 covalently attached to carbon and gold screen printed electrodes by diazonium salt and thiols monolayers. Anal. Chim. Acta 2009, 633, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Bistolas, N.; Wollenberger, U.; Jung, C.; Scheller, F.W. Cytochrome p450 biosensors—A review. Biosens. Bioelectron. 2005, 20, 2408–2423. [Google Scholar] [CrossRef] [PubMed]
- Shumyantseva, V.V.; Bulko, T.V.; Archakov, A.I. Electrochemical reduction of cytochrome p450 as an approach to the construction of biosensors and bioreactors. J. Inorg. Biochem. 2005, 99, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Schenkman, J.B.; Rusling, J.F. Bioelectronic delivery of electrons to cytochrome p450 enzymes. J. Phys. Chem. B 2011, 115, 8371–8380. [Google Scholar] [CrossRef] [PubMed]
- Udit, A.K.; Hill, M.G.; Gray, H.B. Electrochemistry of cytochrome p450 bm3 in sodium dodecyl sulfate films. Langmuir 2006, 22, 10854–10857. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.; Rusling, J.F.; Lvov, Y.M.; Friedberg, T.; Fuhr, U. An amperometric biosensor with human cyp3a4 as a novel drug screening tool. Biochem. Pharmacol. 2003, 65, 1817–1826. [Google Scholar] [CrossRef]
- Shumyantseva, V.V.; Bulko, T.V.; Suprun, E.V.; Chalenko, Y.M.; Vagin, M.Y.; Rudakov, Y.O.; Shatskaya, M.A.; Archakov, A.I. Electrochemical investigations of cytochrome p450. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2011, 1814, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Shumyantseva, V.V.; Bulko, T.V.; Rudakov, Y.O.; Kuznetsova, G.P.; Samenkova, N.F.; Lisitsa, A.V.; Karuzina, I.I.; Archakov, A.I. Electrochemical properties of cytochroms p450 using nanostructured electrodes: Direct electron transfer and electro catalysis. J. Inorg. Biochem. 2007, 101, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Shumyantseva, V.V.; Makhova, A.A.; Bulko, T.V.; Kuzikov, A.V.; Shich, E.V.; Suprun, E.V.; Kukes, V.; Usanov, S.; Archakov, A. The dose-dependent influence of antioxidant vitamins on electrochemically-driven cytochrome p450 catalysis. Oxid. Antioxid Med. Sci. 2013, 2, 113–117. [Google Scholar] [CrossRef]
- Fantuzzi, A.; Capria, E.; Mak, L.H.; Dodhia, V.R.; Sadeghi, S.J.; Collins, S.; Somers, G.; Huq, E.; Gilardi, G. An electrochemical microfluidic platform for human p450 drug metabolism profiling. Anal. Chem. 2010, 82, 10222–10227. [Google Scholar] [CrossRef] [PubMed]
- Tully, D.C.; Frechet, J.M.J. Dendrimers at surfaces and interfaces: Chemistry and applications. Chem. Commun. 2001, 1229–1239. [Google Scholar] [CrossRef]
- Boas, U.; Heegaard, P.M.H. Dendrimers in drug research. Chem. Soc. Rev. 2004, 33, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Bosman, A.W.; Janssen, H.M.; Meijer, E.W. About dendrimers: Structure, physical properties, and applications. Chem. Rev. 1999, 99, 1665–1688. [Google Scholar] [CrossRef] [PubMed]
- Esfand, R.; Tomalia, D.A. Poly(amidoamine) (pamam) dendrimers: From biomimicry to drug delivery and biomedical applications. Drug Discov. Today 2001, 6, 427–436. [Google Scholar] [CrossRef]
- Dufès, C.; Uchegbu, I.F.; Schätzlein, A.G. Dendrimers in gene delivery. Adv. Drug Deliv. Rev. 2005, 57, 2177–2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patri, A.K.; Majoros, I.J.; Baker, J.R., Jr. Dendritic polymer macromolecular carriers for drug delivery. Curr. Opin. Chem. Biol. 2002, 6, 466–471. [Google Scholar] [CrossRef]
- Bosnjakovic, A.; Mishra, M.K.; Han, H.J.; Romero, R.; Kannan, R.M. A dendrimer-based immunosensor for improved capture and detection of tumor necrosis factor-α cytokine. Anal. Chim. Acta 2012, 720, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Vieira, N.C.S.; Figueiredo, A.; de Queiroz, A.A.A.; Zucolotto, V.; Guimarães, F.E.G. Self-assembled films of dendrimers and metallophthalocyanines as fet-based glucose biosensors. Sensors 2011, 11, 9442–9449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sezgintürk, M.K.; Uygun, Z.O. An impedimetric vascular endothelial growth factor biosensor-based pamam/cysteamine-modified gold electrode for monitoring of tumor growth. Anal. Biochem. 2012, 423, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Sutriyo; Mutalib, A.; Ristaniah; Anwar, E.; Radji, M.; Pujiyanto, A.; Purnamasari, P.; Joshita, D.; Adang, H.G. Synthesis of gold nanoparticles with polyamidoamine (pamam) generation 4 dendrimer as stabilizing agent for ct scan contrast agent. Macromol. Symp. 2015, 353, 96–101. [Google Scholar] [CrossRef]
- Yoon, H.C.; Kim, H.-S. Multilayered assembly of dendrimers with enzymes on gold: Thickness-controlled biosensing interface. Anal. Chem. 2000, 72, 922–926. [Google Scholar] [CrossRef] [PubMed]
- Schilder, J.; Ubbink, M. Formation of transient protein complexes. Curr. Opin. Struct. Biol. 2013, 23, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Hasemann, C.A.; Kurumbail, R.G.; Boddupalli, S.S.; Peterson, J.A.; Deisenhofer, J. Structure and function of cytochromes p450: A comparative analysis of three crystal structures. Structure 1995, 3, 41–62. [Google Scholar] [CrossRef]
- Liu, S.; Leech, D.; Ju, H. Application of colloidal gold in protein immobilization, electron transfer, and biosensing. Anal. Lett. 2003, 36, 1–19. [Google Scholar] [CrossRef]
- Kot, M.; Daniel, W.A. The relative contribution of human cytochrome p450 isoforms to the four caffeine oxidation pathways: An in vitro comparative study with cdna-expressed p450s including cyp2c isoforms. Biochem. Pharmacol. 2008, 76, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Kot, M.; Daniel, W.A. Effect of diethyldithiocarbamate (ddc) and ticlopidine on cyp1a2 activity and caffeine metabolism: An in vitro comparative study with human cdna-expressed cyp1a2 and liver microsomes. Pharmacol. Rep. 2009, 61, 1216–1220. [Google Scholar] [CrossRef]
- Turkevich, J.; Stevenson, P.C.; Hillier, J. A study of the nucleation and growth processes in the synthesis of colloidal gold. Discuss. Faraday Soc. 1951, 11, 55–75. [Google Scholar] [CrossRef]
- Frens, G. Controlled nucleation for the regulation of the particle size in monodisperse gold suspensions. Nat. Phys. Sci. 1973, 241, 20–22. [Google Scholar] [CrossRef]
- van Duijvenbode, R.C.; Borkovec, M.; Koper, G.J.M. Acid-base properties of poly(propylene imine)dendrimers. Polymer 1998, 39, 2657–2664. [Google Scholar] [CrossRef]
- Zheng, J.; Petty, J.T.; Dickson, R.M. High quantum yield blue emission from water-soluble au8 nanodots. J. Am. Chem. Soc. 2003, 125, 7780–7781. [Google Scholar] [CrossRef] [PubMed]
- Gilardi, G.; Meharenna, Y.T.; Tsotsou, G.E.; Sadeghi, S.J.; Fairhead, M.; Giannini, S. Molecular lego: Design of molecular assemblies of p450 enzymes for nanobiotechnology. Biosens. Bioelectron. 2002, 17, 133–145. [Google Scholar] [CrossRef]
- Ferrero, V.E.V.; Andolfi, L.; Di Nardo, G.; Sadeghi, S.J.; Fantuzzi, A.; Cannistraro, S.; Gilardi, G. Protein and electrode engineering for the covalent immobilization of p450 bmp on gold. Anal. Chem. 2008, 80, 8438–8446. [Google Scholar] [CrossRef] [PubMed]
- Yarman, A.; Wollenberger, U.; Scheller, F.W. Sensors based on cytochrome p450 and cyp mimicking systems. Electrochimica Acta 2013, 110, 63–72. [Google Scholar] [CrossRef]






© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, M.; Agarwal, N.; Kim, J. A Cytochrome P450 3A4 Biosensor Based on Generation 4.0 PAMAM Dendrimers for the Detection of Caffeine. Biosensors 2016, 6, 44. https://doi.org/10.3390/bios6030044
Müller M, Agarwal N, Kim J. A Cytochrome P450 3A4 Biosensor Based on Generation 4.0 PAMAM Dendrimers for the Detection of Caffeine. Biosensors. 2016; 6(3):44. https://doi.org/10.3390/bios6030044
Chicago/Turabian StyleMüller, Michael, Neha Agarwal, and Jungtae Kim. 2016. "A Cytochrome P450 3A4 Biosensor Based on Generation 4.0 PAMAM Dendrimers for the Detection of Caffeine" Biosensors 6, no. 3: 44. https://doi.org/10.3390/bios6030044
APA StyleMüller, M., Agarwal, N., & Kim, J. (2016). A Cytochrome P450 3A4 Biosensor Based on Generation 4.0 PAMAM Dendrimers for the Detection of Caffeine. Biosensors, 6(3), 44. https://doi.org/10.3390/bios6030044