
A Urea Potentiometric Biosensor Based on a Thiophene Copolymer


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
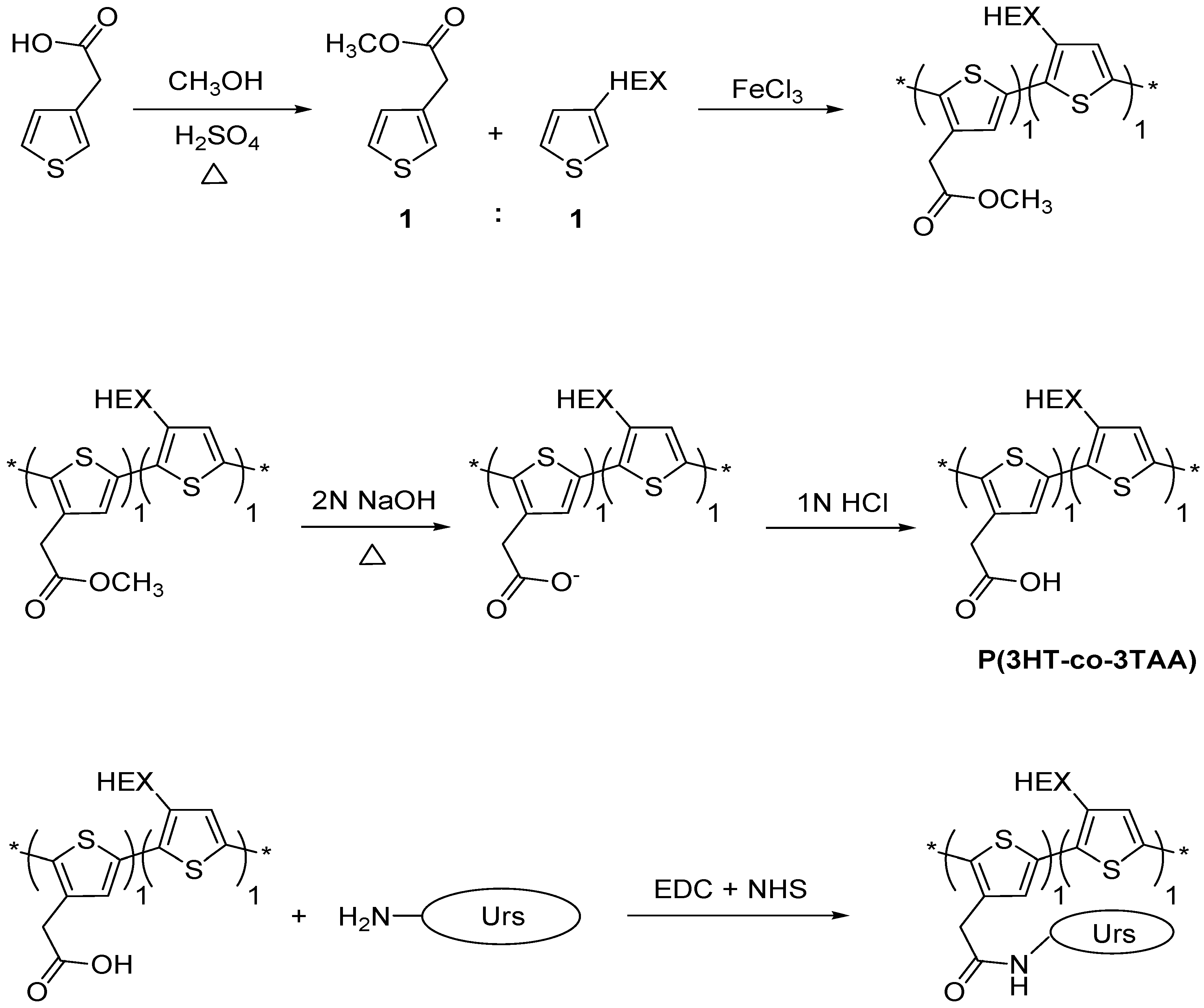
2.2. Synthetic Procedures
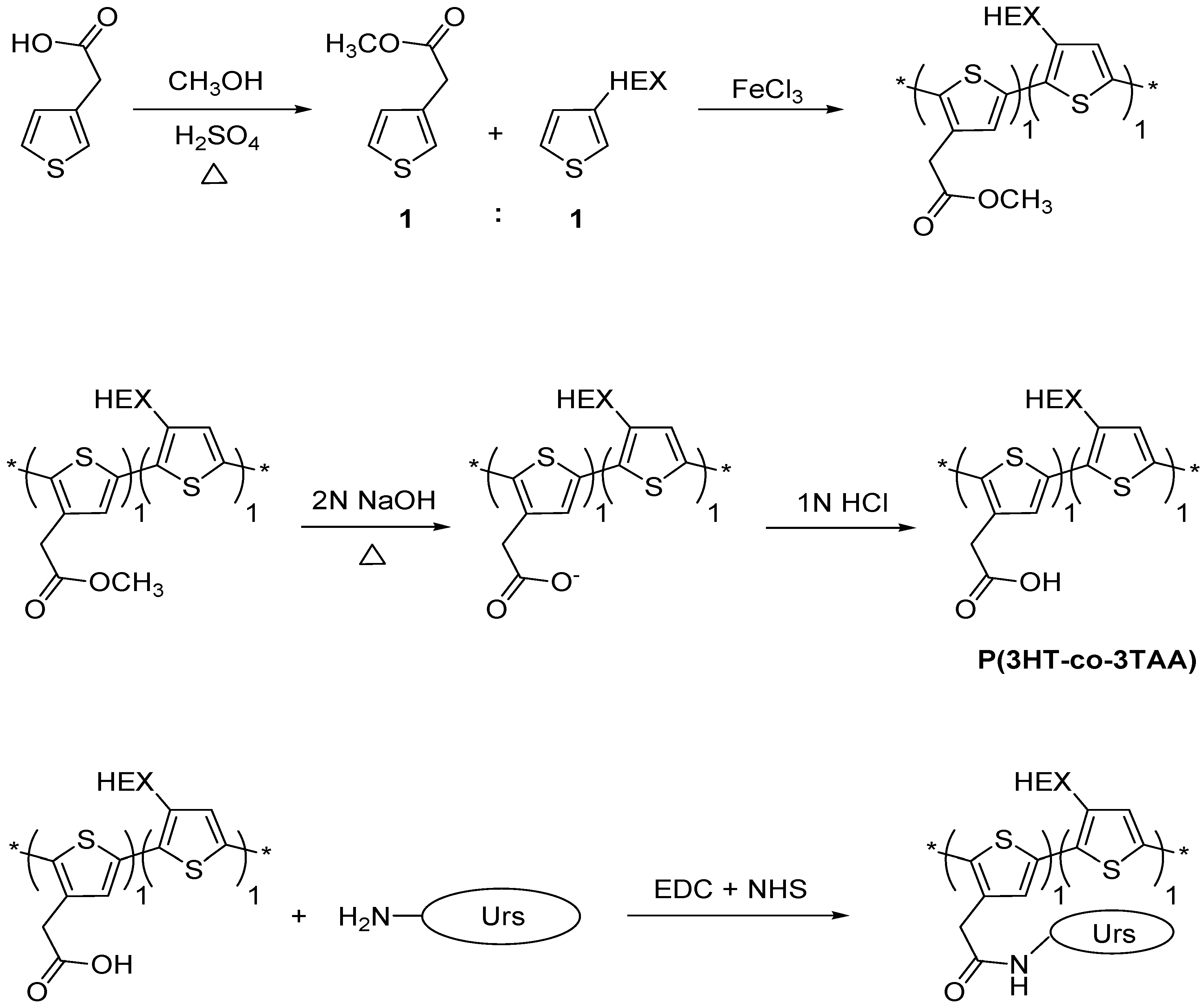
2.2.1. Esterification of 3-thiopheneacetic acid
2.2.2. Synthesis of poly(3-hexylthiophene-co-methyl 2-(thiophene-3-yl)acetate), (P(3HT-co-MTA))
2.2.3. Hydrolysis of poly(3-hexylthiophene-co-methyl-2-(thiophene-3-yl) acetate)
2.2.4. Immobilization of Urease on the Surface of P(3HT-co-3TAA) Using a Carbodiimide Coupling Reaction
2.3. Spectrophotometric Assay of the Urease Immobilized on the Surface of P(3HT-co-3TAA)
2.4. Potentiometric Assay Using the Electrochemical Biosensor System
2.5. Electrochemical Analysis by Cyclic Voltammetry
2.6. Polymer Film Morphology
3. Results and Discussion
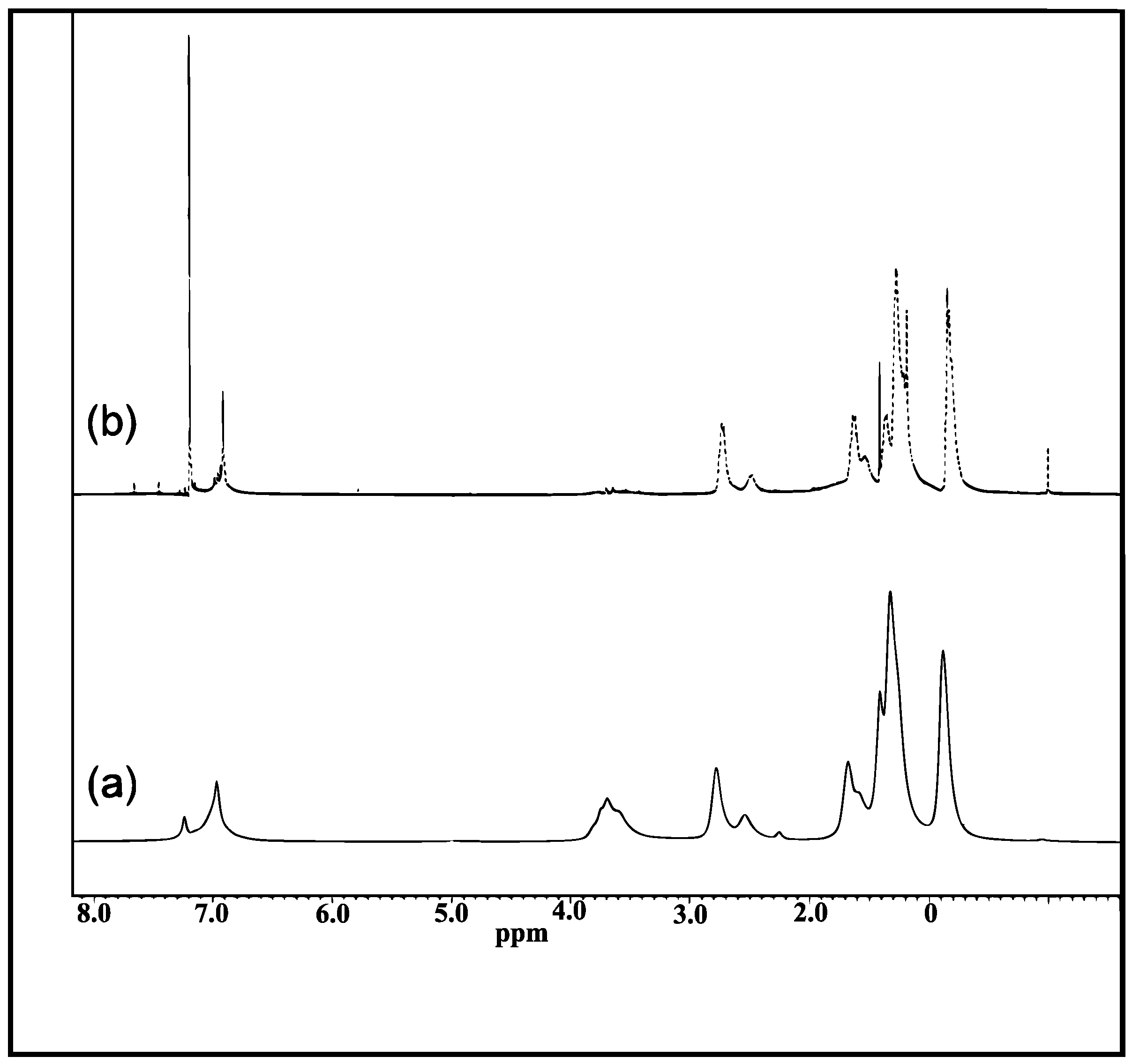
3.1. 1H-NMR Spectra for P(3HT-co-MTA) and P(3HT-co-3TAA)
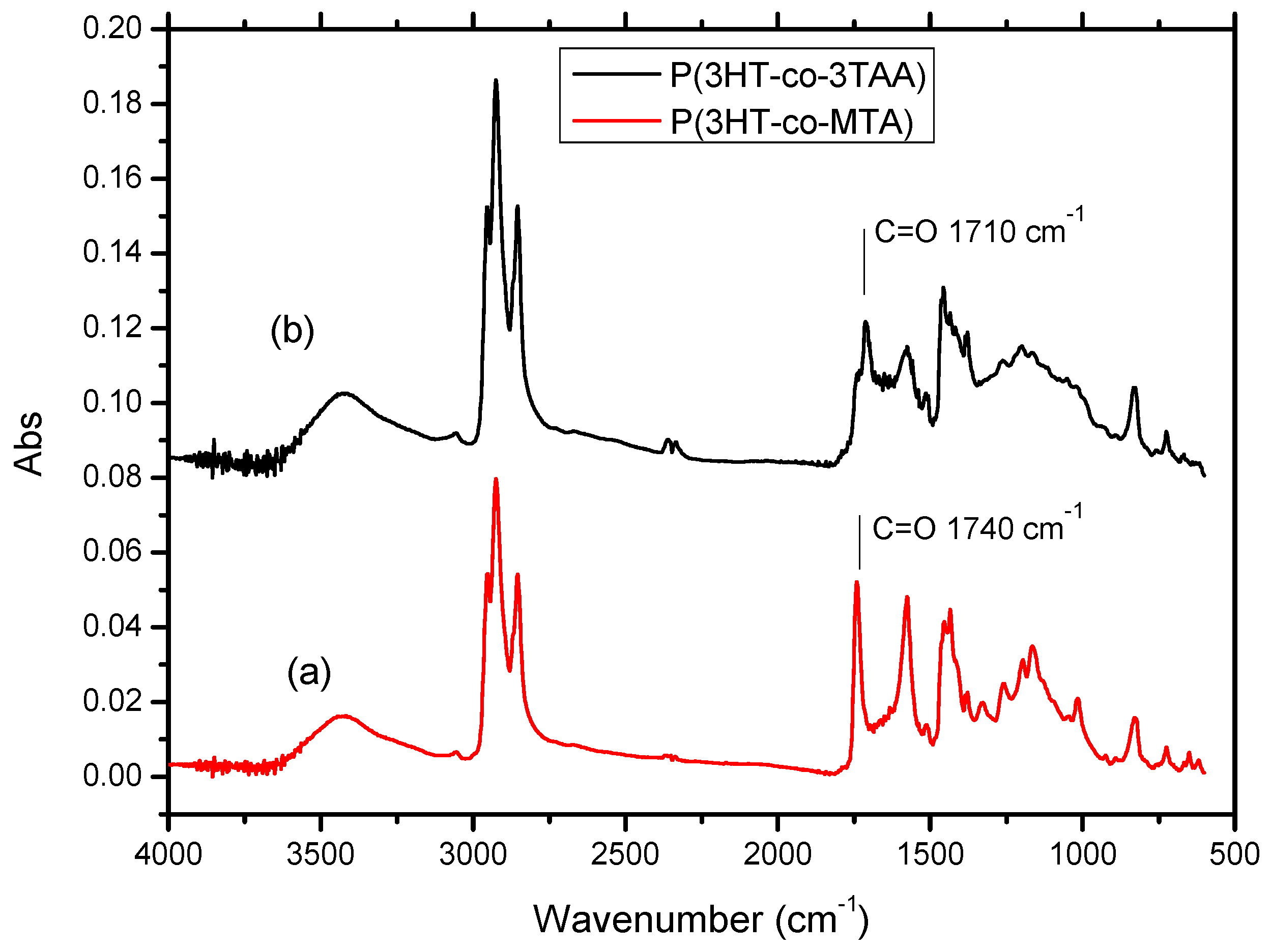
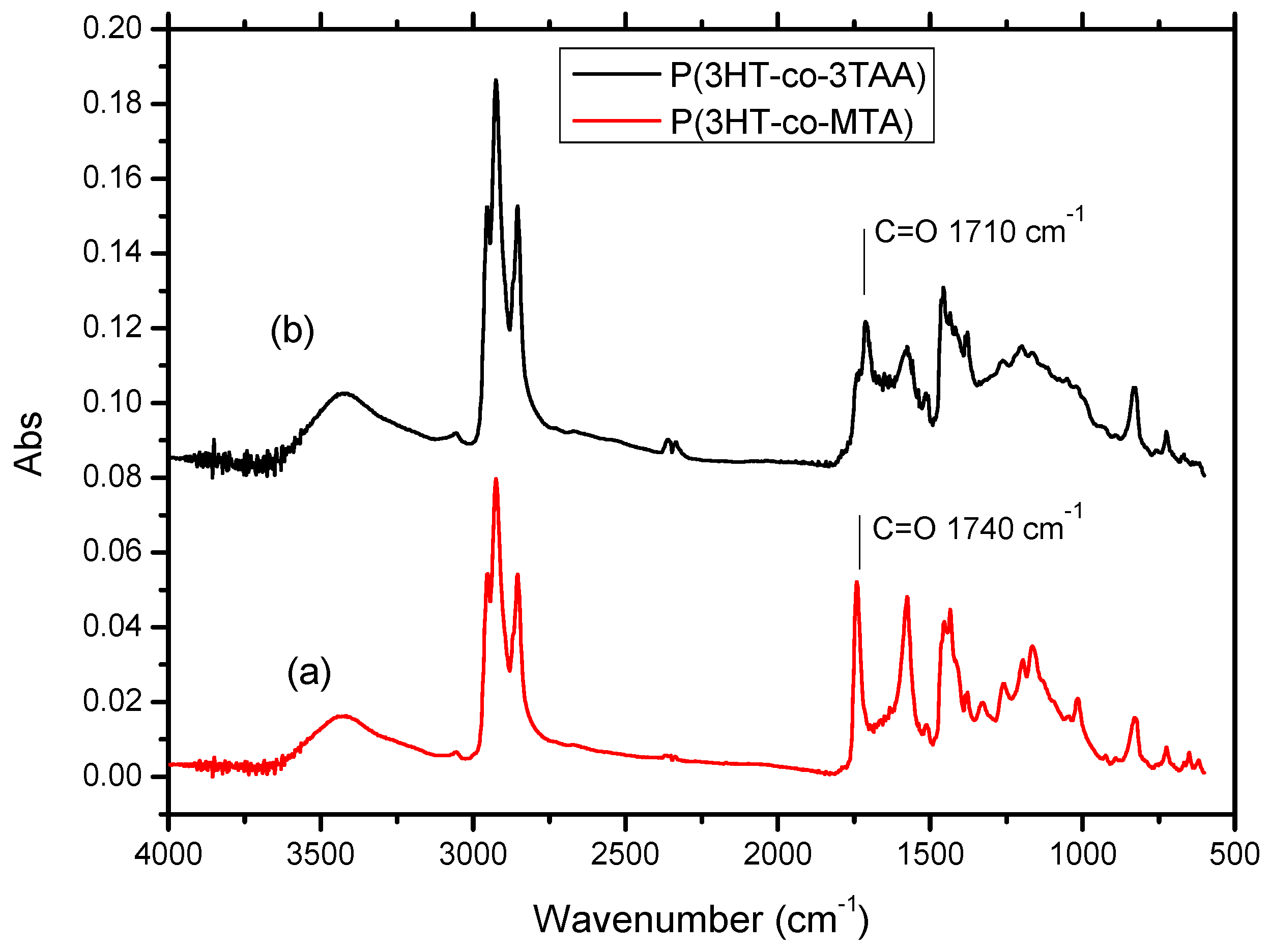
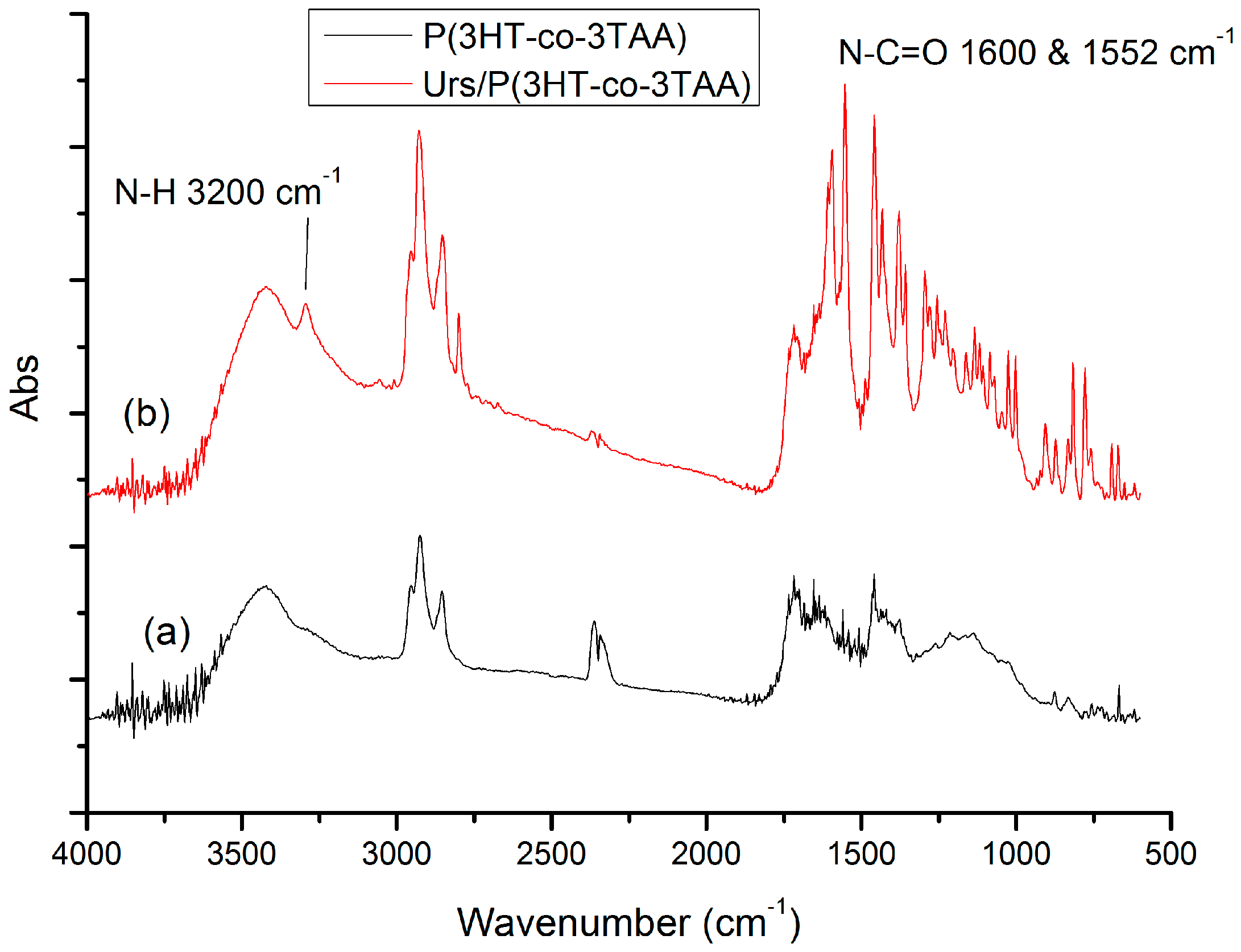
3.2. FT-IR Spectra of P(3HT-co-MTA), P(3HT-co-3TAA) and Urs/P(3HT-co-3TAA)
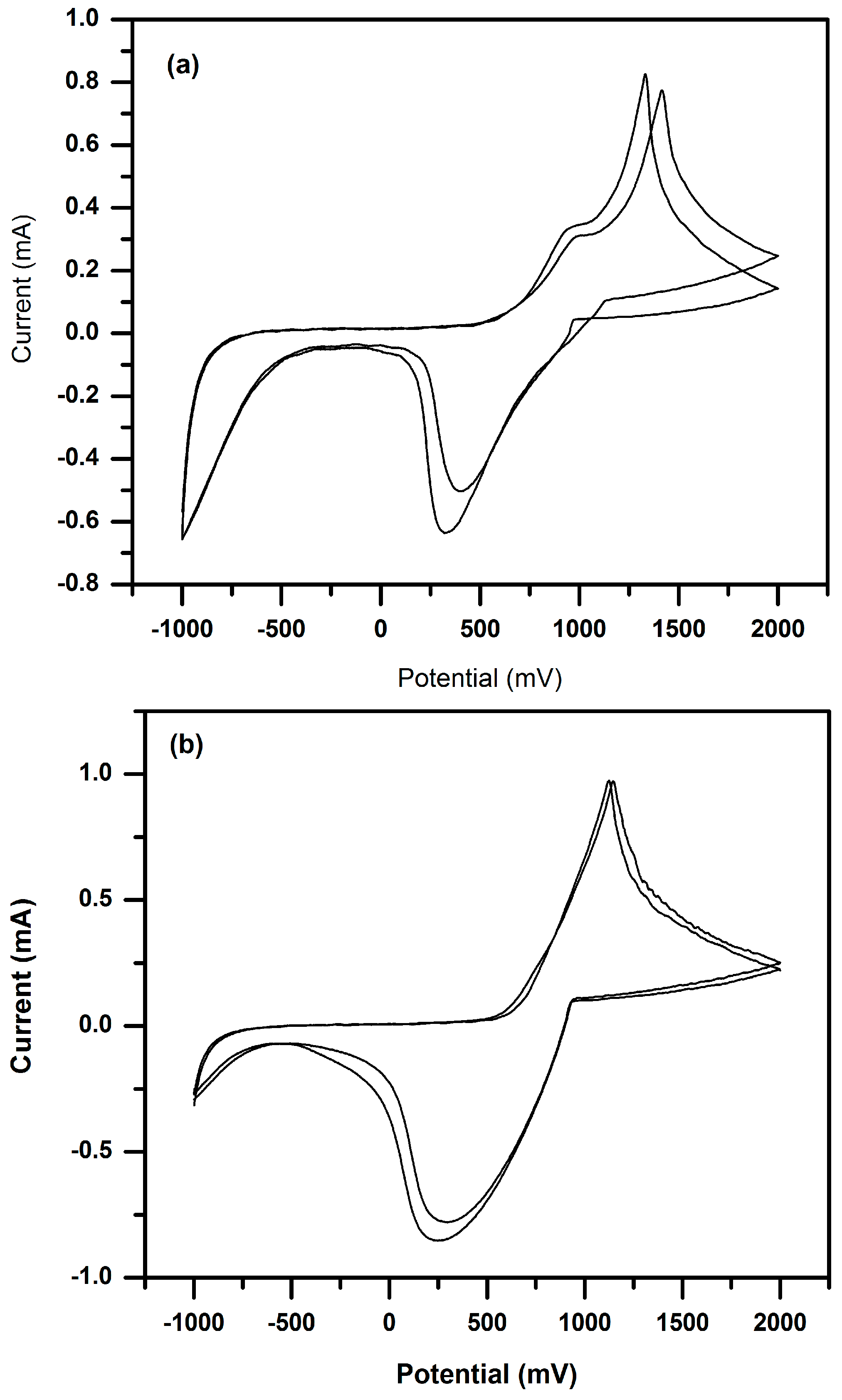
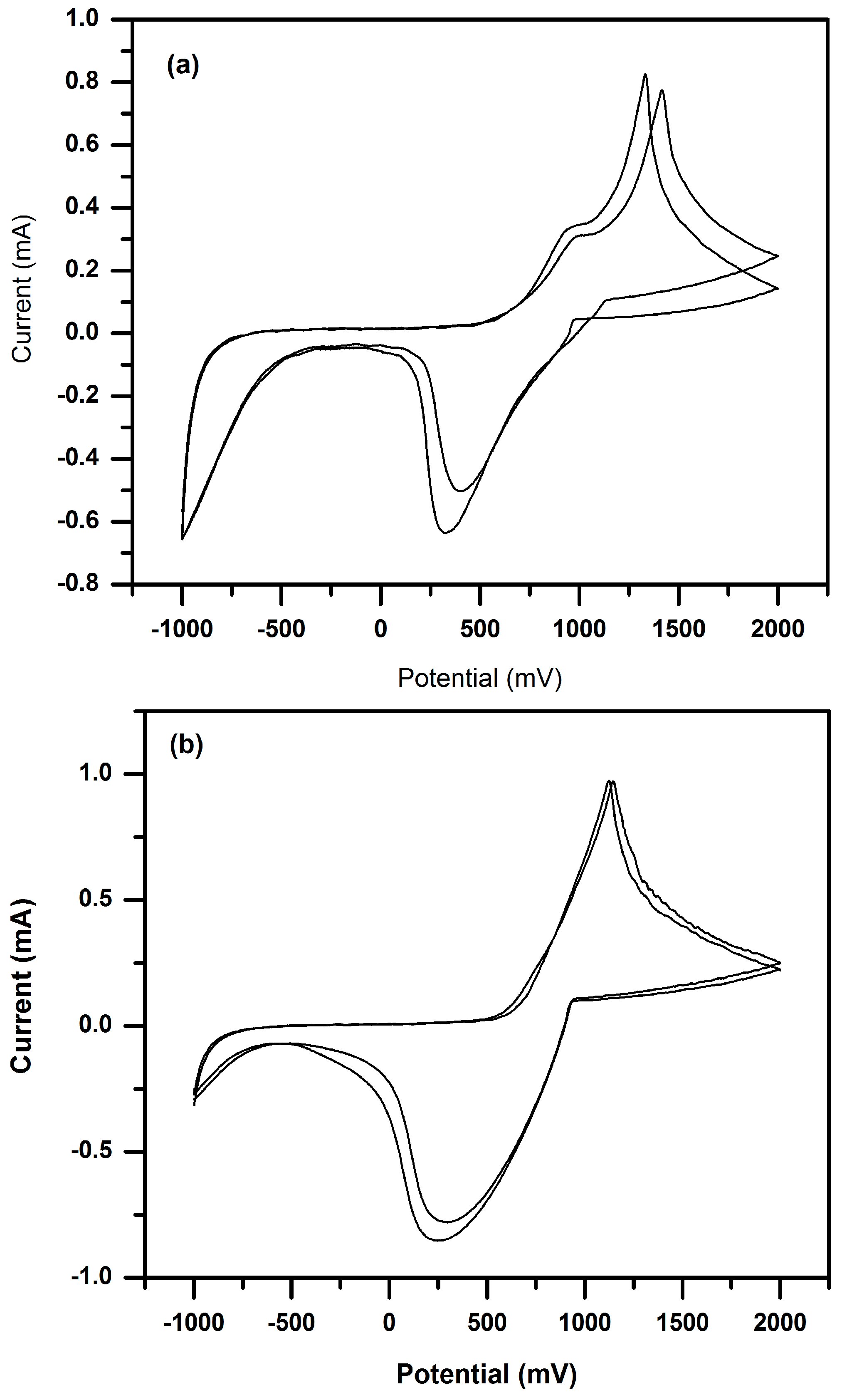
3.3. Cyclic Voltammetry (CV) of P(3HT-co-3TAA) and Urs/P(3HT-co-3TAA) in 0.1 M LiClO4/Propylene Carbonate
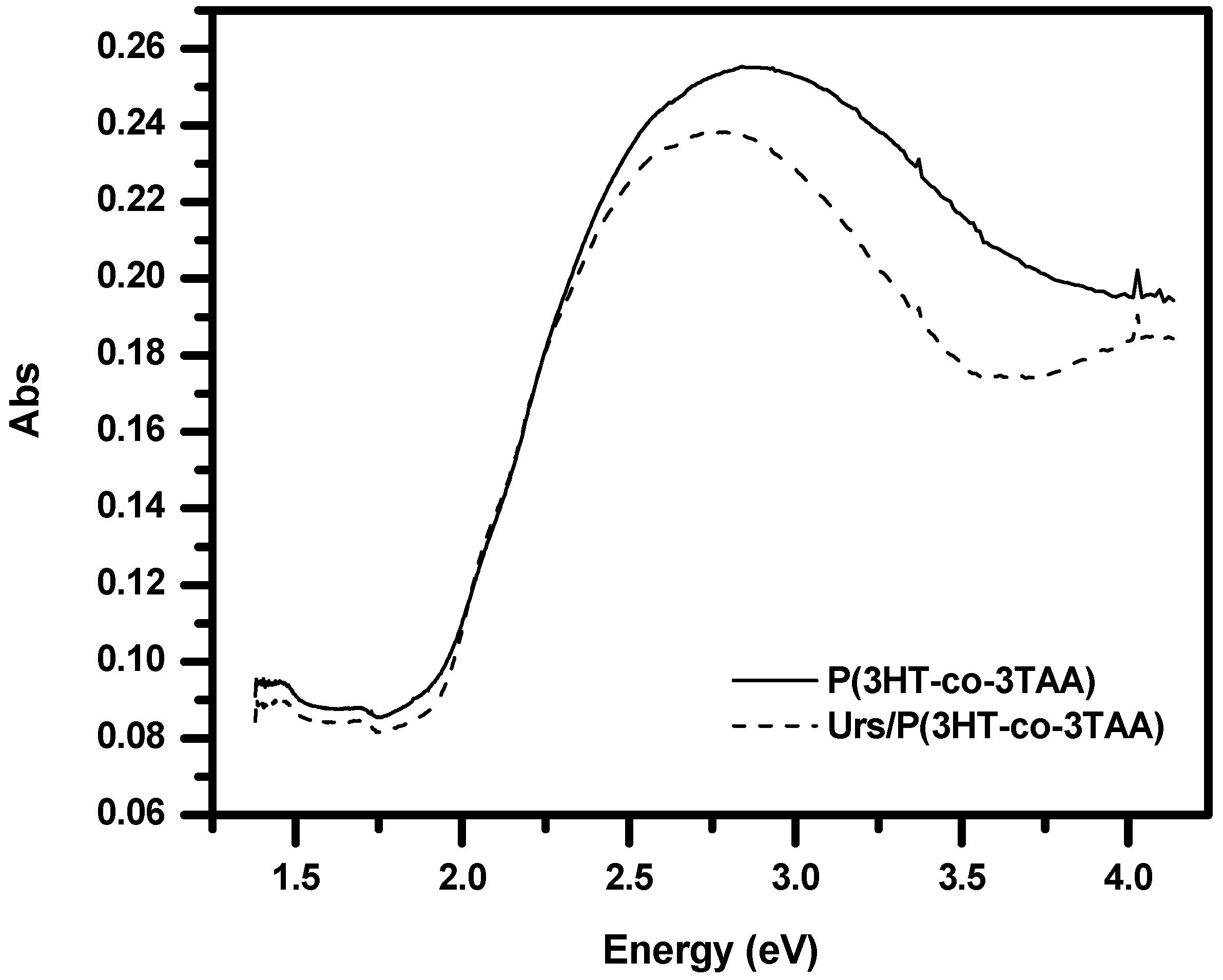
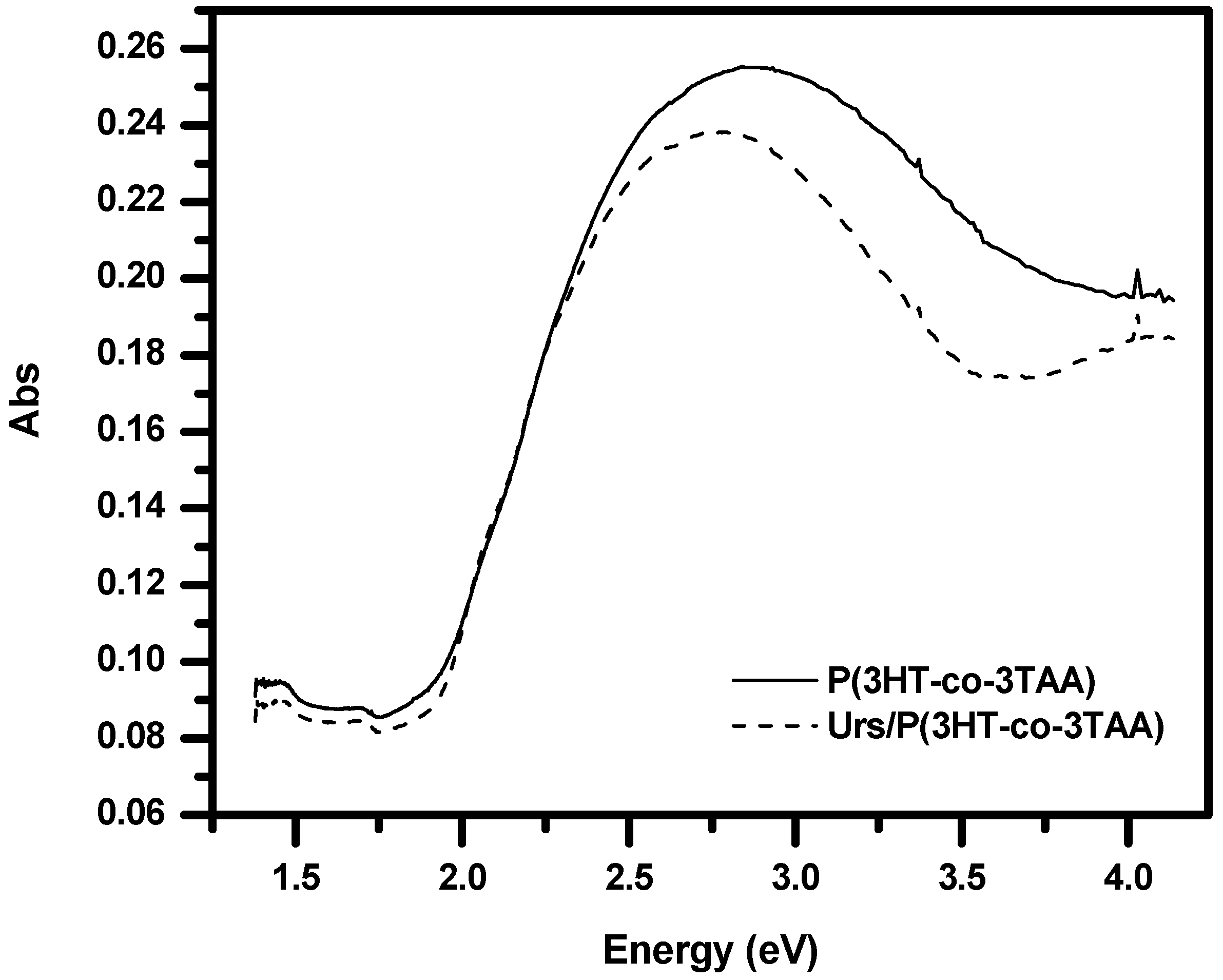
3.4. UV-Visible Absorption Spectra of P(3HT-co-3TAA) and Urs/P(3HT-co-3TAA)
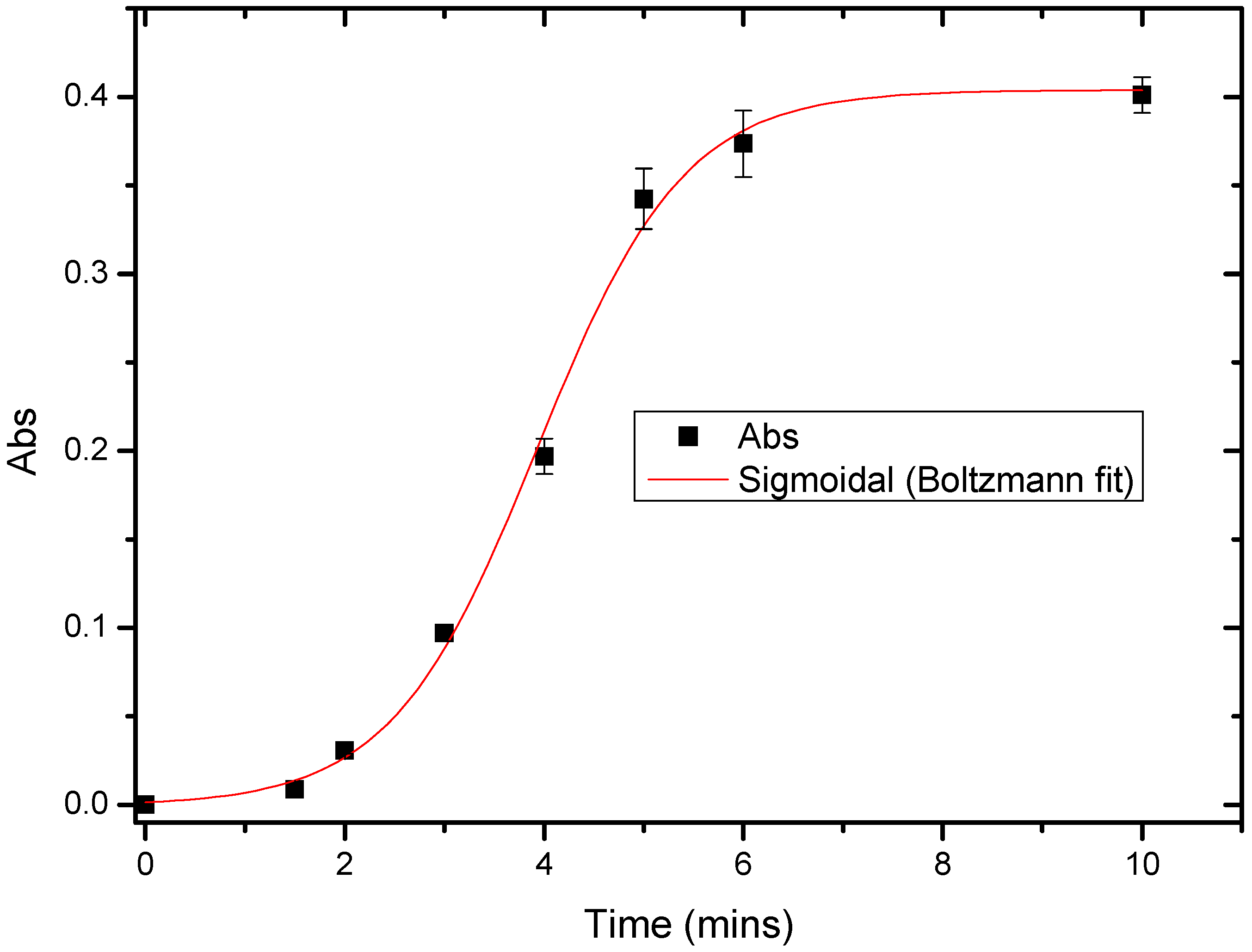
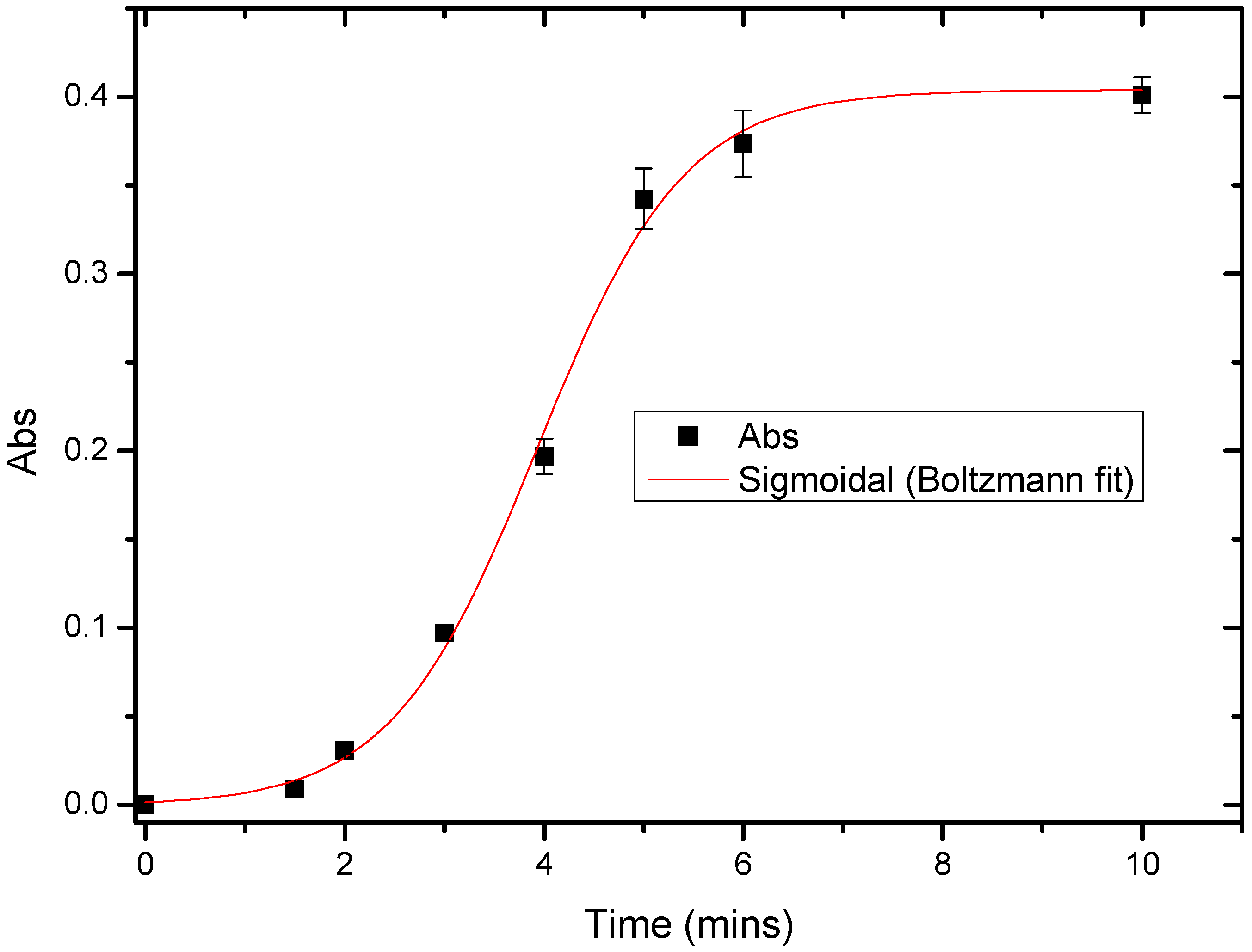
3.5. Response Time of Urs/P(3HT-co-3TAA)/ITO Glass by Spectrophotometric Studies
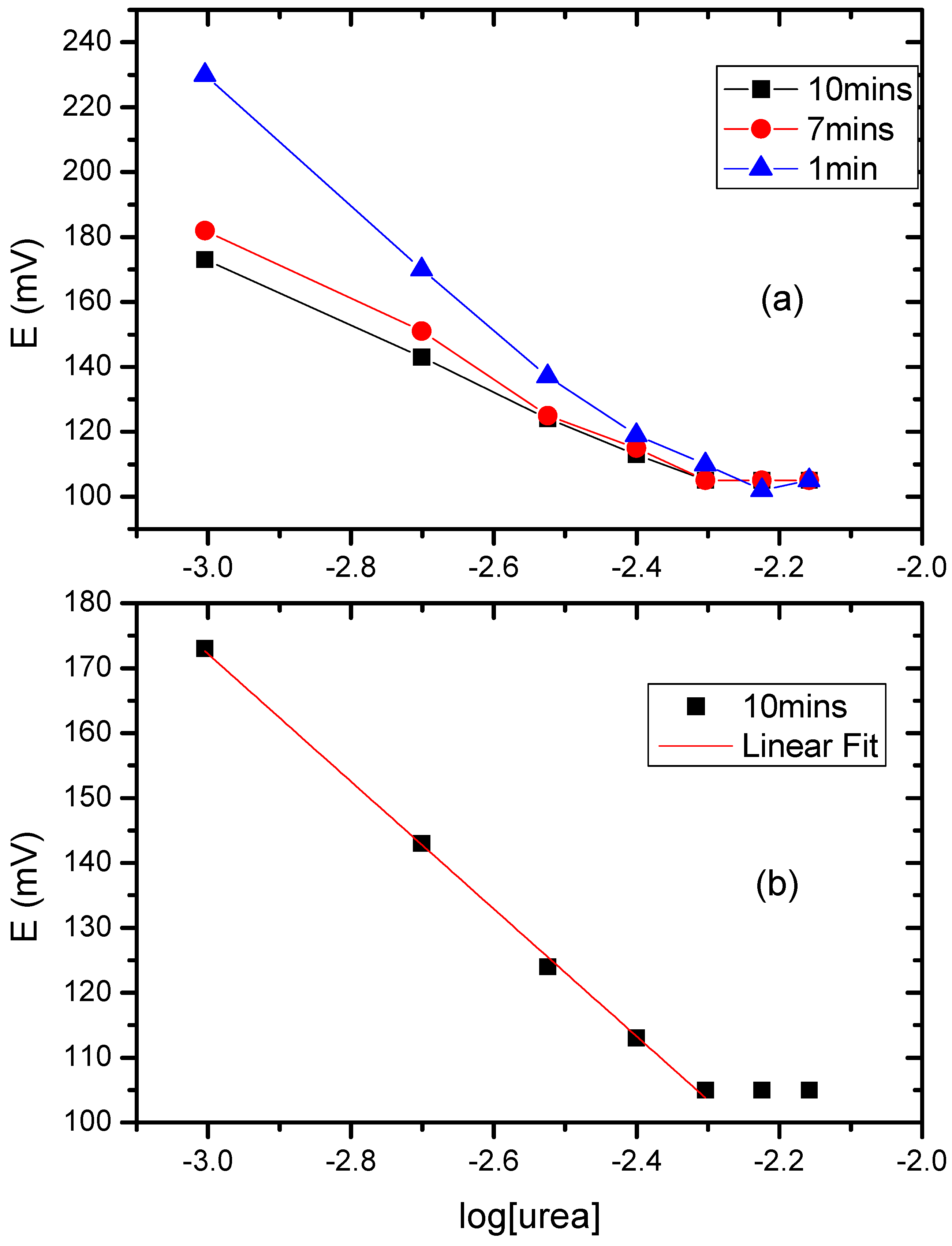
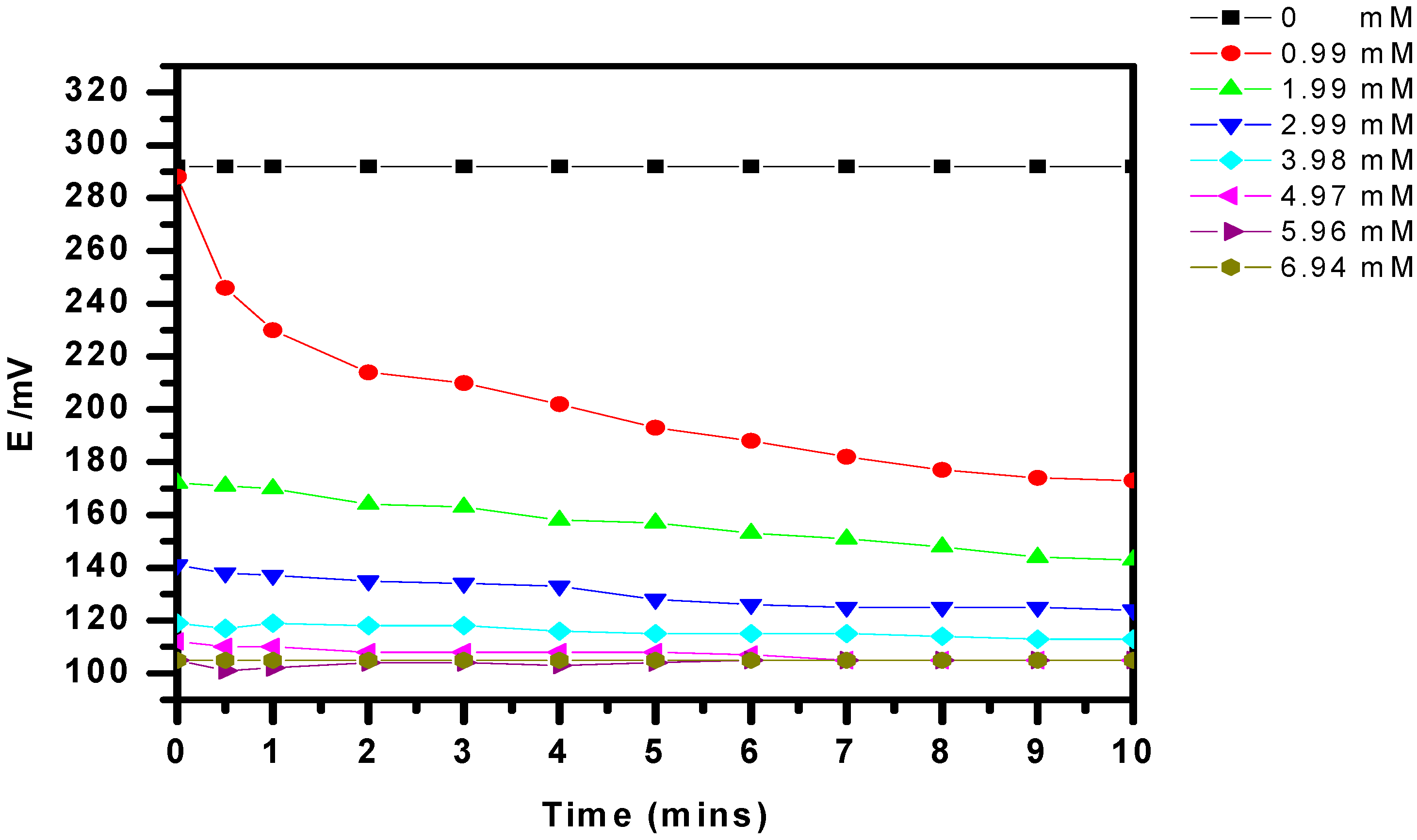
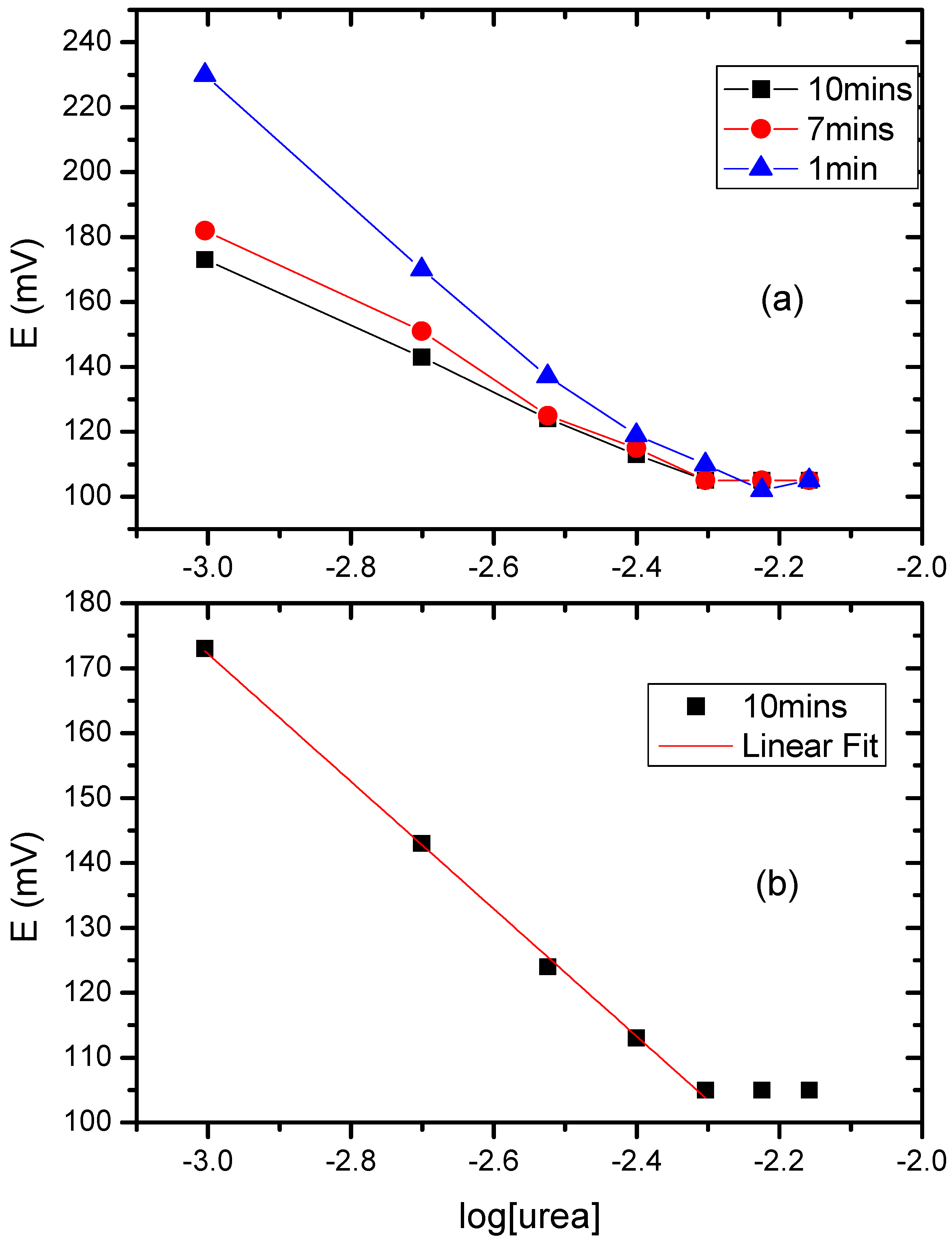
3.6. The Potentiometric Assay of Urease/P(3HT-co-3TAA)/ITO Glass
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Clark, L.C., Jr.; Lyons, C. Electrode systems for continuous monitoring in cardiovascular surgery. Ann. N. Y. Acad. Sci. 1962, 102, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Vidal, J.-C.; Garcia-Ruiz, E.; Castillo, J.-R. Recent advances in electropolymerized conducting polymers in amperometric biosensors. Microchim. Acta 2003, 143, 93–111. [Google Scholar] [CrossRef]
- Lowe, C.R.; Foulds, N.C. Enzyme entrapment in electrically conducting polymers—Immobilization of glucose-oxidase in polypyrrole and its application in amperometric glucose sensors. J. Chem. Soc. Faraday Trans. I 1986, 82, 1259–1264. [Google Scholar]
- Cosnier, S.; Ionescu, R.E.; Herrmann, S.; Bouffier, L.; Demeunynck, M.; Marks, R.S. Electroenzymatic polypyrrole-intercalator sensor for the determination of West Nile virus cDNA. Anal. Chem. 2006, 78, 7054–7057. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Ohta, H.; Kuwahara, T.; Shimomura, M. Amperometric glucose responding property of enzyme electrodes fabricated by covalent immobilization of glucose oxidase on conducting polymer films with macroporous structure. Eur. Polym. J. 2008, 44, 1114–1122. [Google Scholar] [CrossRef]
- Shimomura, M.; Miyata, R.; Kuwahara, T.; Miyauchi, S. Immobilization of glucose oxidase on the films prepared by electrochemical copolymerization of pyrrole and 1-(2-carboxyethyl)pyrrole for glucose sensing. Eur. Polym. J. 2007, 43, 388–394. [Google Scholar] [CrossRef]
- Korri-Youssoufi, H.; Makrouf, B.; Yassar, A. Synthesis of 3-derivatized pyrroles precursors polymers for functionalization with biomolecules toward biosensor devices. Mater. Sci. Eng. C 2001, 15, 265–268. [Google Scholar] [CrossRef]
- Kojima, K.; Yamauchi, T.; Oshima, K.; Shimomura, M.; Miyauchi, S. Covalent immobilization of glucose oxidase on poly[1-(2-carboxyethyl)pyrrole] film for glucose sensing. Polymer 1998, 39, 2079–2082. [Google Scholar] [CrossRef]
- Malhotra, B.D.; Chaubey, A.; Singh, S.P. Prospects of conducting polymers in biosensors. Anal. Chim. Acta 2006, 578, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Foot, P.J.S.; Kaiser, A.B. Conducting Polymers. In Kirk-Othmer Encyclopedia of Chemical Technology; Seidel, A., Bickford, M., Eds.; John Wiley & Sons, Inc.: New York, NY, USA, 2004. [Google Scholar]
- Chaplin, M.F. Biosensors. In Molecular Biology and Biotechnology, 6th ed.; Rapley, R., Whitehouse, D., Eds.; Royal Society of Chemistry: Cambridge, UK, 2015; pp. 225–256. [Google Scholar]
- Dhawan, G.; Sumana, G.; Malhotra, B.D. Recent developments in urea biosensors. Biochem. Eng. J. 2009, 44, 42–52. [Google Scholar] [CrossRef]
- Tian, F.M.; Xu, B.; Zhu, L.D.; Zhu, G.Y. Hydrogen peroxide biosensor with enzyme entrapped within electrodeposited polypyrrole based on mediated sol-gel derived composite carbon electrode. Anal. Chim. Acta 2001, 443, 9–16. [Google Scholar] [CrossRef]
- McGraw, S.K.; Alocilja, E.; Senecal, A.; Senecal, K. Synthesis of a functionalized polypyrrole coated electrotextile for use in biosensors. Biosensors 2012, 2, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Datta, K.; Ghosh, P.; Rushi, A.; Mulchandani, A.; Shirsat, M. Fe nanoparticle tailored poly (N-methyl pyrrole) nanowire matrix: A CHEMFET study from the perspective of discrimination among electron donating analytes. J. Phys. D Appl. Phys. 2015, 48, 195301. [Google Scholar] [CrossRef]
- Sehgal, D.; Vijay, I.K. A method for the high-efficiency of water-soluble carbodiimide-mediated amidation. Anal. Biochem. 1994, 218, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, T.; Mir, I.A.; Kumar, D.; Rajesh. Potentiometric urea biosensor based on BSA embedded surface modified polypyrrole film. Sens. Actuators B Chem. 2008, 134, 140–145. [Google Scholar] [CrossRef]
- Rajesh; Bisht, V.; Takashima, W.; Kaneto, K. An amperometric urea biosensor based on covalent immobilization of urease onto an electrochemically prepared copolymer, poly(N-3-aminopropyl pyrrole-co-pyrrole) film. Biomaterials 2005, 26, 3685–3690. [Google Scholar] [CrossRef]
- Gambhir, A.; Kumar, A.; Malhotra, B.D.; Miksa, B.; Slomkowski, S. Covalent immobilization of urease on polypyrrole microspheres for application as a urea biosensor. e-Polymers 2002, 52, 1–9. [Google Scholar] [CrossRef]
- Ivanova, S.; Ivanov, Y.; Godjevargova, T. Urea Amperometric Biosensors Based on Nanostructured Polypyrrole and Poly Ortho-Phenylenediamine. Open J. Appl. Biosens. 2013, 2, 12–19. [Google Scholar] [CrossRef]
- Bourigua, S.; Hafaid, I.; Korri-Youssoufi, H.; Maaref, A.; Jafferezic-Renault, N. A novel urea biosensor based on modified electrodes with urease immobilized on poly(N-hydroxyphthalimide-pyrrole-co-pyrrole) film incorporating ethylamine ferrocene as redox marker. Sens. Lett. 2009, 7, 731–738. [Google Scholar] [CrossRef]
- Ethanol content in must grape by alcohol dehydrogenase biosensor based on doped-polyaniline modified screen printed electrodes. Available online: http://s3.amazonaws.com/academia.edu.documents/37493809/2015_Grape_Quality.pdf?AWSAccessKeyId=AKIAIWOWYYGZ2Y53UL3A&Expires=1488469178&Signature=SY8QVyohJXwbIjYqZVQkU8tWhQ0%3D&response-content-disposition=inline%3B%20filename%3DEthanol_content_in_must_grape_by_Alcohol.pdf (accessed on 14 January 2017).
- Lakard, B.; Magnin, D.; Deschaume, O.; Vanlancker, G.; Glinel, K.; Demoustier-Champagne, S.; Nysten, B.; Jonas, A.M.; Bertrand, P.; Yunus, S. Urea potentiometric enzymatic biosensor based on charged biopolymers and electrodeposited polyaniline. Biosens. Bioelectron. 2011, 26, 4139–4145. [Google Scholar] [CrossRef] [PubMed]
- Das, G.; Yoon, H.H. Amperometric urea biosensors based on sulfonated graphene/polyaniline nanocomposite. Int. J. Nanomed. 2015, 10, 55–66. [Google Scholar]
- Çevik, E.; Şenel, M.; Abasiyanik, M.F. An amperometric urea biosensor based on covalent immobilization of urease on copolymer of glycidyl methacrylate and vinylferrocene. J. Solid State Electrochem. 2012, 16, 367–373. [Google Scholar] [CrossRef]
- Hiller, M.; Kranz, C.; Huber, J.; Bauerle, P.; Schuhmann, W. Amperometic biosensors produced by immobilization of redox enzymes at polythiophene-modified electrode surfaces. Adv. Mater. 1996, 8, 219–222. [Google Scholar] [CrossRef]
- Kuwahara, T.; Oshima, K.; Miyauchi, S.; Shimomura, M. Immobilization of glucose oxidase and electron-mediating groups on the film of 3-methylthiophene/thiophene-3-acetic acid copolymer and its application to reagentless sensing of glucose. Polymer 2005, 46, 8091–8097. [Google Scholar] [CrossRef]
- Kros, A.; Sommerdijk, N.A.; Nolte, R.J.M. Polypyrrole versus poly(3,4-ethylenedioxythiophene): Implications for biosensor applications. Sens. Actuators B Chem. 2005, 106, 289–295. [Google Scholar] [CrossRef]
- Liu, C.; Kuwahara, T.; Yamazaki, R.; Shimomura, M. Covalent immobilization of glucose oxidase on films prepared by electrochemical copolymerization of 3-methylthiophene and thiophene-3-acetic acid for amperometric sensing of glucose: Effects of polymerization conditions on sensing properties. Eur. Polym. J. 2007, 43, 3264–3276. [Google Scholar] [CrossRef]
- Çil, M.; Böyükbayram, A.E.; Kiralp, S.; Toppare, L.; Yagci, Y. Various applications of immobilised glucose oxidase and polyphenol oxidase in a conducting polymer matrix. Int. J. Biol. Macromol. 2007, 41, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Abasiyanik, M.F.; Senel, M. Immobilization of glucose oxidase on reagentless ferrocene-containing polythiophene derivative and its glucose sensing application. J. Electroanal. Chem. 2010, 639, 21–26. [Google Scholar] [CrossRef]
- Welzel, H.P.; Kossmehl, G.; Engelmann, G.; Neumann, B.; Wollenberger, U.; Scheller, F.; Schröder, W. Reactive groups on polymer covered electrodes, 4a. Lactate-oxidase-biosensor based on electrodes modified by polythiophene. Macromol. Chem. Phys. 1996, 197, 3355–3363. [Google Scholar] [CrossRef]
- Rahman, M.M.; Shiddiky, M.J.; Rahman, M.A.; Shim, Y.B. A lactate biosensor based on lactate dehydrogenase/nictotinamide adenine dinucleotide (oxidized form) immobilized on a conducting polymer/multiwall carbon nanotube composite film. Anal. Biochem. 2009, 384, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Jiang, Y.; Yu, J.; Xie, G. Choline oxidase immobilized into conductive poly(3,4-ethylenedioxythiophene) film for choline detection. Appl. Surf. Sci. 2008, 254, 6337–6340. [Google Scholar] [CrossRef]
- Rahman, M.A.; Kwon, N.-H.; Won, M.-S.; Choe, E.S.; Shim, Y.-B. Functionalized conducting polymer as an enzyme-immobilizing substrate: An amperometric glutamate microbiosensor for in vivo measurements. Anal. Chem. 2005, 77, 4854–4860. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-Y.; Hung, Y.-T.; Chen, S.-M.; Tsai, T.-H.; Lou, B.-S.; Liu, X. Fabrication of silver hexacyanoferrate and functionalized MWCNT with poly(3,4-ethylenedioxythiophene) hybrid film modified electrode for selectively determination of ascorbic acid and hydrazine. Int. J. Electrochem. Sci. 2015, 10, 1128–1135. [Google Scholar]
- Kim, H.-J.; Choi, S.-H.; Lee, K.-P.; Gopalan, A.I.; Oh, S.-H.; Woo, J.-C. Fabrication of functional poly(thiophene) electrode for biosensors. Ultramicroscopy 2008, 108, 1360–1364. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.C.; Tsai, T.H.; Chen, S.M. Performing enzyme-free H2O2 biosensor and simultaneous determination for AA, DA, and UA by MWCNT–PEDOT film. Biosens. Bioelectron. 2010, 26, 608–614. [Google Scholar] [CrossRef] [PubMed]
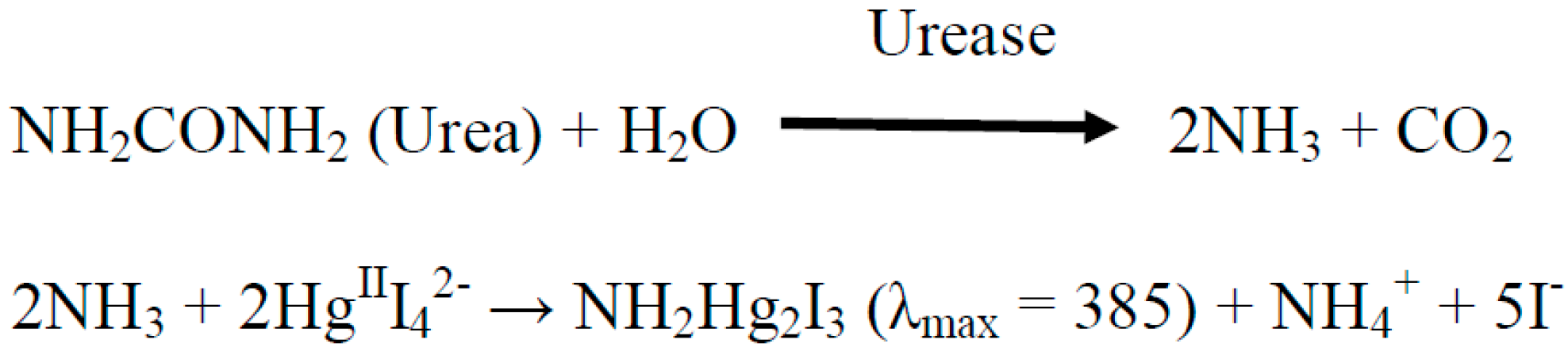



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
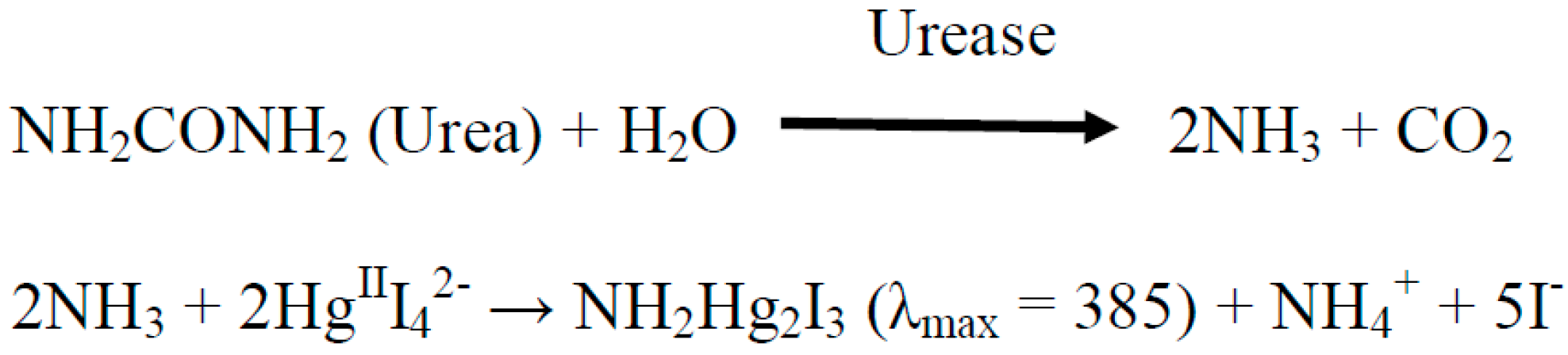{kind=link}
| [urea] (mM) | E (V) | RT/βnF (n = 1) | ln{[urea]*α*2} (Predicted) | ln{[urea]*2} | α |
|---|---|---|---|---|---|
| 0.99 | 0.182 | 0.0426 | −6.3915 | −6.2247 | 0.8462 |
| 1.99 | 0.152 | 0.0426 | −5.6703 | −5.5256 | 0.8660 |
| 2.99 | 0.125 | 0.0426 | −5.0653 | −5.1193 | 1.0555 |
| 3.98 | 0.115 | 0.0426 | −4.8326 | −4.8333 | 1.0007 |
| 4.97 | 0.105 | 0.0426 | −4.5999 | −4.6112 | 1.0113 |
| 5.96 | 0.105 | 0.0426 | −4.5999 | −4.4295 | (0.8433) |
| 6.95 | 0.105 | 0.0426 | −4.5999 | −4.2758 | (0.6338) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, C.-Y.; Foot, P.J.S.; Brown, J.W.; Spearman, P. A Urea Potentiometric Biosensor Based on a Thiophene Copolymer. Biosensors 2017, 7, 13. https://doi.org/10.3390/bios7010013
Lai C-Y, Foot PJS, Brown JW, Spearman P. A Urea Potentiometric Biosensor Based on a Thiophene Copolymer. Biosensors. 2017; 7(1):13. https://doi.org/10.3390/bios7010013
Chicago/Turabian StyleLai, Cheng-Yuan (Kevin), Peter J. S. Foot, John W. Brown, and Peter Spearman. 2017. "A Urea Potentiometric Biosensor Based on a Thiophene Copolymer" Biosensors 7, no. 1: 13. https://doi.org/10.3390/bios7010013
APA StyleLai, C.-Y., Foot, P. J. S., Brown, J. W., & Spearman, P. (2017). A Urea Potentiometric Biosensor Based on a Thiophene Copolymer. Biosensors, 7(1), 13. https://doi.org/10.3390/bios7010013