Detection of Metabolic Changes Induced via Drug Treatments in Live Cancer Cells and Tissue Using Raman Imaging Microscopy

Abstract
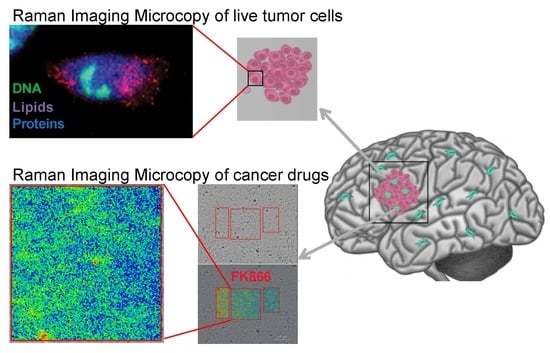
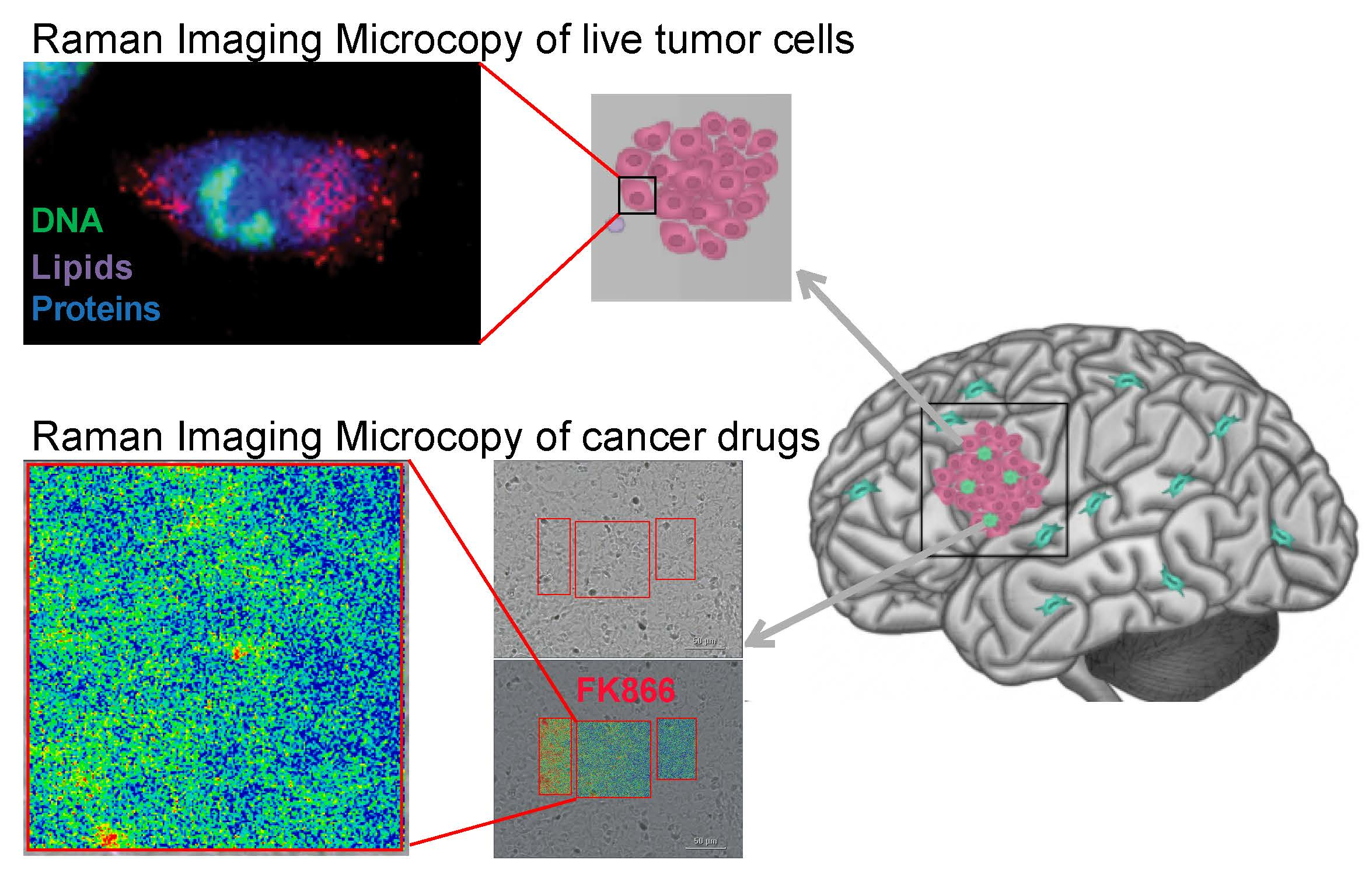
:
1. Introduction
2. Materials and Methods
2.1. Raman Imaging Microscopy
2.2. Cell Culture and Viability Measurements
2.3. Sample Preparation for Nuclear Magneti Resonance (NMR) Spectroscopy
2.4. 1H NMR Spectral Acquisition and Analysis
2.5. Lipidomics Using Ultra Performance Liquid Chromatography and Quadrupole Time-of-Flight Mass Spectrometry (UPLC-QTOF-MS)
2.6. Mice Experiments
3. Results
3.1. Raman Spectroscopy of Live HT1080 Cells Revealed Decreased Lipids upon FK866 Drug Treatment
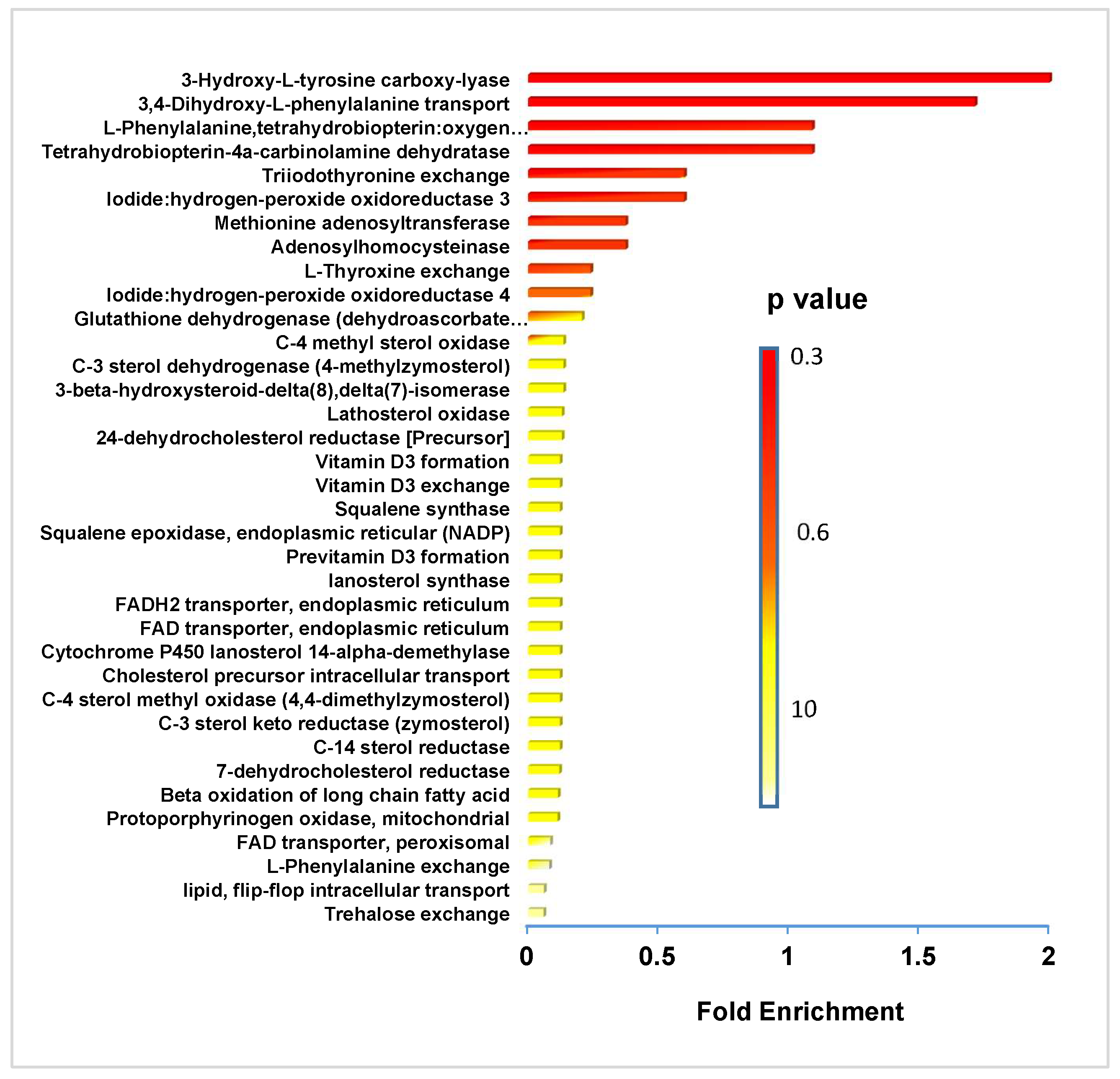
3.2. Mass Spectrometry and NMR Identify Classes of Lipids Altered by FK866 Drug Treatment
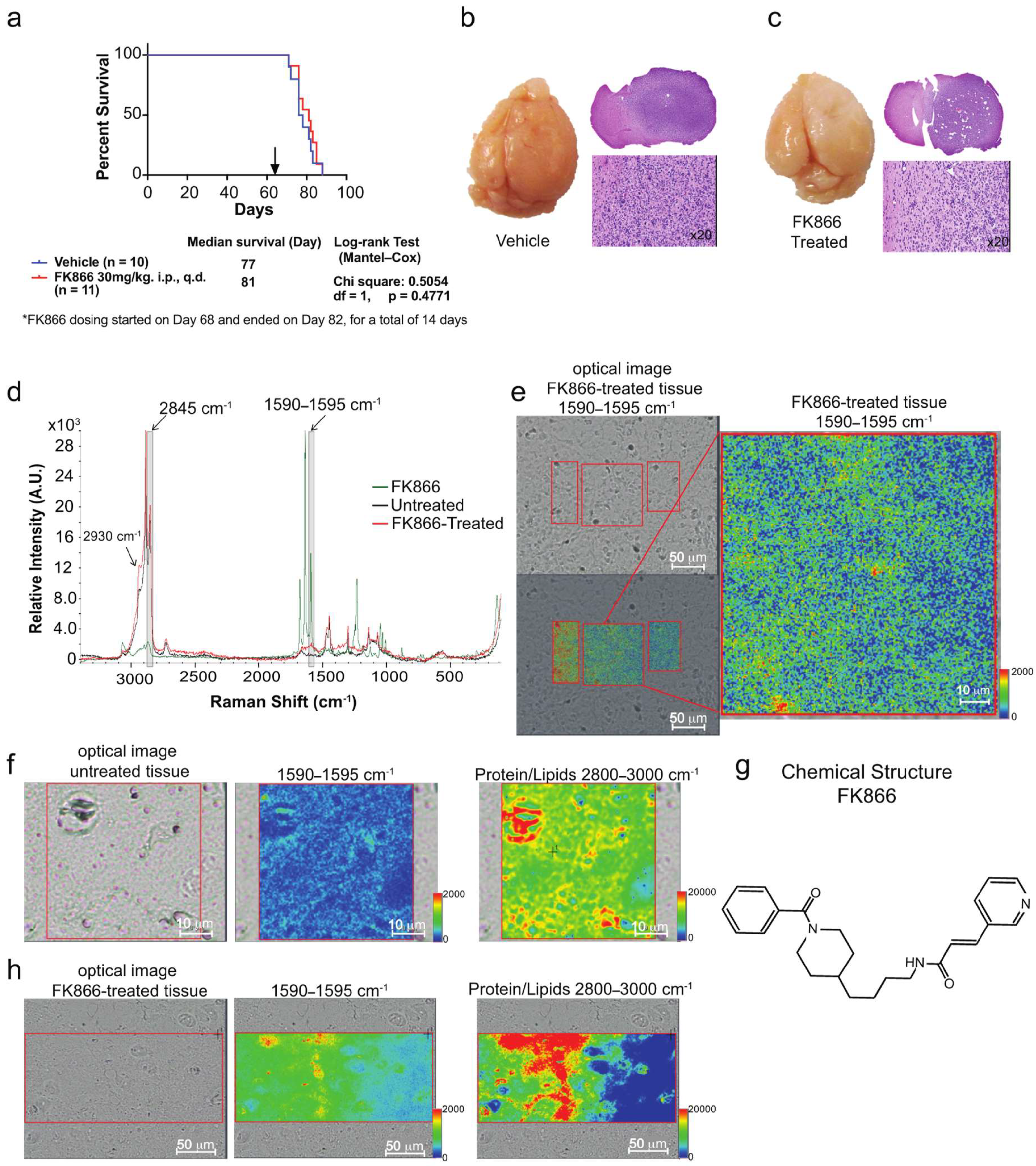
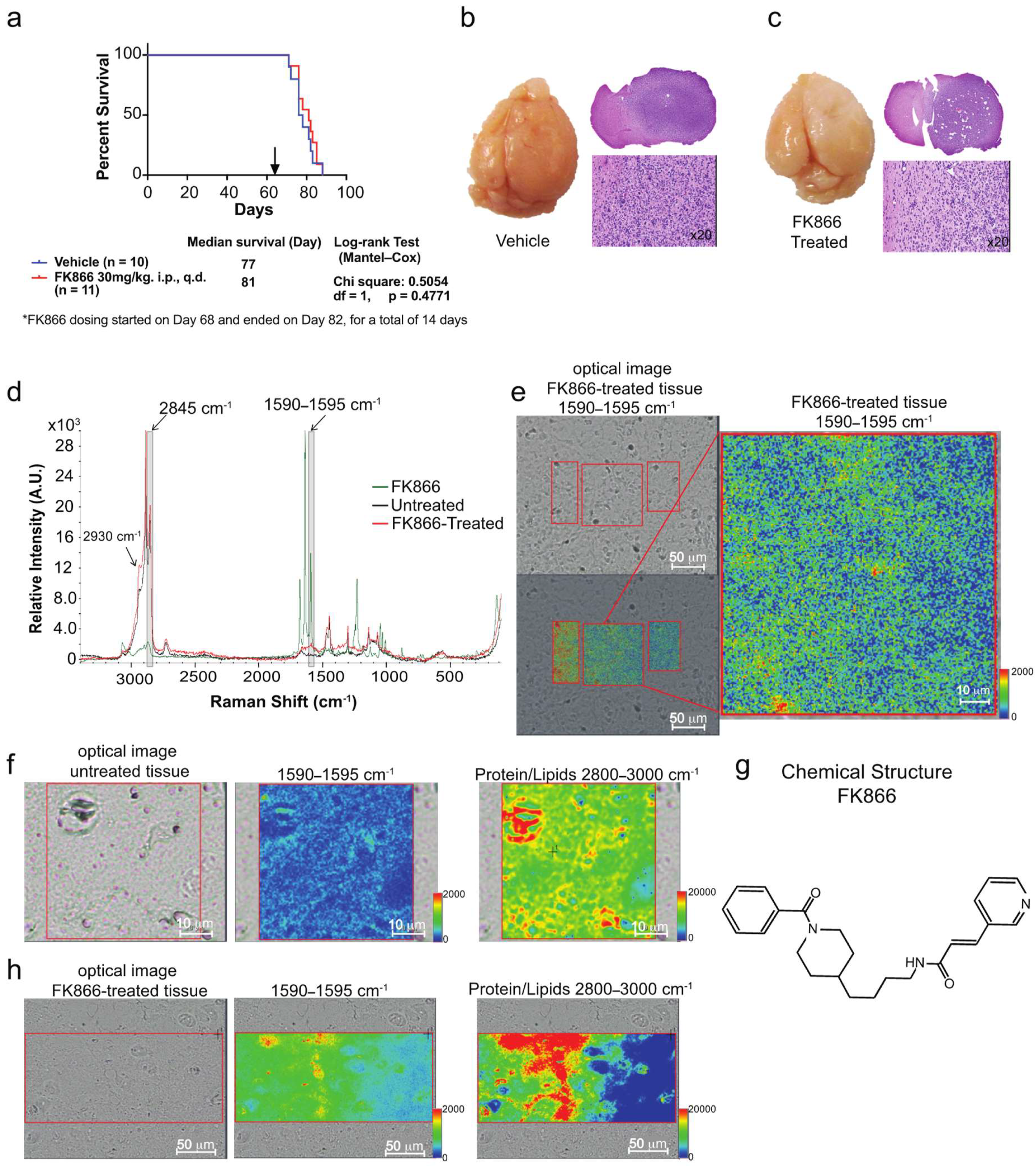
3.3. Raman Imaging Microscopy Reveals Increased Lipids in Tissue
4. Discussion
4.1. Live Cell Imaging via Raman Imaging Microscopy
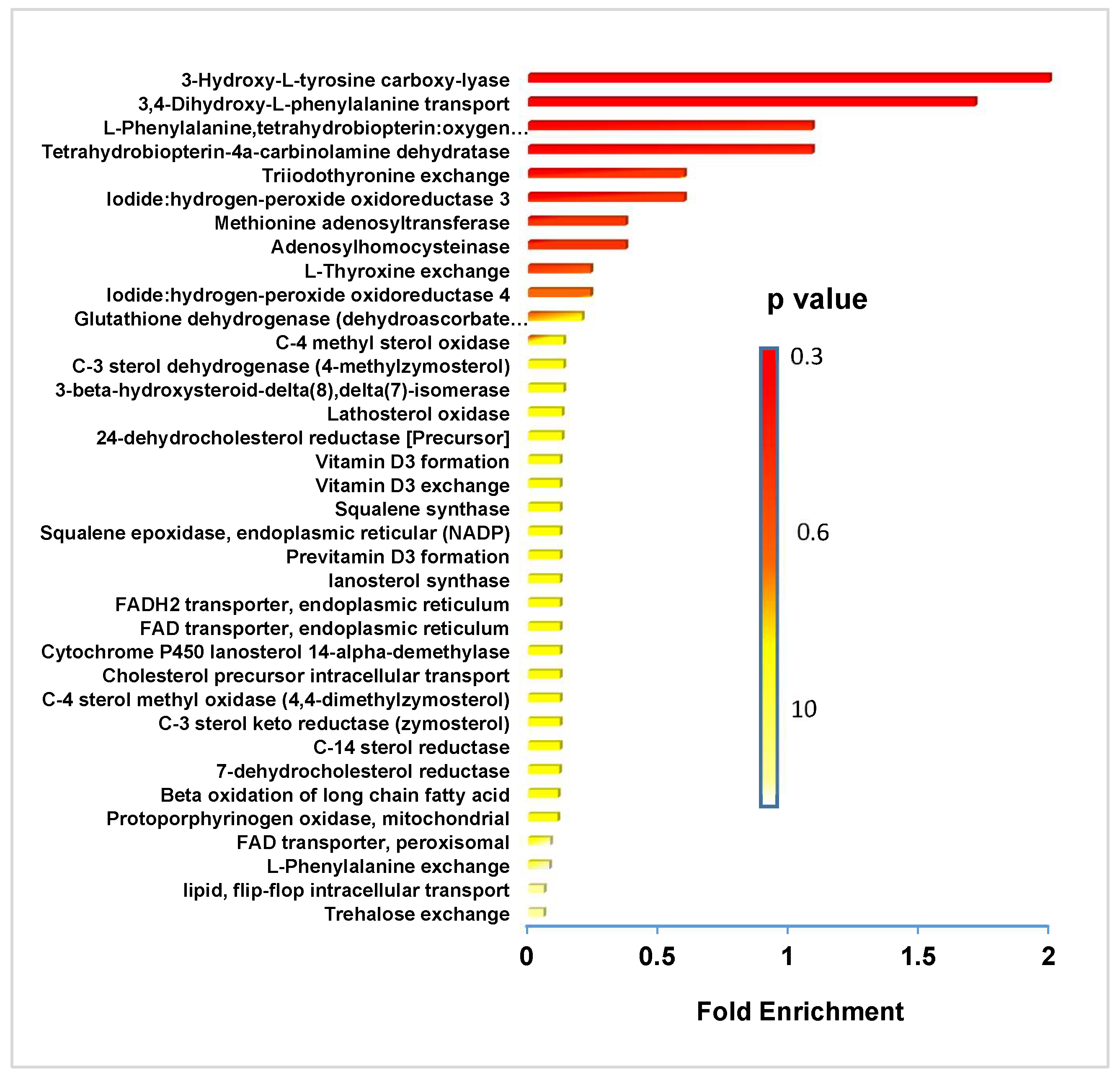
4.2. Link between NAD+ Depletion and Decreased Lipid and Catecholamine Synthesis
4.3. Mapping of FK866 in Mice Tissue via Raman Imaging Microscopy
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Raman, C.V. A new radiation. Proc. Indian Acad. Sci. Sect. A 1953, 37, 333–341. [Google Scholar] [CrossRef]
- Petibois, C. Imaging methods for elemental, chemical, molecular, and morphological analyses of single cells. Anal. Bioanal. Chem. 2010, 397, 2051–2065. [Google Scholar] [CrossRef] [PubMed]
- Krafft, C.; Schie, I.W.; Meyer, T.; Schmitt, M.; Popp, J. Developments in spontaneous and coherent Raman scattering microscopic imaging for biomedical applications. Chem. Soc. Rev. 2016, 45, 1819–1849. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, A.N.; Pliss, A.; Prasad, P.N. Ramanomics: New Omics Disciplines Using Micro Raman Spectrometry with Biomolecular Component Analysis for Molecular Profiling of Biological Structures. Biosensors 2017, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Devpura, S.; Barton, K.N.; Brown, S.L.; Palyvoda, O.; Kalkanis, S.; Naik, V.M.; Siddiqui, F.; Naik, R.; Chetty, I.J. Vision 20/20: The role of Raman spectroscopy in early stage cancer detection and feasibility for application in radiation therapy response assessment. Med. Phys. 2014, 41, 050901. [Google Scholar] [CrossRef] [PubMed]
- Kast, R.; Auner, G.; Yurgelevic, S.; Broadbent, B.; Raghunathan, A.; Poisson, L.M.; Mikkelsen, T.; Rosenblum, M.L.; Kalkanis, S.N. Identification of regions of normal grey matter and white matter from pathologic glioblastoma and necrosis in frozen sections using Raman imaging. J. Neuro-Oncol. 2015, 125, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Kalkanis, S.N.; Kast, R.E.; Rosenblum, M.L.; Mikkelsen, T.; Yurgelevic, S.M.; Nelson, K.M.; Raghunathan, A.; Poisson, L.M.; Auner, G.W. Raman spectroscopy to distinguish grey matter, necrosis, and glioblastoma multiforme in frozen tissue sections. J. Neuro-Oncol. 2014, 116, 477–485. [Google Scholar] [CrossRef]
- Kuzmin, A.N.; Levchenko, S.M.; Pliss, A.; Qu, J.; Prasad, P.N. Molecular profiling of single organelles for quantitative analysis of cellular heterogeneity. Sci. Rep. 2017, 7, 6512. [Google Scholar] [CrossRef]
- Levchenko, S.M.; Kuzmin, A.N.; Pliss, A.; Qu, J.; Prasad, P.N. Macromolecular Profiling of Organelles in Normal Diploid and Cancer Cells. Anal. Chem. 2017, 89, 10985–10990. [Google Scholar] [CrossRef] [Green Version]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.M.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; Su, S.-S.M. Isocitrate Dehydrogenase Mutation and (R)-2-Hydroxyglutarate: From Basic Discovery to Therapeutics Development. Annu. Rev. Biochem. 2017, 86, 305–331. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015, 28, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Hasmann, M.; Schemainda, I. FK866, a Highly Specific Noncompetitive Inhibitor of Nicotinamide Phosphoribosyltransferase, Represents a Novel Mechanism for Induction of Tumor Cell Apoptosis. Cancer Res. 2003, 63, 7436–7442. [Google Scholar] [PubMed]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Altadill, T.; Dowdy, T.M.; Gill, K.; Reques, A.; Menon, S.S.; Moiola, C.P.; Lopez-Gil, C.; Coll, E.; Matias-Guiu, X.; Cabrera, S.; et al. Metabolomic and Lipidomic Profiling Identifies The Role of the RNA Editing Pathway in Endometrial Carcinogenesis. Sci. Rep. 2017, 7, 8803. [Google Scholar] [CrossRef] [Green Version]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, S.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E.; et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef]
- Cantó, C.; Menzies, K.; Auwerx, J. NAD(+) metabolism and the control of energy homeostasis—A balancing act between mitochondria and the nucleus. Cell MeTable 2015, 22, 31–53. [Google Scholar] [CrossRef] [PubMed]
- Delikatny, E.J.; Chawla, S.; Leung, D.J.; Poptani, H. MR-visible lipids and the tumor microenvironment. NMR Biomed. 2011, 24, 592–611. [Google Scholar] [CrossRef] [PubMed]
- Freudiger, C.W.; Min, W.; Saar, B.G.; Lu, S.; Holtom, G.R.; He, C.; Tsai, J.C.; Kang, J.X.; Xie, X.S. Label-Free Biomedical Imaging with High Sensitivity by Stimulated Raman Scattering Microscopy. Science 2008, 322, 1857–1861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzmin, A.N.; Pliss, A.; Rzhevskii, A.; Lita, A.; Larion, M. BCAbox Algorithm Expands Capabilities of Raman Microscope for Single Organelles Assessment. Biosensors 2018, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342 Pt 2, 249–268. [Google Scholar] [CrossRef]
- Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxid. Redox Signal. 2008, 10, 179–206. [Google Scholar] [CrossRef]
- Bosch-Presegué, L.; Vaquero, A. The Dual Role of Sirtuins in Cancer. Genes Cancer 2011, 2, 648–662. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.-C.; Guarente, L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. 2014, 25, 138–145. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Chen, H.; He, X.; Nie, H.; Hong, Y.; Sheng, C.; Wang, Q.; Xia, W.; Ying, W. NAD+ Metabolism and NAD+-Dependent Enzymes: Promising Therapeutic Targets for Neurological Diseases. Curr. Drug Targets 2012, 13, 222–229. [Google Scholar] [CrossRef]
- Li, Z.; Agellon, L.B.; Allen, T.M.; Umeda, M.; Jewell, L.; Mason, A.; Vance, D.E. The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis. Cell Metab. 2006, 3, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K. Role of Catecholamine Signaling in Brain and Nervous System Functions: New Insights from Mouse Molecular Genetic Study. J. Investig. Dermatol. Symp. Proc. 2001, 6, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Almuhaideb, A.; Papathanasiou, N.; Bomanji, J. (18)F-FDG PET/CT Imaging In Oncology. Ann. Saudi Med. 2011, 31, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Yamakoshi, H.; Dodo, K.; Palonpon, A.; Ando, J.; Fujita, K.; Kawata, S.; Sodeoka, M. Alkyne-Tag Raman Imaging for Visualization of Mobile Small Molecules in Live Cells. J. Am. Chem. Soc. 2012, 134, 20681–20689. [Google Scholar] [CrossRef] [PubMed]
- Andreou, C.; Kishore, S.A.; Kircher, M.F. Surface-Enhanced Raman Spectroscopy: A New Modality for Cancer Imaging. J. Nucl. Med. 2015, 56, 1295–1299. [Google Scholar] [CrossRef] [PubMed]
- Harmsen, S.; Huang, R.; Wall, M.A.; Karabeber, H.; Samii, J.M.; Spaliviero, M.; White, J.R.; Monette, S.; O’Connor, R.; Pitter, K.L.; et al. Surface-enhanced resonance Raman scattering nanostars for high-precision cancer imaging. Sci. Transl. Med. 2015, 7, 271ra7. [Google Scholar] [CrossRef] [PubMed]
- Karabeber, H.; Huang, R.; Iacono, P.; Samii, J.M.; Pitter, K.; Holland, E.C.; Kircher, M.F. Guiding brain tumor resection using surface-enhanced Raman scattering nanoparticles and a hand-held Raman scanner. ACS Nano 2014, 8, 9755–9766. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Hu, F.; Shen, Y.; Chen, Z.; Yu, Y.; Lin, C.-C.; Wang, M.C.; Min, W. Live-cell imaging of alkyne-tagged small biomolecules by stimulated Raman scattering. Nat. Methods 2014, 11, 410–412. [Google Scholar] [CrossRef] [Green Version]
- Jermyn, M.; Mok, K.; Mercier, J.; Desroches, J.; Pichette, J.; Saint-Arnaud, K.; Bernstein, L.; Guiot, M.-C.; Petrecca, K.; Leblond, F. Intraoperative brain cancer detection with Raman spectroscopy in humans. Sci. Transl. Med. 2015, 7, 274ra19. [Google Scholar] [CrossRef]
- Ji, M.; Orringer, D.A.; Freudiger, C.W.; Ramkissoon, S.; Liu, X.; Lau, D.; Golby, A.J.; Norton, I.; Hayashi, M.; Agar, N.Y.R.; et al. Rapid, Label-Free Detection of Brain Tumors with Stimulated Raman Scattering Microscopy. Sci. Transl. Med. 2013, 5, 201ra119. [Google Scholar] [CrossRef]
- Ji, M.; Lewis, S.; Camelo-Piragua, S.; Ramkissoon, S.H.; Snuderl, M.; Venneti, S.; Fisher-Hubbard, A.; Garrard, M.; Fu, D.; Wang, A.C.; et al. Detection of human brain tumor infiltration with quantitative stimulated Raman scattering microscopy. Sci. Transl. Med. 2015, 7, 309ra163. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Band Position (cm−1) | Assignment | ||
|---|---|---|---|
| 785–788 | Pyrimidine Bases | Ring Breathing | Nucleic Acids |
| 1655–1662 | Amide I | C=O Stretching Mode | Proteins |
| 2850–2880 | CH | Stretching | Lipids |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larion, M.; Dowdy, T.; Ruiz-Rodado, V.; Meyer, M.W.; Song, H.; Zhang, W.; Davis, D.; Gilbert, M.R.; Lita, A. Detection of Metabolic Changes Induced via Drug Treatments in Live Cancer Cells and Tissue Using Raman Imaging Microscopy. Biosensors 2019, 9, 5. https://doi.org/10.3390/bios9010005
Larion M, Dowdy T, Ruiz-Rodado V, Meyer MW, Song H, Zhang W, Davis D, Gilbert MR, Lita A. Detection of Metabolic Changes Induced via Drug Treatments in Live Cancer Cells and Tissue Using Raman Imaging Microscopy. Biosensors. 2019; 9(1):5. https://doi.org/10.3390/bios9010005
Chicago/Turabian StyleLarion, Mioara, Tyrone Dowdy, Victor Ruiz-Rodado, Matthew W. Meyer, Hua Song, Wei Zhang, Dionne Davis, Mark R. Gilbert, and Adrian Lita. 2019. "Detection of Metabolic Changes Induced via Drug Treatments in Live Cancer Cells and Tissue Using Raman Imaging Microscopy" Biosensors 9, no. 1: 5. https://doi.org/10.3390/bios9010005