Molecular Targets of β-Lactam-Based Antimicrobials: Beyond the Usual Suspects

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
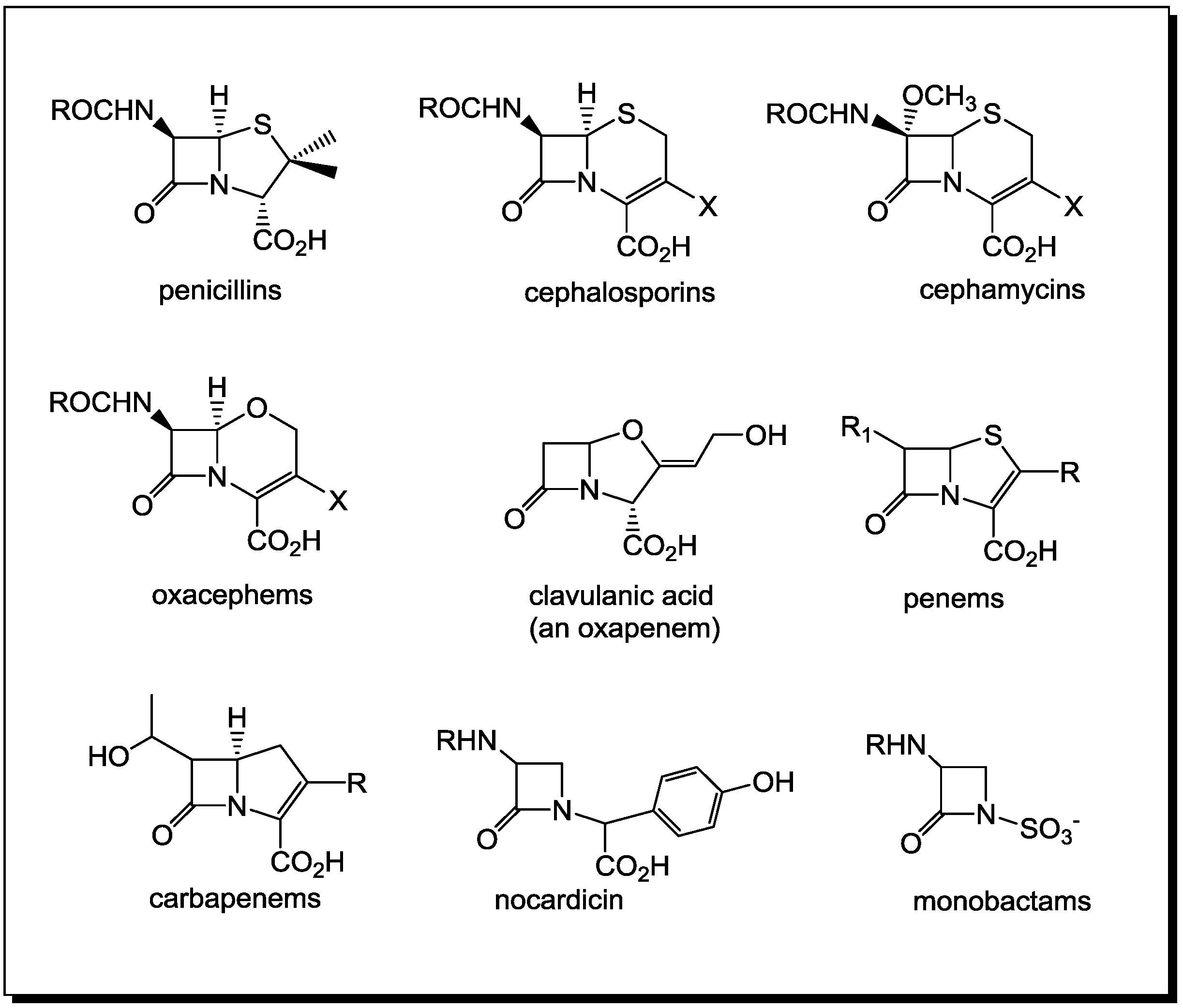
Ingenuity of the β-Lactam as the Acylating Agent

2. PBPs and β-Lactamases: the Two Main Molecular Targets for Drug Development, So Far


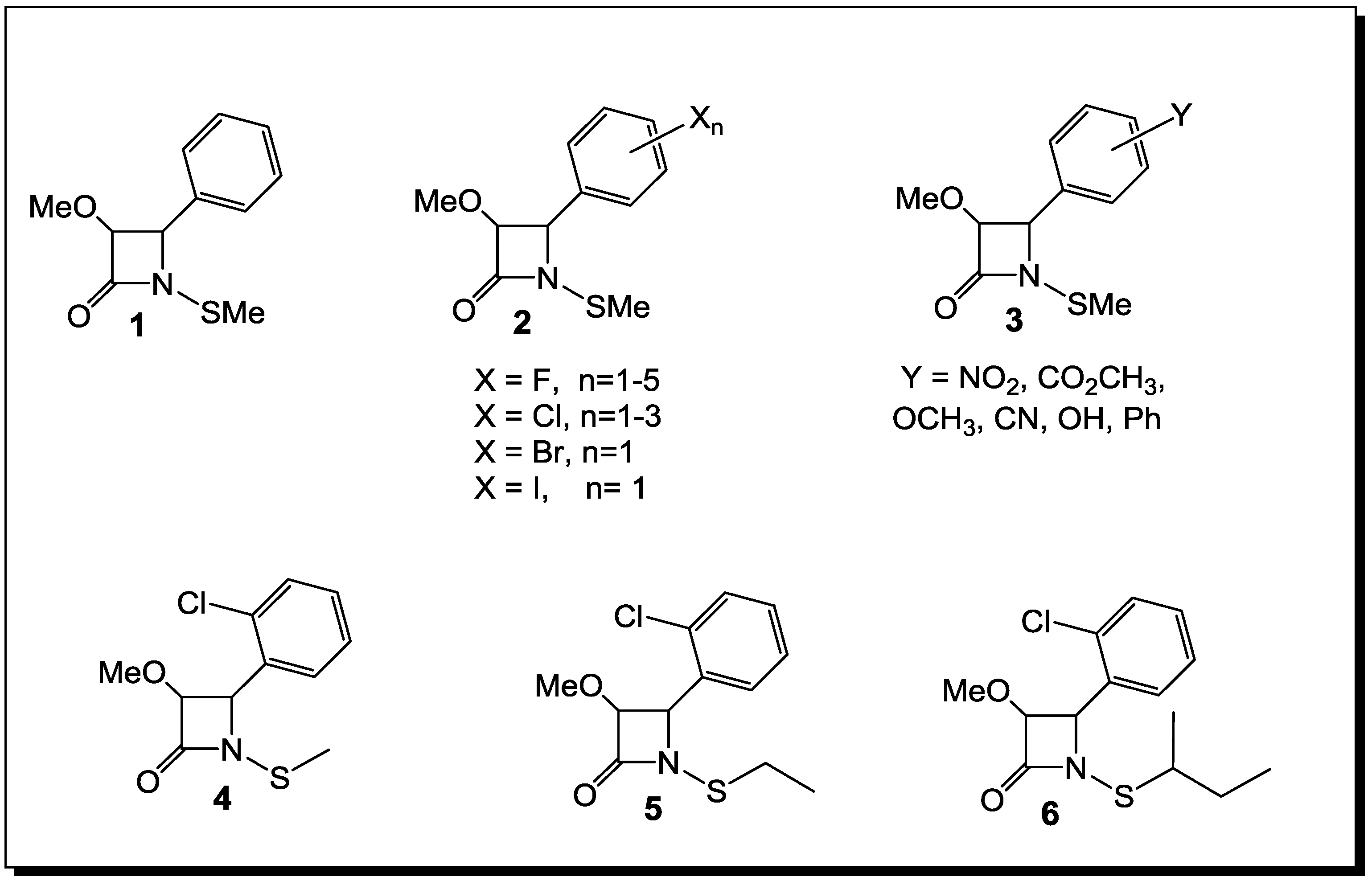
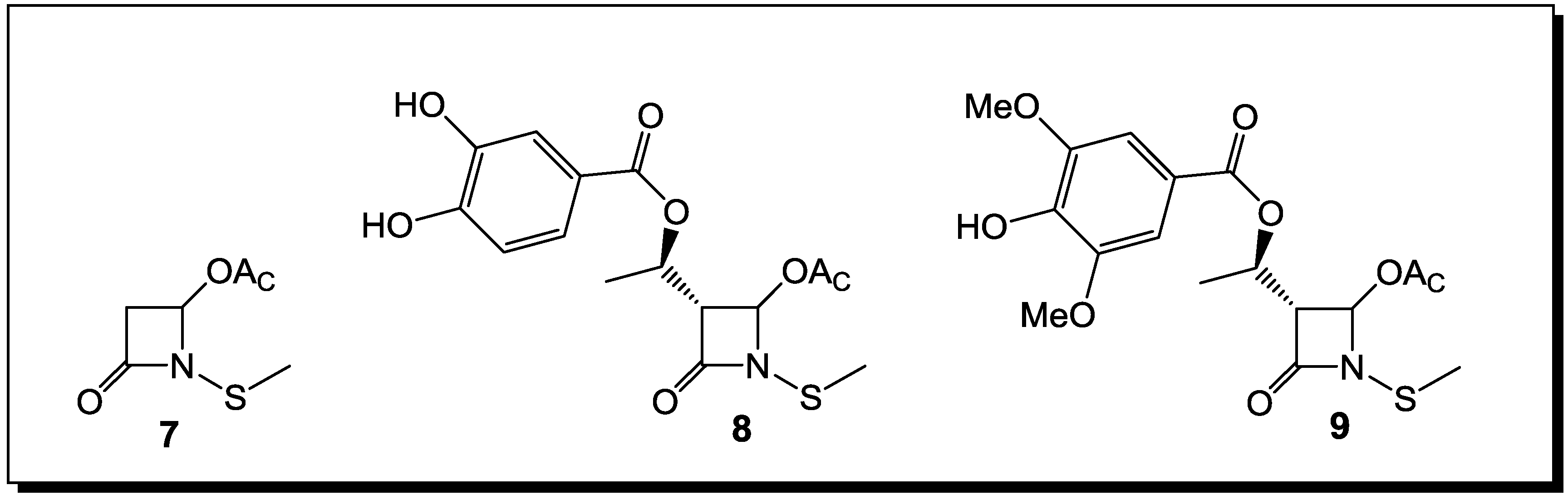
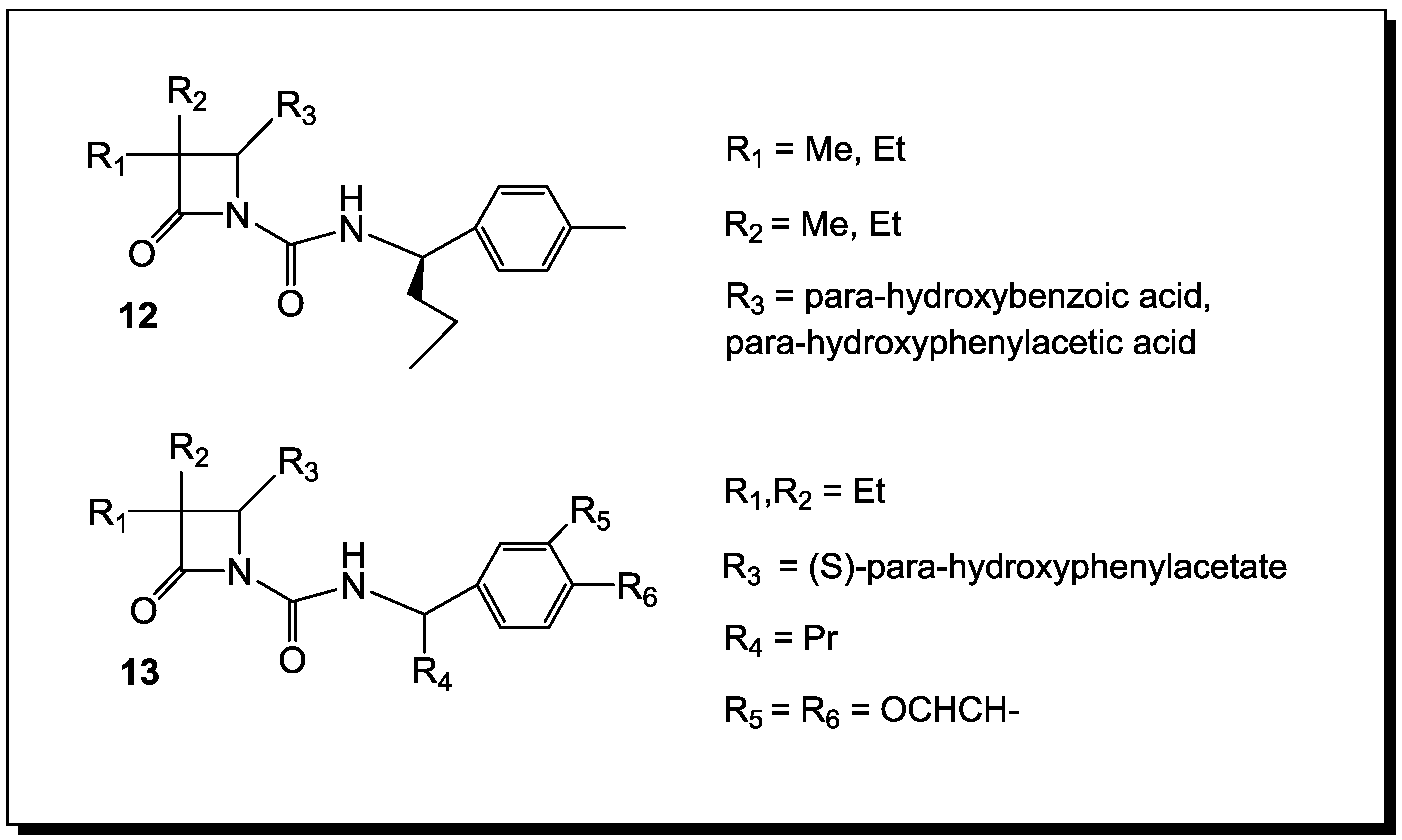
3. β-Lactams Lacking the Ionizable Residue at the Lactam Nitrogen: A New Direction of Antimicrobial Compounds against Bacteria



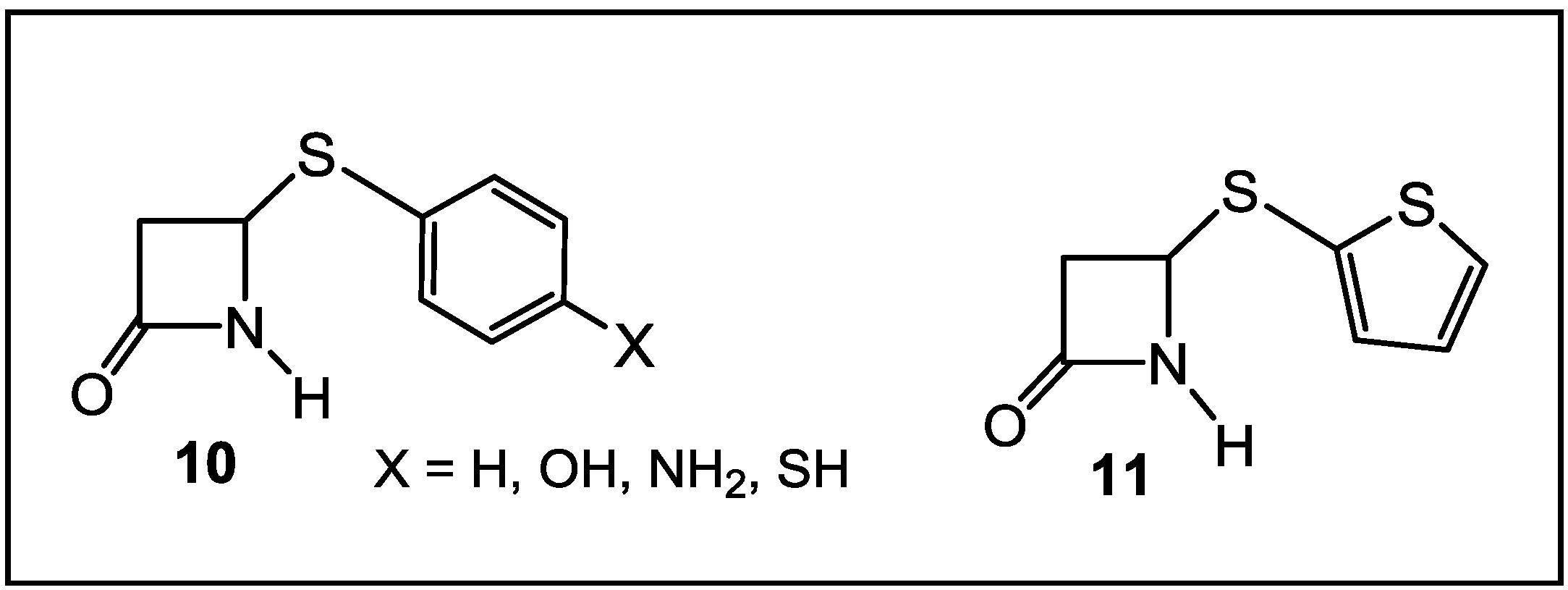
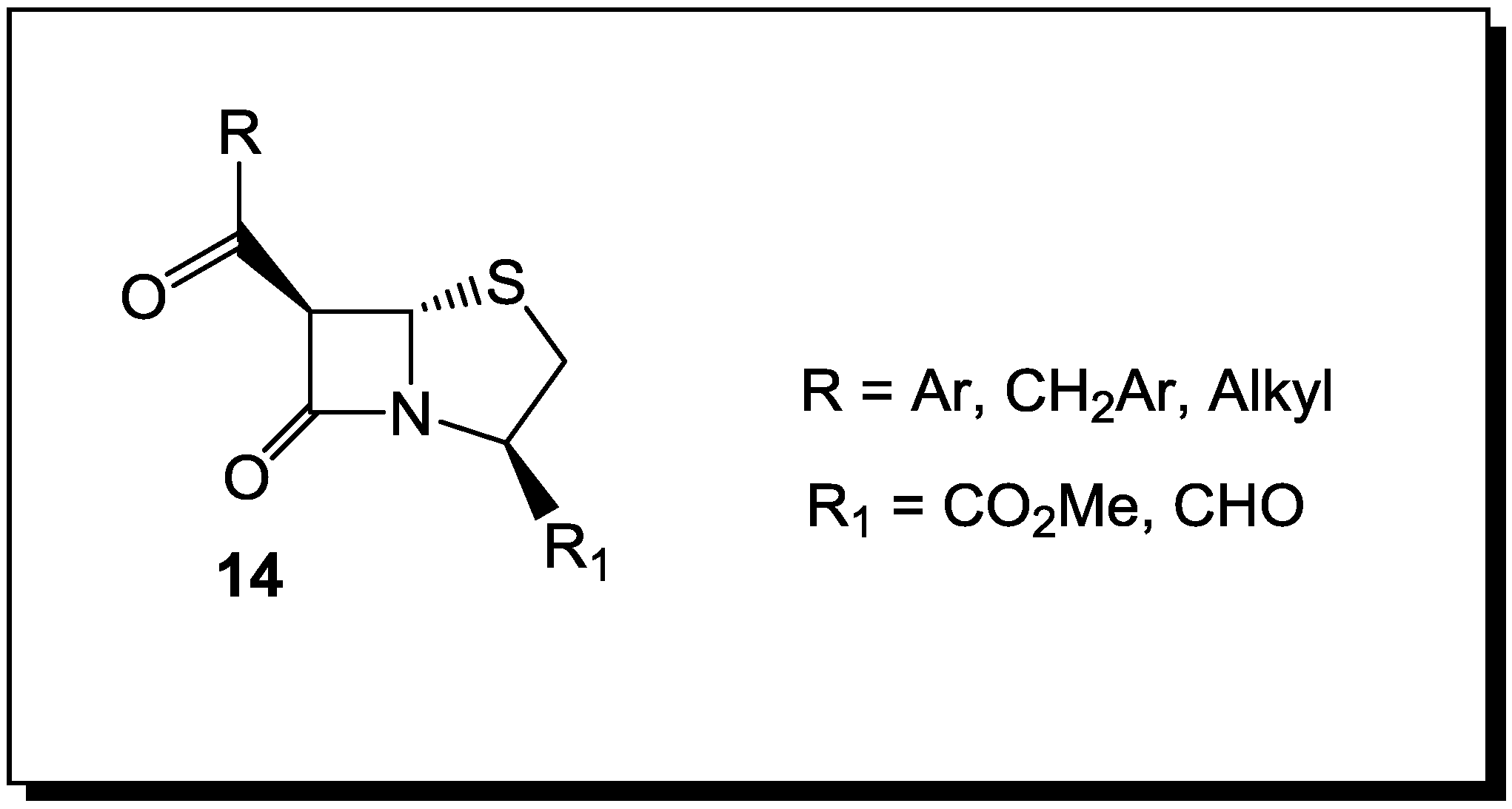
4. β-Lactams with Carboxylic Acid Bioisosteres as Leader Peptidase and Pilus Assembly Inhibitors


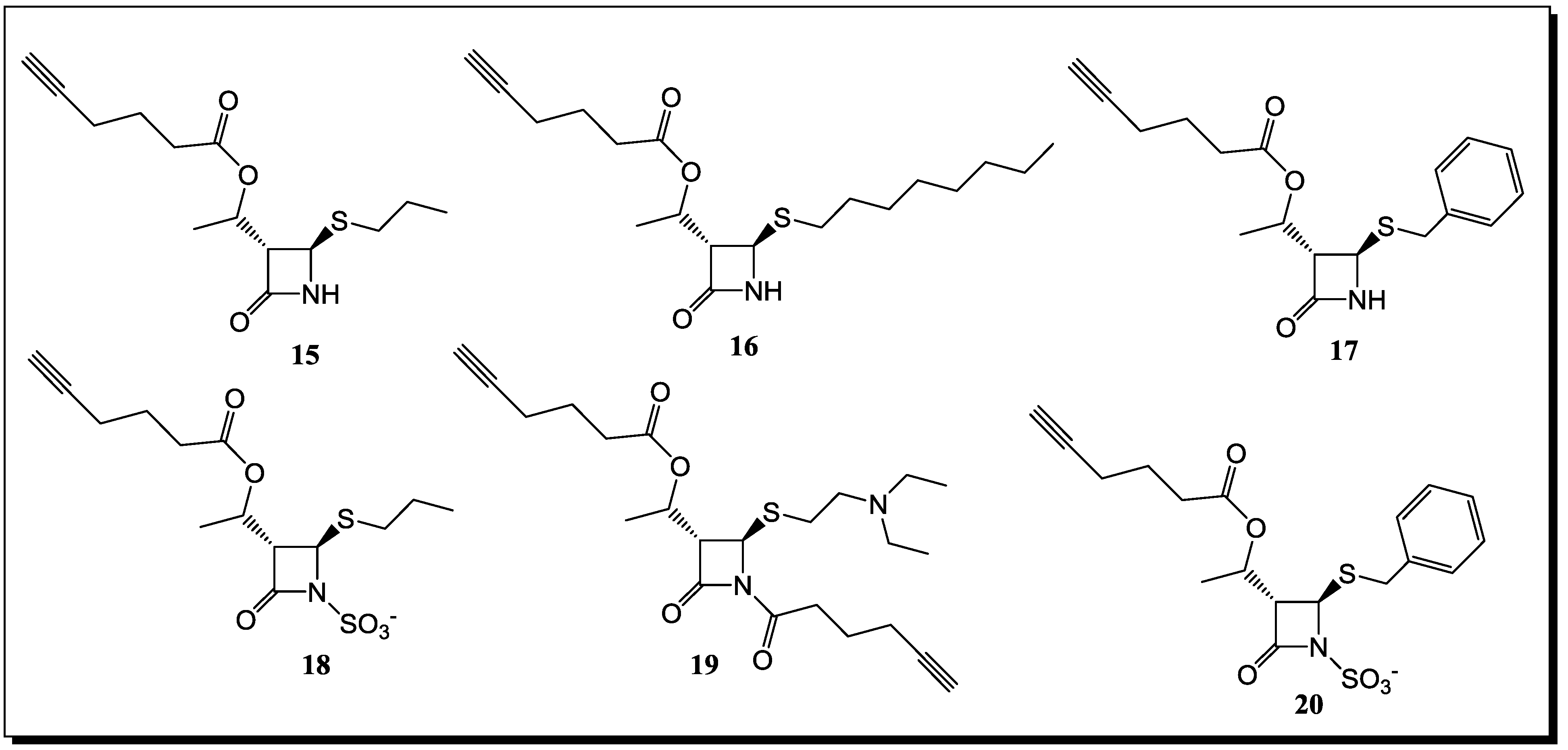
5. β-Lactams as Chemical Probes for Bacterial Enzymes

6. Conclusions
Conflicts of Interest
References
- Strominger, J.L. Enzymatic reactions in bacterial cell wall synthesis sensitive to penicillins, cephalosprins and other antibacterial agents. Antibiotics 1967, 1, 705–713. [Google Scholar] [CrossRef]
- Woodward, R.B. The Chemistry of Penicillin; Clarke, H.T., Johnson, J.R., Robinson, R., Eds.; Princeton University Press: Princeton, NJ, USA, 1949; p. 443. [Google Scholar]
- Page, M.I. The mechanisms of reactions of β-lactam antibiotics. Acc. Chem. Res. 1984, 17, 144–151. [Google Scholar] [CrossRef]
- Collings, A.J.; Jackson, P.F.; Morgan, K.J. Carbonyl group frequency. III. Thiocarboxylic S-esters. J. Chem. Soc. B 1970, 1970, 581–584. [Google Scholar] [CrossRef]
- Page, M.I. The reactivity of β-lactams, the mechanism of catalysis and the inhibition of β-lactamases. Curr. Pharm. Des. 1999, 5, 895–913. [Google Scholar]
- Rhazi, N.; Galleni, M.; Page, M.I.; Frere, J.-M. Peptidase activity of β-lactamases. Biochem. J. 1999, 341, 409–413. [Google Scholar] [CrossRef]
- Page, M.I.; Laws, A.P. The chemical reactivity of β-lactams, β-sultams and β-pospholactams. Tetrahedron 2000, 56, 5631–5638. [Google Scholar] [CrossRef]
- Page, M.I.; Laws, A.P. The mechanism of catalysis and the inhibition of β-lactamases. J. Chem. Soc. Chem. Commun. 1998, 331, 1609–1617. [Google Scholar] [CrossRef]
- Page, M.I. The mechanisms of reactions of β-lactam antibiotics. Adv. Phys. Org. Chem. 1987, 23, 165–270. [Google Scholar]
- Crowfoot, D.; Bunn, C.W.; Rogers-Low, B.W.; Turner-Jones, A. The Chemistry of Penicillin; Clarke, H.T., Johnson, J.R., Robinson, R., Eds.; Princeton University Press: Princeton, NJ, USA, 1949; pp. 310–367. [Google Scholar]
- Woodward, R.B. Recent advances in the chemistry of natural products. Science 1966, 153, 487–493. [Google Scholar]
- Mucsi, Z.; Chass, G.A.; Abranyi-Balogh, P.; Jozart, B.; Fang, D.-C.; Ramirez-Cuesta, A.J.; Viskolcz, B.; Csizmadia, I.G. Penicillin’s catalytic mechanism revealed by inelastic neutrons and quantum chemical theory. Phys. Chem. Chem. Phys. 2013, 15, 20447–20455. [Google Scholar] [CrossRef]
- Mucsi, Z.; Tsai, A.; Szori, M.; Chass, G.A.; Viscolcz, B.; Csizmadia, I.G. A quanitative scale for the extent of conjugation of the amide bond. Amidity percentage as chemical driving force. J. Phys. Chem. A 2007, 111, 13245–13254. [Google Scholar]
- Mucsi, Z.; Chass, G.A.; Csizmadia, I.G. Amidicity change as a significant driving force and thermodynamic selection rule of transamidation reactions. A synergy between experiment and theory. J. Phys. Chem. B 2008, 112, 7885–7893. [Google Scholar]
- Mucsi, Z.; Chass, G.A.; Csizmadia, I.G. Systemic energy management by strategically located functional components within molecular frameworks, determined by systems chemistry. J. Phys. Chem. B 2009, 113, 10308–10314. [Google Scholar]
- Glover, S.A.; Rosser, A.A. Reliable determination of amidicity in Acyclic Amides and Lactams. J. Org. Chem. 2012, 77, 5492–5502. [Google Scholar] [CrossRef]
- The Organic Chemistry of β-Lactams; Georg, G.I. (Ed.) VCH: New York, NY, USA, 1993.
- Chemistry and Biology of β-Lactam Antibiotics; Morin, R.B.; Gorman, M. (Eds.) Academic Press: New York, NY, USA, 1982; Volume: 1–3.
- Chin, G.J.; Marx, J. Resistance to antibiotics. Science 1994, 264, 359–393. [Google Scholar]
- Antimicrobial Drug Resistance; Bryan, L.E. (Ed.) Academic Press: New York, NY, USA, 1984.
- Gunda, E.T.; Jaszberenyi, J.C. Functional modifications and nuclear analogs of β-lactam antibiotics—Part II. Prog. Med. Chem. 1977, 14, 181–248. [Google Scholar] [CrossRef]
- Fisher, J.F.; Meroueh, S.O.; Mobashery, S. Bacterial resistance to beta-lactam antibiotics: compelling opportunism, compelling opportunity. Chem. Rev. 2005, 105, 395–424. [Google Scholar] [CrossRef]
- Llarrull, L.I.; Testero, S.A.; Fisher, J.F.; Mobashery, S. The future of the β-lactams. Curr. Opin. Microbiol. 2010, 13, 551–557. [Google Scholar] [CrossRef]
- Kong, K.F.; Schneper, L.; Mathee, K. Beta-lactam antibiotics: From antibiosis to resistance and bacteriology. APMIS 2010, 118, 1–36. [Google Scholar]
- Sassiver, M.L.; Lewis, A. Structure-Activity Relationships Among the Semisynthetic Antibiotics; Perlman, D., Ed.; Academic Press: New York, NY, USA, 1977; pp. 87–160. [Google Scholar]
- Recent Advances in the Chemistry of Anti-Infective Agents; Bentley, P.H.; Ponsford, R. (Eds.) The Royal Society of Chemistry: Cambridge, UK, 1993.
- Guthikonda, R.N.; Cama, L.D.; Quesada, M.; Woods, M.F.; Salzmann, T.N.; Christensen, B.G. Modification of natural products to improve their biological properties. Pure Appl. Chem. 1987, 59, 455–458. [Google Scholar]
- Knott-Hunziker, V.; Petursson, S.; Waley, S.G.; Jaurin, B.; Grundstroem, T. The acyl-enzyme mechanism of β-lactamase action. The evidence for C β-lactamases. Biochem. J. 1982, 207, 315–322. [Google Scholar]
- Fuad, N.; Frère, J.M.; Ghuysen, J.M.; Duez, C.; Iwatsubo, M. Mode of interaction between beta-lactam antibiotics and the exocellular DD-carboxypeptidase-transpeptidase from Streptomyces R39. Biochem. J. 1976, 155, 623–629. [Google Scholar]
- Bush, K. Excitement in the beta-lactamase arena. J. Antimicrob. Chemother. 1989, 24, 831–836. [Google Scholar]
- Tipper, D.J.; Strominger, J.L. Mechanism of action of penicillins: A proposal based on their structural similarity to acyl-d-alanyl-d-alanine. Proc. Natl. Acad. Sci. USA 1965, 54, 1113–1141. [Google Scholar]
- Kelly, J.A.; Diedeberg, O.; Charlier, P.; Wery, J.; Libert, M.; Moews, P.; Knox, J.; Duez, C.; Fraipont, C.; Joris, B.; et al. On the origin of bacterial resistance to penicillin: Comparison of a beta-lactamase and a penicillin target. Science 1986, 231, 1429–1431. [Google Scholar]
- Samraoui, B.; Sutton, B.; Todd, R.; Artimyuk, P.; Waley, S.G.; Phillips, D. Tertiary structural similarity between a class A β-lactamase and a penicillin-sensitive d-alanyl-carboxypeptidase-transpeptidase. Nature 1986, 320, 378–380. [Google Scholar] [CrossRef]
- Joris, B.; Ghuysen, J.-M.; Dive, G.; Renard, A.; Diedeberg, O.; Charlier, P.; Frere, J.-M.; Kelly, J.A.; Boyington, J.C.; Moews, P.C.; et al. The active-site serine penicillin-recognizing enzymes as members of the Streptomyces R61 DD-peptidase family. Biochem. J. 1988, 250, 313–324. [Google Scholar]
- Pratt, R.F. The Chemistry of β-Lactams; Page, M.I., Ed.; Blackie: Glasgow, UK, 1993; p. 351. [Google Scholar]
- Pratt, R.F.; Govardhan, C.P. beta-Lactamase-catalyzed hydrolysis of acyclic depsipeptides and acyl transfer to specific amino acid acceptors. Proc. Natl. Acad. Sci. USA 1984, 84, 1302–1306. [Google Scholar] [CrossRef]
- Bush, K.; Mobashery, S. Resolving the Antibiotic Paradox: Progress in Understanding Drug Resistance and Development of New Antibiotics; Rosen, B.P., Mobashery, S., Eds.; Plenum Press: New York, NY, USA, 1998; pp. 71–98. [Google Scholar]
- Bush, K.; Jacoby, G.A.; Medeiros, A.A. A functional classification scheme for beta-lactamases and its correlation with molecular structure. Antimicrob. Agents Chemother. 1995, 39, 1211–1233. [Google Scholar] [CrossRef]
- Massova, I.; Mobashery, S. Kinship and diversification of bacterial penicillin-binding proteins and β-lactamases. Antimicrob. Agents Chemother. 1998, 42, 1–17. [Google Scholar] [CrossRef]
- Massova, I.; Mobashery, S. Molecular Bases for Interactions between β-Lactam Antibiotics and β-Lactamases. Acc. Chem. Res. 1997, 30, 162–168. [Google Scholar] [CrossRef]
- Bulychev, A.; Massova, I.; Miyashita, K.; Mobashery, S. Nuances of mechanisms and their implications for evolution of the versatile β-lactamase activity: from biosynthetic enzymes to drug resistance factors. J. Am. Chem. Soc. 1997, 119, 7619–7625. [Google Scholar] [CrossRef]
- Golemi, D.; Maveyraud, L.; Vakulenko, S.; Tranier, S.; Ishiwata, A.; Kotra, L.P.; Samam, J.-P.; Mobashery, S. The first structural and mechanistic insights for class D β-lactamases: Evidence for a novel catalytic process for turnover of β-lactam antibiotics. J. Am. Chem. Soc. 2000, 122, 6132–6133. [Google Scholar] [CrossRef]
- Barrett, A.J.; Rawlings, N.D. Families and clans of serine peptidases. Arch. Biochem. Biophys. 1995, 318, 247–250. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J. Evolutionary families of peptidases. Biochem. J. 1993, 290, 205–218. [Google Scholar]
- Krem, M.M.; di Cera, E. Molecular markers of serine protease evolution. EMBO J. 2001, 20, 3036–3045. [Google Scholar] [CrossRef]
- Krem, M.M.; Rose, T.; di Cera, E. Sequence determinants of function and evolution in serine proteases. Trends Cardiovasc. Med. 2000, 10, 171–176. [Google Scholar] [CrossRef]
- Neu, H.C. The crisis of antibiotic resistance. Science 1992, 257, 1064–1072. [Google Scholar]
- Bush, K. Classification of β-lactamases: Group 1, 2a, 2b, and 2b'. Antimicrob. Agents Chemother. 1989, 33, 264–270. [Google Scholar]
- Avorn, J.L.; Barrett, J.F.; Davey, P.G.; McEwen, S.A.; O’Brien, T.F.; Levy, S.B. World Health Organization, Alliance for the Prudent Use of Antibiotics. Available online: http://whqlibdoc.who.int/hq/2000/who_cds_csr_aph_2000.4.pdf/ (accessed on 20 December 2013).
- Bush, K.; Macielag, M.; Clancy, J. “Superbugs”. New antibacterials in the pipeline. Emerg. Drugs 2000, 5, 347–365. [Google Scholar] [CrossRef]
- Levy, S.B. The Antibiotic Paradox: How Miracle Drugs Are Destroying the Miracle; Plenum Press: New York, NY, USA, 1992. [Google Scholar]
- Garett, L. The Coming Plague; Farrar, Straus and Giroux Press: New York, NY, USA, 1994; pp. 411–456. [Google Scholar]
- Cooper, M.A.; Shlaes, D. Fix the antibiotic pipeline. Nature 2011, 472, 32. [Google Scholar] [CrossRef]
- Sanders, C.C.; Sanders, W.E. beta-Lactam resistance in gram-negative bacteria: Global trends and clinical impact. Clin. Infect. Dis. 1992, 15, 824–839. [Google Scholar] [CrossRef]
- Knowles, J.R. Penicillin resistance: The chemistry of β-lactamase inhibition. Acc. Chem. Res. 1985, 18, 97–104. [Google Scholar] [CrossRef]
- Therrien, C.; Levesque, R.C. Molecular basis of antibiotic resistance and β-lactamase inhibition by mechanism-based inactivators: Perspectives and future directions. FEMS Microbiol. Rev. 2000, 24, 251–262. [Google Scholar]
- Williams, J.D. β-Lactamases and β-lactamase inhibition. Int. J. Antimicrob. Agents 1999, 12, S3–S7. [Google Scholar] [CrossRef]
- Maiti, S.N.; Phillips, O.A.; Micetich, R.G.; Livermore, D.M. Beta-lactamase inhibitors: Agents to overcome bacterial resistance. Curr. Med. Chem. 1998, 5, 441–456. [Google Scholar]
- Zhanel, G.G.; Lawson, C.D.; Adam, H.; Schweizer, F.; Zelenitsky, S.; Lagace-Wiens, P.R.S.; Denisuik, A.; Rubinstein, E.; Gin, A.S.; Hoban, D.J.; et al. Ceftazidime-Avibactam: A novel cephalosporin/β-lactamase inhibitor combination. Drugs 2013, 73, 159–177. [Google Scholar] [CrossRef]
- Chen, J.; Shang, X.; Hu, F.; Lao, X.; Gao, X.; Zheng, H.; Yao, W. β-Lactamase inhibition: An update. Mini Rev. Med. Chem. 2013, 13, 1846–1861. [Google Scholar] [CrossRef]
- Pratt, R.F. The Chemistry of β-Lactams; Page, M.I., Ed.; Blackie Academic & Professional: Glasgow, UK, 1993; pp. 229–271. [Google Scholar]
- Buynak, J.D.; Doppalapudi, V.R.; Frotan, M.; Kumar, R.; Chambers, A. Catalytic approaches to the synthesis of β-lactamase inhibitors. Tetrahedron 2000, 56, 5709–5718. [Google Scholar] [CrossRef]
- Medeiros, A.A. Evolution and dissemination of beta-lactamases accelerated by generations of beta-lactam antibiotics. Rev. Infect. Dis. 1997, 24, S19–S45. [Google Scholar] [CrossRef]
- Leonard, D.A.; Bonomo, R.A.; Powers, R.A. Class D β-Lactamases: A reappraisal after five decades. Acc. Chem. Res. 2013, 46, 2407–2415. [Google Scholar] [CrossRef]
- Gutkind, G.O.; di Conza, J.; Power, P.; Radice, M. β-lactamase-mediated resistance: A biochemical, epidemiological and genetic overview. Curr. Pharm. Des. 2013, 19, 164–208. [Google Scholar] [CrossRef]
- Worthington, R.J.; Melander, C. Overcoming resistance to β-lactam antibiotics. J. Org. Chem. 2013, 78, 4207–4213. [Google Scholar] [CrossRef]
- Bush, K. Improving known classes of antibiotics: An optimistic approach for the future. Curr. Opin. Pharmacol. 2012, 12, 527–534. [Google Scholar] [CrossRef]
- Shlaes, D.M. New β-lactam-β-lactamase inhibitor combinations in clinical development. Ann. NY Acad. Sci. 2013, 1277, 105–114. [Google Scholar]
- Turos, E.; Konaklieva, M.I.; Ren, R.; Shi, H.; Gonzalez, J.; Dickey, S.; Lim, D.V. N-thiolated bicyclic and monocyclic β-lactams. Tetrahedron 2000, 56, 5571–5578. [Google Scholar] [CrossRef]
- Mehta, P.D.; Sengar, N.P.S.; Pathak, A.K. 2-Azetidinone—A new profile of various pharmacological activities. Eur. J. Med. Chem. 2010, 45, 5541–5560. [Google Scholar] [CrossRef]
- Galletti, P.; Giacomini, D. Monocyclic β-lactams: New structures for new biological activities. Curr. Med. Chem. 2011, 18, 4265–4283. [Google Scholar] [CrossRef]
- Toraskar, M.; Kulkarni, V.; Kadam, V. Azetidinone: A bioactive moiety. J. Pharm. Res. 2010, 3, 169–173. [Google Scholar]
- Turos, E.; Long, T.E.; Konaklieva, M.I.; Coates, C.; Shim, Y.-J.; Dickey, S.; Lim, D.V.; Cannons, A. N-Thiolated β-Lactams: Novel antibacterial agents for methicillin-resistant Staphylococcus aureus. Bioorg. Med. Chem. Lett. 2002, 12, 2229–2231. [Google Scholar] [CrossRef]
- Long, T.E.; Turos, E.; Konaklieva, M.I.; Blum, A.E.; Amry, A.; Baker, E.A.; Suwandi, L.S.; McCain, M.D.; Rahman, M.F.; Dickey, S.; et al. Effect of aryl ring fluorination on the antibacterial properties of C4 aryl-substituted N-methylthio β-Lactams. Bioorg. Med. Chem. 2003, 11, 1859–1863. [Google Scholar] [CrossRef]
- Bhattacharya, B.; Turos, E. Synthesis and biology of N-thiolated β-lactams. Tetrahedron 2012, 68, 10665–10685. [Google Scholar] [CrossRef]
- Revell, K.D.; Heldreth, B.; Long, T.E.; Jang, S.; Turos, E. N-thiolated β-lactams: Studies on the mode of action and identification of a primary cellular target in Staphylococcus aureus. Biorg. Med. Chem. 2007, 15, 2453–2467. [Google Scholar] [CrossRef]
- Galletti, P.; Quintayalla, A.; Ventrici, C.; Giannini, G.; Cabri, W.; Penco, S.; Gallo, G.; Vincenti, S.; Giacomini, D. Azetidinones as zinc-binding groups to design selective HDAC8 inhibitors. Chem. Med. Chem. 2009, 4, 1991–2201. [Google Scholar] [CrossRef]
- Cervellati, R.; Galletti, P.; Greco, E.; Cocuzza, C.E.A.; Musumeci, R.; Bardini, L.; Paolucci, F.; Pori, M.; Soldati, R.; Giacomini, D. Monocyclic β-lactams as antibacterial agents: Facing antioxidant activity of N-methylthio-azetidinones. Eur. J. Med. Chem. 2013, 60, 340–349. [Google Scholar] [CrossRef]
- Kostova, M.B.; Myers, C.J.; Beck, T.N.; Plotkin, B.J.; Green, J.M.; Boshoff, H.I.; Barry, C.E., III; Deschamps, J.; Konaklieva, M.I. C4-alkylthiols with activity against Moraxella catarrhalis and Mycobacterium tuberculosis. Bioorg. Med. Chem. 2011, 19, 6842–6852. [Google Scholar] [CrossRef]
- Randall, L.L.; Hardy, J.S.; Thom, J.R. Export of protein: a biochemical view. Annu. Rev. Microb. 1987, 41, 507–541. [Google Scholar] [CrossRef]
- Kuo, D.; Weidner, J.; Griffin, P.; Shah, S.K.; Knight, W.B. Determination of the kinetic parameters of Escherichia coli leader peptidase activity using a continuous assay: The pH dependence and time-dependent inhibition by β-lactams are consistent with a novel serine protease mechanism. Biochemistry 1994, 33, 8347–8354. [Google Scholar] [CrossRef]
- Hultgren, S.J.; Abraham, S.; Caparon, M.; Falk, P.; St. Geme, J.W., III; Normark, S. Pilus and nonpilus bacterial adhesins: Assembly and function in cell recognition. Cell 1993, 73, 887–901. [Google Scholar] [CrossRef]
- Emtenas, H.; Soto, G.; Hultgren, S.J.; Marshall, G.R.; Almqvist, F. Stereoselective synthesis of optically active β-lactams, potential inhibitors of pilus assembly in pathogenic bacteria. Org. Lett. 2000, 2, 2065–2067. [Google Scholar] [CrossRef]
- Flemmer Karlsson, K.; Walse, B.; Drakenberg, T.; Roy, S.; Bergqust, K.-E.; Pinkner, J.S.; Hultgren, S.J.; Kihlberg, J. Peptides inhibit complexation of the bacterial chaperone PapD and reveal potential to block assembly of virulence associated pili. Bioorg. Med. Chem. 1995, 5, 927–932. [Google Scholar] [CrossRef]
- Staub, I.; Sieber, S.A. β-Lactams as selective chemical probes for the in vivo labeling of bacterial enzymes involved in cell wall biosynthesis, antibiotic resistance, and virulence. J. Am. Chem. Soc. 2008, 130, 13400–13409. [Google Scholar] [CrossRef]
- Staub, I.; Sieber, S.A. β-Lactam Probes as selective chemical-proteomic tools for the identification and functional characterization of resistance associated enzymes in MRSA. J. Am. Chem. Soc. 2009, 131, 6271–6276. [Google Scholar] [CrossRef]
- Wang, J.; Kodali, S.; Lee, S.H.; Galgoci, A.; Painter, R.; Dorso, K.; Racine, F.; Motyl, M.; Hernandez, L.; Tinney, E.; et al. Discovery of platencin, a dual FabF and FabH inhibitor with in vivo antibiotic properties. Proc. Natl. Acad. Sci. USA 2007, 104, 7612–7616. [Google Scholar] [CrossRef]
- Chae, H.Z.; Robison, K.; Poole, L.B.; Church, G.; Storz, G.; Rhee, S.G. Cloning and sequencing of thiol-specific antioxidant from mammalian brain: Alkyl hydroperoxide reductase and thiol-specific antioxidant define a large family of antioxidant enzymes. Proc. Natl. Acad. Sci. USA 1994, 91, 7017–7021. [Google Scholar]
- Michel, A.; Agerer, F.; Hauck, C.R.; Herrmann, M.; Ullrich, J.; Hacker, J.; Ohlsen, K. Global regulatory impact of ClpP protease of Staphylococcus aureus on regulons involved in virulence, oxidative stress response, autolysis, and DNA repair. J. Bacteriol. 2006, 188, 5783–5796. [Google Scholar] [CrossRef]
- Conlon, B.P.; Nakayasu, E.S.; Fleck, L.E.; LaFleur, M.D.; Isabella, V.M.; Coleman, K.; Leonard, S.N.; Smith, R.D.; Adkins, J.N.; Lewis, K. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 2013, 503, 365–370. [Google Scholar] [CrossRef]
- Brötz-Oesterhelt, H.; Sass, P. Bacterial caseinolytic proteases as novel targets for antibacterial treatment. Int. J. Med. Microbiol. 2014, 304, 23–30. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Konaklieva, M.I. Molecular Targets of β-Lactam-Based Antimicrobials: Beyond the Usual Suspects. Antibiotics 2014, 3, 128-142. https://doi.org/10.3390/antibiotics3020128
Konaklieva MI. Molecular Targets of β-Lactam-Based Antimicrobials: Beyond the Usual Suspects. Antibiotics. 2014; 3(2):128-142. https://doi.org/10.3390/antibiotics3020128
Chicago/Turabian StyleKonaklieva, Monika I. 2014. "Molecular Targets of β-Lactam-Based Antimicrobials: Beyond the Usual Suspects" Antibiotics 3, no. 2: 128-142. https://doi.org/10.3390/antibiotics3020128