
Antimicrobial Resistance and Reduced Susceptibility in Clostridium difficile: Potential Consequences for Induction, Treatment, and Recurrence of C. difficile Infection

Abstract
:1. Introduction

2. Ribosomally Active Antimicrobial Agents
2.1. Clindamycin
2.2. Erythromycin
2.3. Tetracyclines
2.4. Linezolid
3. DNA and DNA/RNA Accessory Enzyme Inhibitors
3.1. Fluoroquinolones
3.2. Rifamycins
3.3. Fidaxomicin
3.4. Metronidazole

{kind=link}
{kind=link}
| Gene/Protein implicated | Potential Contribution to Metronidazole Resistance | Ref. |
|---|---|---|
| Ferric uptake regulator (fur) | Point mutation could lead to altered binding of Fur to SOD therefore reduced oxidative stress in C. difficile in response to MTZ exposure. | [97,98] |
| Putative nitroreductase | Frameshift mutation could affect activation of MTZ. | [98] |
| Coproporphyrinogen III Oxidase (hemN) | Frameshift mutation could disrupt heme biosynthesis/metabolism, defective electron transport and reduced MTZ activation. | [98] |
| Ferritin | Absence in MTZR strain under MTZ pressure therefore deficient iron storage | [99] |
| Butyryl CoA dehydrogenase (Bcd) | Significant reduction under MTZ pressure, therefore possible reduced ferredoxin reduction and consequent reduction in MTZ activation. | [99] |
| Ferredoxin (2 proteins) | Reduced expression in MTZR and revertant strains, possible reduction in MTZ activation. Another ferredoxin protein was increased in expression in MTZR and revertant strains. Unclear significance. | [97] |
4. Cell Wall Synthesis Inhibitors
4.1. Vancomycin
4.2. Penicillins
5. Antimicrobial Susceptibility of the Indigenous Gut Microflora
5.1. Antimicrobial Agents Associated with CDI Induction
| Negatively Impacted Populations | Positively Impacted Populations | Refs | ||||
|---|---|---|---|---|---|---|
| Antimicrobial Agent | Anaerobes | Facultative Anaerobes | Anaerobes | Facultative Anaerobes | ||
| Common inducers of CDI | Clindamycin | Bifidobacteria, Bacteroides, Eubacteria, Clostridia | Lactobacilli | No effect | Enterobacteria, Enterococci | [53,107,110,111,112,114,117,130] |
| Ciprofloxacin | Anaerobes overall, Bifidobacteria, Bacteroides spp., Clostridia | E. coli (LFE), Lactobacilli, Enterococci | No effect | Enterococci (PD) | [62,131] | |
| Moxifloxacin | Bifidobacteria, Bacteroides fragilis group, Clostridia | LFE, Enterococci | No effect | Enterococci (PD) | [62] | |
| Levofloxacin * | Bifidobacteria, Bacteroides fragilis group | LFE, Enterococci, Lactobacilli | No effect | Facultative anaerobes overall | [62] | |
| Co-amoxyclav | Bifidobacteria, Bacteroides fragilis group, Clostridia | No effect | No effect | Enterococci, LFE | [132] | |
| Infrequent inducers of CDI | Piperacillin tazobactam | Bifidobacteria, Anaerobic cocci | Lactobacilli, Enterococci | No effect | Enterococci, Lactobacilli, Clostridia (PD) | [133] |
| Piperacillin tazobactam | Anaerobes overall, Bifidobacteria, Bacteroides fragilis group, | Lactobacilli, LFE, | No Effect | Enterococci, Lactobacilli, Clostridia (PD) | [108] | |
| Mecillinam | Bifidobacteria | LFE | No effect | No effect | [113] | |
| Erythromycin | Bifidobacteria, Bacteroides, Clostridia | E. coli, Streptococci, Lactobacilli, Enterococci | Eubacteria | No effect | [134] | |
5.2. Antimicrobial Agents to Treat CDI: Metronidazole, Vancomycin, and Fidaxomicin
| Antimicrobial Agent | Negatively Impacted Populations | Positively Impacted Populations | Refs | ||
|---|---|---|---|---|---|
| Anaerobes | Facultative Anaerobes | Anaerobes | Facultative Anaerobes | ||
| Vancomycin | Bifidobacteria, Bacteroides, Clostridia | Lactobacilli, Enterococci | No effect | LFE PD. Lactobacilli (PD) | [111,151] |
| Metronidazole+ | Bifidobacteria, Bacteroides, Clostridia | No effect (one study E. coli) | No effect | LFE | [110,152] |
| Fidaxomicin | Bifidobacteria | Enterococci | No effect | LFE | [146] |
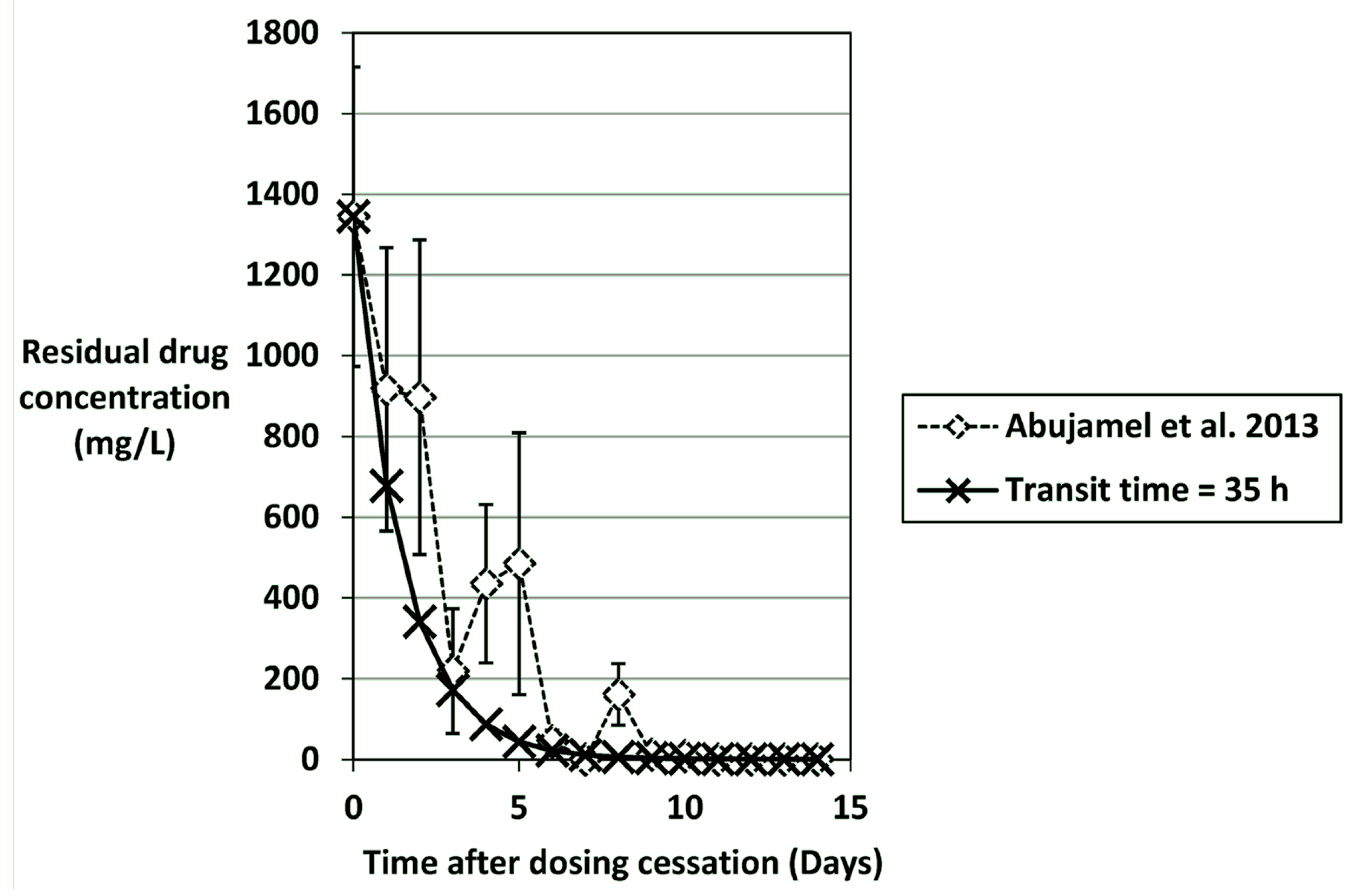
6. Role of C. difficile Antimicrobial Susceptibility in CDI Induction and Treatment

| Antimicrobial Agent | Representative Steady-State Concentration (mg/L) | Washout Time (days) to Achieve Residual Antimicrobial Concentration | Faecal/bile (B) Concentrations in vivo (mg/kg) | Refs | |||||
|---|---|---|---|---|---|---|---|---|---|
| 32 mg/L | 16 mg/L | 8 mg/L | 4 mg/L | 2 mg/L | 1 mg/L | ||||
| Metronidazole | 15 | NR | NR | 1 | 2 | 3 | 4 | 0, 9.3, 26 | [123,144,148,149,162] |
| Vancomycin Fidaxomicin | 1350 1400 | 6 | 7 | 8 | 9 | 10 | 11 | 1345,1406 1396 | [123,150] [154] |
| Clindamycin Ciprofloxacin | 150 | 3 | 4 | 5 | 6 | 7 | 8 | 33.9(B), 97, 147.4, 203.8 136.8, 168.5, 891 | [130,163,164,165] [131,165,166] |
| Erythromycin Moxifloxacin | 500 | 5 | 6 | 7 | 8 | 9 | 10 | 330, 978 573.3 | [134,164] [167] |
| Rifaximin | 8000 | 9 | 10 | 11 | 12 | 13 | 14 | 7961 | [168] |
7. Conclusions
Author Contributions
Conflicts of Interest
References
- Bartlett, J.G.; Moon, N.; Chang, T.W.; Taylor, N.; Onderdonk, A.B. Role of Clostridium difficile in antibiotic-associated pseudomembranous colitis. Gastroenterology 1978, 75, 778–782. [Google Scholar] [PubMed]
- George, R.H.; Symonds, J.M.; Dimock, F.; Brown, J.D.; Arabi, Y.; Shinagawa, N.; Keighley, M.R.; Alexander-Williams, J.; Burdon, D.W. Identification of Clostridium difficile as a cause of pseudomembranous colitis. Br. Med. J. 1978, 1, 695. [Google Scholar] [CrossRef] [PubMed]
- Owens, R.C., Jr.; Donskey, C.J.; Gaynes, R.P.; Loo, V.G.; Muto, C.A. Antimicrobial-associated risk factors for Clostridium difficile infection. Clin. Infect. Dis. 2008, 46, S19–S31. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.A.; Lahue, B.J.; Caro, J.J.; Davidson, D.M. The emerging infectious challenge of Clostridium difficile-associated disease in Massachusetts hospitals: Clinical and economic consequences. Infect. Control Hosp. Epidemiol. 2007, 28, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Kyne, L.; Hamel, M.B.; Polavaram, R.; Kelly, C.P. Health care costs and mortality associated with nosocomial diarrhea due to Clostridium difficile. Clin. Infect. Dis. 2002, 34, 346–353. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Miyajima, F.; Roberts, P.; Ellison, L.; Pickard, D.J.; Martin, M.J.; Connor, T.R.; Harris, S.R.; Fairley, D.; Bamford, K.B.; et al. Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nat. Genet. 2013, 45, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, F.J.; Barton, R.W.; Alpers, D.H. Clindamycin-associated colitis. A prospective study. Ann. Intern. Med. 1974, 81, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Fawley, W.N.; Parnell, P.; Verity, P.; Freeman, J.; Wilcox, M.H. Molecular epidemiology of endemic Clostridium difficile infection and the significance of subtypes of the United Kingdom epidemic strain (PCR ribotype 1). J. Clin. Microbiol. 2005, 43, 2685–2696. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.; Vernon, J.; Morris, K.; Nicholson, S.; Todhunter, S.; Longshaw, C.; Wilcox, M.H. Pan-European longitudinal surveillance of antibiotic resistance among prevalent Clostridium difficile ribotypes. Clin. Microbiol. Infect. 2015, 21. [Google Scholar] [CrossRef] [PubMed]
- Reil, M.; Hensgens, M.P.; Kuijper, E.J.; Jakobiak, T.; Gruber, H.; Kist, M.; Borgmann, S. Seasonality of Clostridium difficile infections in Southern Germany. Epidemiol. Infect. 2012, 140, 1787–1793. [Google Scholar] [CrossRef] [PubMed]
- Terhes, G.; Maruyama, A.; Latkoczy, K.; Szikra, L.; Konkoly-Thege, M.; Princz, G.; Nagy, E.; Urban, E. In vitro antibiotic susceptibility profile of Clostridium difficile excluding PCR ribotype 027 outbreak strain in Hungary. Anaerobe 2014, 30C, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Spigaglia, P.; Barbanti, F.; Mastrantonio, P. Multidrug resistance in European Clostridium difficile clinical isolates. J. Antimicrob. Chemother. 2011, 66, 2227–2234. [Google Scholar] [CrossRef] [PubMed]
- Lachowicz, D.; Pituch, H.; Obuch-Woszczatynski, P. Antimicrobial susceptibility patterns of Clostridium difficile strains belonging to different polymerase chain reaction ribotypes isolated in Poland in 2012. Anaerobe 2015, 31, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, Y.; Lee, K.; Riley, T.V.; Kim, H. The changes of PCR ribotype and antimicrobial resistance of Clostridium difficile in a tertiary care hospital over 10 years. J. Med. Microbiol. 2014, 63, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Fang, H.; Weintraub, A.; Nord, C.E. Distinct ribotypes and rates of antimicrobial drug resistance in Clostridium difficile from Shanghai and Stockholm. Clin. Microbiol. Infect. 2009, 15, 1170–1173. [Google Scholar] [CrossRef] [PubMed]
- Usui, M.; Nanbu, Y.; Oka, K.; Takahashi, M.; Inamatsu, T.; Asai, T.; Kamiya, S.; Tamura, Y. Genetic relatedness between Japanese and European isolates of Clostridium difficile originating from piglets and their risk associated with human health. Front. Microbiol. 2014, 5, e513. [Google Scholar] [CrossRef] [PubMed]
- Pelaez, T.; Alcala, L.; Blanco, J.L.; Alvarez-Perez, S.; Marin, M.; Martin-Lopez, A.; Catalan, P.; Reigadas, E.; Garcia, M.E.; Bouza, E. Characterization of swine isolates of Clostridium difficile in Spain: A potential source of epidemic multidrug resistant strains? Anaerobe 2013, 22, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Pirs, T.; Avbersek, J.; Zdovc, I.; Krt, B.; Andlovic, A.; Lejko-Zupanc, T.; Rupnik, M.; Ocepek, M. Antimicrobial susceptibility of animal and human isolates of Clostridium difficile by broth microdilution. J. Med. Microbiol. 2013, 62, 1478–1485. [Google Scholar] [CrossRef] [PubMed]
- Spigaglia, P.; Mastrantonio, P. Comparative analysis of Clostridium difficile clinical isolates belonging to different genetic lineages and time periods. J. Med. Microbiol. 2004, 53, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Spigaglia, P.; Drigo, I.; Barbanti, F.; Mastrantonio, P.; Bano, L.; Bacchin, C.; Puiatti, C.; Tonon, E.; Berto, G.; Agnoletti, F. Antibiotic resistance patterns and PCR-ribotyping of Clostridium difficile strains isolated from swine and dogs in Italy. Anaerobe 2015, 31, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Solomon, K.; Fanning, S.; McDermott, S.; Murray, S.; Scott, L.; Martin, A.; Skally, M.; Burns, K.; Kuijper, E.; Fitzpatrick, F.; et al. PCR ribotype prevalence and molecular basis of macrolide-lincosamide-streptogramin B (MLSB) and fluoroquinolone resistance in Irish clinical Clostridium difficile isolates. J. Antimicrob. Chemother. 2011, 66, 1976–1982. [Google Scholar] [CrossRef] [PubMed]
- Tenover, F.C.; Tickler, I.A.; Persing, D.H. Antimicrobial-resistant strains of Clostridium difficile from North America. Antimicrob. Agents Chemother. 2012, 56, 2929–2932. [Google Scholar] [CrossRef] [PubMed]
- Goudarzi, M.; Goudarzi, H.; Alebouyeh, M.; Azimi Rad, M.; Shayegan Mehr, F.S.; Zali, M.R.; Aslani, M.M. Antimicrobial susceptibility of Clostridium difficile clinical isolates in Iran. Iran. Red Crescent Med. J. 2013, 15, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Kuijper, E.J.; Barbut, F.; Brazier, J.S.; Kleinkauf, N.; Eckmanns, T.; Lambert, M.L.; Drudy, D.; Fitzpatrick, F.; Wiuff, C.; Brown, D.J.; et al. Update of Clostridium difficile infection due to PCR ribotype 027 in Europe, 2008. Euro Surveill. 2008, 13, 1–7. [Google Scholar]
- Bacci, S.; St-Martin, G.; Olesen, B.; Bruun, B.; Olsen, K.E.; Nielsen, E.M.; Molbak, K. Outbreak of Clostridium difficile 027 in North Zealand, Denmark, 2008–2009. Euro Surveill. 2009, 14, 1–3. [Google Scholar]
- Johnson, S.; Samore, M.H.; Farrow, K.A.; Killgore, G.E.; Tenover, F.C.; Lyras, D.; Rood, J.I.; DeGirolami, P.; Baltch, A.L.; Rafferty, M.E.; et al. Epidemics of diarrhea caused by a clindamycin-resistant strain of Clostridium difficile in four hospitals. N. Engl. J. Med. 1999, 341, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Farrow, K.A.; Lyras, D.; Rood, J.I. Genomic analysis of the erythromycin resistance element Tn5398 from Clostridium difficile. Microbiology 2001, 147, 2717–2128. [Google Scholar] [PubMed]
- Spigaglia, P.; Carucci, V.; Barbanti, F.; Mastrantonio, P. ErmB determinants and Tn916-Like elements in clinical isolates of Clostridium difficile. Antimicrob. Agents Chemother. 2005, 49, 2550–2553. [Google Scholar] [CrossRef] [PubMed]
- Tang-Feldman, Y.J.; Henderson, J.P.; Ackermann, G.; Feldman, S.S.; Bedley, M.; Silva, J., Jr.; Cohen, S.H. Prevalence of the ermB gene in Clostridium difficile strains isolated at a university teaching hospital from 1987 through 1998. Clin. Infect. Dis. 2005, 40, 1537–1540. [Google Scholar] [CrossRef] [PubMed]
- Spigaglia, P.; Barbanti, F.; Dionisi, A.M.; Mastrantonio, P. Clostridium difficile isolates resistant to fluoroquinolones in Italy: Emergence of PCR ribotype 018. J. Clin. Microbiol. 2010, 48, 2892–2896. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Loffler, B.; Ackermann, G. Antimicrobial phenotypes and molecular basis in clinical strains of Clostridium difficile. Diagn. Microbiol. Infect. Dis. 2007, 59, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Lebel, S.; Bouttier, S.; Lambert, T. The cme gene of Clostridium difficile confers multidrug resistance in Enterococcus faecalis. FEMS Microbiol. Lett. 2004, 238, 93–100. [Google Scholar] [PubMed]
- Keessen, E.C.; Hensgens, M.P.; Spigaglia, P.; Barbanti, F.; Sanders, I.M.; Kuijper, E.J.; Lipman, L.J. Antimicrobial susceptibility profiles of human and piglet Clostridium difficile PCR-ribotype 078. Antimicrob. Resist. Infect. Control 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Ilchmann, C.; Zaiss, N.H.; Speicher, A.; Christner, M.; Ackermann, G.; Rohde, H. Comparison of resistance against erythromycin and moxifloxacin, presence of binary toxin gene and PCR ribotypes in Clostridium difficile isolates from 1990 and 2008. Eur. J. Clin. Microbiol. Infect. Dis. 2010, 29, 1571–1573. [Google Scholar] [CrossRef] [PubMed]
- Wasels, F.; Spigaglia, P.; Barbanti, F.; Mastrantonio, P. Clostridium difficile erm(B)-containing elements and the burden on the in vitro fitness. J. Med. Microbiol. 2013, 62, 1461–1467. [Google Scholar] [CrossRef] [PubMed]
- Chopra, I.; Roberts, M. Tetracycline antibiotics: Mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J.; Markowitz, S.M.; Macrina, F.L. Transferable tetracycline resistance in Clostridium difficile. Antimicrob. Agents Chemother. 1981, 19, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Wust, J.; Hardegger, U. Transferable resistance to clindamycin, erythromycin, and tetracycline in Clostridium difficile. Antimicrob. Agents Chemother. 1983, 23, 784–786. [Google Scholar] [CrossRef] [PubMed]
- Jasni, A.S.; Mullany, P.; Hussain, H.; Roberts, A.P. Demonstration of conjugative transposon (Tn5397)-mediated horizontal gene transfer between Clostridium difficile and Enterococcus faecalis. Antimicrob. Agents Chemother. 2010, 54, 4924–4926. [Google Scholar] [CrossRef] [PubMed]
- Barbut, F.; Mastrantonio, P.; Delmee, M.; Brazier, J.; Kuijper, E.; Poxton, I. Prospective study of Clostridium difficile infections in Europe with phenotypic and genotypic characterisation of the isolates. Clin. Microbiol. Infect. 2007, 13, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Noren, T.; Alriksson, I.; Akerlund, T.; Burman, L.G.; Unemo, M. In vitro susceptibility to 17 antimicrobials of clinical Clostridium difficile isolates collected in 1993–2007 in Sweden. Clin. Microbiol. Infect. 2010, 16, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Weintraub, A.; Fang, H.; Nord, C.E. Antimicrobial resistance in Clostridium difficile. Int. J. Antimicrob. Agents 2009, 34, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Spigaglia, P.; Barbanti, F.; Mastrantonio, P. Tetracycline resistance gene tet(W) in the pathogenic bacterium Clostridium difficile. Antimicrob. Agents Chemother. 2008, 52, 770–773. [Google Scholar] [CrossRef] [PubMed]
- Mullany, P.; Wilks, M.; Lamb, I.; Clayton, C.; Wren, B.; Tabaqchali, S. Genetic analysis of a tetracycline resistance element from Clostridium difficile and its conjugal transfer to and from Bacillus subtilis. J. Gen. Microbiol. 1990, 136, 1343–1349. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Zhang, L.; Chen, X.; Jiang, C.; Yu, B.; Wang, X.; Peng, Y. Antimicrobial susceptibility and resistance mechanisms of clinical Clostridium difficile from a Chinese tertiary hospital. Int. J. Antimicrob. Agents 2013, 41, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Hachler, H.; Berger-Bachi, B.; Kayser, F.H. Genetic characterization of a Clostridium difficile erythromycin-clindamycin resistance determinant that is transferable to Staphylococcus aureus. Antimicrob. Agents Chemother. 1987, 31, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Corver, J.; Bakker, D.; Brouwer, M.S.; Harmanus, C.; Hensgens, M.P.; Roberts, A.P.; Lipman, L.J.; Kuijper, E.J.; van Leeuwen, H.C. Analysis of a Clostridium difficile PCR ribotype 078 100 kilobase island reveals the presence of a novel transposon, Tn6164. BMC Microbiol. 2012, 12, e130. [Google Scholar] [CrossRef] [PubMed]
- Knetsch, C.; Connor, T.; Mutreja, A.; van Dorp, S.; Sanders, I.; Browne, H.; Harris, D.; Lipman, L.; Keessen, E.; Corver, J.; et al. Whole genome sequencing reveals potential spread of Clostridium difficile between humans and farm animals in the Netherlands, 2002 to 2011. Euro Surveill. 2014, 19, 1–12. [Google Scholar] [CrossRef]
- Bakker, D.; Corver, J.; Harmanus, C.; Goorhuis, A.; Keessen, E.C.; Fawley, W.N.; Wilcox, M.H.; Kuijper, E.J. Relatedness of human and animal Clostridium difficile PCR ribotype 078 isolates determined on the basis of multilocus variable-number tandem-repeat analysis and tetracycline resistance. J. Clin. Microbiol. 2010, 48, 3744–3749. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, D.Y.; Unal, S. New antimicrobial agents for the treatment of Gram-positive bacterial infections. Clin. Microbiol. Infect. 2008, 14, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.L.; Ma, Y.; Salmi, D.B.; McIndoo, E.; Wallace, R.J.; Bryant, A.E. Impact of antibiotics on expression of virulence-associated exotoxin genes in methicillin-sensitive and methicillin-resistant Staphylococcus aureus. J. Infect. Dis. 2007, 195, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Gemmell, C.G.; Ford, C.W. Virulence factor expression by Gram-positive cocci exposed to subinhibitory concentrations of linezolid. J. Antimicrob. Chemother. 2002, 50, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Baines, S.D.; Noel, A.R.; Huscroft, G.S.; Todhunter, S.L.; O’Connor, R.; Hobbs, J.K.; Freeman, J.; Lovering, A.M.; Wilcox, M.H. Evaluation of linezolid for the treatment of Clostridium difficile infection caused by epidemic strains using an in vitro human gut model. J. Antimicrob. Chemother. 2011, 66, 1537–1546. [Google Scholar] [CrossRef] [PubMed]
- Pelaez, T.; Alonso, R.; Perez, C.; Alcala, L.; Cuevas, O.; Bouza, E. In vitro activity of linezolid against Clostridium difficile. Antimicrob. Agents Chemother. 2002, 46, 1617–1618. [Google Scholar] [CrossRef] [PubMed]
- Citron, D.M.; Merriam, C.V.; Tyrrell, K.L.; Warren, Y.A.; Fernandez, H.; Goldstein, E.J. In vitro activities of ramoplanin, teicoplanin, vancomycin, linezolid, bacitracin, and four other antimicrobials against intestinal anaerobic bacteria. Antimicrob. Agents Chemother. 2003, 47, 2334–2338. [Google Scholar] [CrossRef] [PubMed]
- Marin, M.; Martin, A.; Alcala, L.; Cercenado, E.; Iglesias, C.; Reigadas, E.; Bouza, E. Clostridium difficile isolates with high linezolid MICs harbor the multiresistance gene cfr. Antimicrob. Agents Chemother. 2015, 59, 586–589. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.U.; Dalhoff, A.; Weintraub, A.; Nord, C.E. In vitro activity of MCB3681 against Clostridium difficile strains. Anaerobe 2014, 28, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.I.; MacGowan, A.P. Development of the quinolones. J. Antimicrob. Chemother. 2003, 51, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Drudy, D.; Quinn, T.; O’Mahony, R.; Kyne, L.; O’Gaora, P.; Fanning, S. High-level resistance to moxifloxacin and gatifloxacin associated with a novel mutation in gyrB in toxin-A-negative, toxin-B-positive Clostridium difficile. J. Antimicrob. Chemother. 2006, 58, 1264–1267. [Google Scholar] [CrossRef] [PubMed]
- Dridi, L.; Tankovic, J.; Burghoffer, B.; Barbut, F.; Petit, J.C. gyrA and gyrB mutations are implicated in cross-resistance to Ciprofloxacin and moxifloxacin in Clostridium difficile. Antimicrob. Agents Chemother. 2002, 46, 3418–3421. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, G.; Tang, Y.J.; Kueper, R.; Heisig, P.; Rodloff, A.C.; Silva, J., Jr.; Cohen, S.H. Resistance to moxifloxacin in toxigenic Clostridium difficile isolates is associated with mutations in gyrA. Antimicrob. Agents Chemother. 2001, 45, 2348–2353. [Google Scholar] [CrossRef] [PubMed]
- Saxton, K.; Baines, S.D.; Freeman, J.; O’Connor, R.; Wilcox, M.H. Effects of exposure of Clostridium difficile PCR ribotypes 027 and 001 to fluoroquinolones in a human gut model. Antimicrob. Agents Chemother. 2009, 53, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Dridi, L.; Tankovic, J.; Petit, J.C. CdeA of Clostridium difficile, a new multidrug efflux transporter of the MATE family. Microb. Drug Resist. 2004, 10, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Spigaglia, P.; Barbanti, F.; Mastrantonio, P.; Brazier, J.S.; Barbut, F.; Delmee, M.; Kuijper, E.; Poxton, I.R. Fluoroquinolone resistance in Clostridium difficile isolates from a prospective study of C. difficile infections in Europe. J. Med. Microbiol. 2008, 57, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Walkty, A.; Boyd, D.A.; Gravel, D.; Hutchinson, J.; McGeer, A.; Moore, D.; Simor, A.; Suh, K.; Taylor, G.; Miller, M.; Mulvey, M.R. Molecular characterization of moxifloxacin resistance from Canadian Clostridium difficile clinical isolates. Diagn. Microbiol. Infect. Dis. 2010, 66, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Drudy, D.; Kyne, L.; O’Mahony, R.; Fanning, S. gyrA Mutations in fluoroquinolone-resistant Clostridium difficile PCR-027. Emerg. Infect. Dis. 2007, 13, 504–505. [Google Scholar] [CrossRef] [PubMed]
- Kuwata, Y.; Tanimoto, S.; Sawabe, E.; Shima, M.; Takahashi, Y.; Ushizawa, H.; Fujie, T.; Koike, R.; Tojo, N.; Kubota, T.; et al. Molecular epidemiology and antimicrobial susceptibility of Clostridium difficile isolated from a university teaching hospital in Japan. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Mena, A.; Riera, E.; López-Causapé, C.; Weber, I.; Pérez, J.L.; Oliver, A. In vivo selection of moxifloxacin-resistant Clostridium difficile. Antimicrob. Agents Chemother. 2012, 56, 2788–2789. [Google Scholar] [CrossRef] [PubMed]
- Linder, J.A.; Huang, E.S.; Steinman, M.A.; Gonzales, R.; Stafford, R.S. Fluoroquinolone prescribing in the United States: 1995 to 2002. Am. J. Med. 2005, 118, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Garey, K.W.; Salazar, M.; Shah, D.; Rodrigue, R.; DuPont, H.L. Rifamycin antibiotics for treatment of Clostridium difficile-associated diarrhea. Ann. Pharmacother. 2008, 42, 827–835. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, J.R.; Galang, M.A.; Sambol, S.P.; Hecht, D.W.; Vedantam, G.; Gerding, D.N.; Johnson, S. Rifampin and rifaximin resistance in clinical isolates of Clostridium difficile. Antimicrob. Agents Chemother. 2008, 52, 2813–2817. [Google Scholar] [CrossRef] [PubMed]
- Curry, S.R.; Marsh, J.W.; Shutt, K.A.; Muto, C.A.; O’Leary, M.M.; Saul, M.I.; Pasculle, A.W.; Harrison, L.H. High frequency of rifampin resistance identified in an epidemic Clostridium difficile clone from a large teaching hospital. Clin. Infect. Dis. 2009, 48, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.A.; Korzheva, N.; Mustaev, A.; Murakami, K.; Nair, S.; Goldfarb, A.; Darst, S.A. Structural Mechanism for Rifampicin Inhibition of Bacterial RNA Polymerase. Cell 2001, 104, 901–912. [Google Scholar] [CrossRef]
- Johnson, S.; Schriever, C.; Galang, M.; Kelly, C.P.; Gerding, D.N. Interruption of recurrent Clostridium difficile-associated diarrhea episodes by serial therapy with vancomycin and rifaximin. Clin. Infect. Dis. 2007, 44, 846–848. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Schriever, C.; Patel, U.; Patel, T.; Hecht, D.W.; Gerding, D.N. Rifaximin Redux: Treatment of recurrent Clostridium difficile infections with rifaximin immediately post-vancomycin treatment. Anaerobe 2009, 15, 290–291. [Google Scholar] [CrossRef] [PubMed]
- Carman, R.J.; Boone, J.H.; Grover, H.; Wickham, K.N.; Chen, L. In vivo selection of rifamycin resistant Clostridium difficile during rifaximin therapy. Antimicrob. Agents Chemother. 2012, 56, 6019–6020. [Google Scholar] [CrossRef] [PubMed]
- Debast, S.B.; Bauer, M.P.; Kuijper, E.J. European Society of Clinical Microbiology and Infectious Diseases: Update of the treatment guidance document for Clostridium difficile infection. Clin. Microbiol. Infect. 2014, 20, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, A.A.; Johnson, S. Fidaxomicin: A novel macrocyclic antibiotic approved for treatment of Clostridium difficile infection. Clin. Infect. Dis. 2012, 54, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Babakhani, F.; Seddon, J.; Sears, P. Comparative microbiological studies of transcription inhibitors fidaxomicin and the rifamycins in Clostridium difficile. Antimicrob. Agents Chemother. 2014, 58, 2934–2937. [Google Scholar] [CrossRef] [PubMed]
- Finegold, S.M.; Molitoris, D.; Vaisanen, M.L.; Song, Y.; Liu, C.; Bolanos, M. In vitro activities of OPT-80 and comparator drugs against intestinal bacteria. Antimicrob. Agents Chemother. 2004, 48, 4898–4902. [Google Scholar] [CrossRef] [PubMed]
- Leeds, J.A.; Sachdeva, M.; Mullin, S.; Barnes, S.W.; Ruzin, A. In vitro selection, via serial passage, of Clostridium difficile mutants with reduced susceptibility to fidaxomicin or vancomycin. J. Antimicrob. Chemother. 2014, 69, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, E.J.; Citron, D.M.; Sears, P.; Babakhani, F.; Sambol, S.P.; Gerding, D.N. Comparative susceptibilities to fidaxomicin (OPT-80) of isolates collected at baseline, recurrence, and failure from patients in two phase III trials of fidaxomicin against Clostridium difficile infection. Antimicrob. Agents Chemother. 2011, 55, 5194–5199. [Google Scholar] [CrossRef] [PubMed]
- Kaihovaara, P.; Hook-Nikanne, J.; Uusi-Oukari, M.; Kosunen, T.U.; Salaspuro, M. Flavodoxin-dependent pyruvate oxidation, acetate production and metronidazole reduction by Helicobacter pylori. J. Antimicrob. Chemother. 1998, 41, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Musher, D.M.; Aslam, S.; Logan, N.; Nallacheru, S.; Bhaila, I.; Borchert, F.; Hamill, R.J. Relatively poor outcome after treatment of Clostridium difficile colitis with metronidazole. Clinical infectious diseases 2005, 40, 1586–1590. [Google Scholar] [CrossRef] [PubMed]
- Pepin, J.; Alary, M.E.; Valiquette, L.; Raiche, E.; Ruel, J.; Fulop, K.; Godin, D.; Bourassa, C. Increasing risk of relapse after treatment of Clostridium difficile colitis in Quebec, Canada. Clin. Infect. Dis. 2005, 40, 1591–1597. [Google Scholar] [CrossRef] [PubMed]
- Baines, S.D.; O’Connor, R.; Freeman, J.; Fawley, W.N.; Harmanus, C.; Mastrantonio, P.; Kuijper, E.J.; Wilcox, M.H. Emergence of reduced susceptibility to metronidazole in Clostridium difficile. J. Antimicrob. Chemother. 2008, 62, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Pelaez, T.; Alcala, L.; Alonso, R.; Rodriguez-Creixems, M.; Garcia-Lechuz, J.M.; Bouza, E. Reassessment of Clostridium difficile susceptibility to metronidazole and vancomycin. Antimicrob. Agents Chemother. 2002, 46, 1647–1650. [Google Scholar] [CrossRef] [PubMed]
- Brazier, J.S.; Fawley, W.; Freeman, J.; Wilcox, M.H. Reduced susceptibility of Clostridium difficile to metronidazole. J. Antimicrob. Chemother. 2001, 48, 741–742. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.S.; Hansen, L.M.; Breher, J.E.; Riley, D.A.; Magdesian, K.G.; Madigan, J.E.; Tang, Y.J.; Silva, J., Jr.; Hirsh, D.C. Antimicrobial susceptibilities of equine isolates of Clostridium difficile and molecular characterization of metronidazole-resistant strains. Clin. Infect. Dis. 1997, 25, S266–S267. [Google Scholar] [CrossRef] [PubMed]
- Barbut, F.; Decre, D.; Burghoffer, B.; Lesage, D.; Delisle, F.; Lalande, V.; Delmee, M.; Avesani, V.; Sano, N.; Coudert, C.; Petit, J.C. Antimicrobial susceptibilities and serogroups of clinical strains of Clostridium difficile isolated in France in 1991 and 1997. Antimicrob. Agents Chemother. 1999, 43, 2607–2611. [Google Scholar] [PubMed]
- Pelaez, T.; Cercenado, E.; Alcala, L.; Marin, M.; Martin-Lopez, A.; Martinez-Alarcon, J.; Catalan, P.; Sanchez-Somolinos, M.; Bouza, E. Metronidazole resistance in Clostridium difficile is heterogeneous. J. Clin. Microbiol. 2008, 46, 3028–3032. [Google Scholar] [CrossRef] [PubMed]
- Moura, I.; Spigaglia, P.; Barbanti, F.; Mastrantonio, P. Analysis of metronidazole susceptibility in different Clostridium difficile PCR ribotypes. J. Antimicrob. Chemother. 2012, 68, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Purdell, J.; Fawley, W.N.; Freeman, J.; Wilcox, M.H. Investigation of outcome in cases of Clostridium difficile infection due to isolates with reduced susceptibility to metronidazole. In Proceedings of the 21st European Congress of Clinical Microbiology and Infectious Diseases, Milan, Italy, 7–10 May 2011. Abstract O499.
- Gal, M.; Brazier, J.S. Metronidazole resistance in Bacteroides spp. carrying nim genes and the selection of slow-growing metronidazole-resistant mutants. J. Antimicrob. Chemother. 2004, 54, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Husain, F.; Veeranagouda, Y.; Hsi, J.; Meggersee, R.; Abratt, V.; Wexler, H.M. Two multidrug-resistant clinical isolates of Bacteroides fragilis carry a novel metronidazole resistance nim gene (nimJ). Antimicrob. Agents Chemother. 2013, 57, 3767–3774. [Google Scholar] [CrossRef] [PubMed]
- Carlier, J.P.; Sellier, N.; Rager, M.N.; Reysset, G. Metabolism of a 5-nitroimidazole in susceptible and resistant isogenic strains of Bacteroides fragilis. Antimicrob. Agents Chemother. 1997, 41, 1495–1499. [Google Scholar] [PubMed]
- Chong, P.M.; Lynch, T.; McCorrister, S.; Kibsey, P.; Miller, M.; Gravel, D.; Westmacott, G.R.; Mulvey, M.R. Proteomic analysis of a NAP1 Clostridium difficile clinical isolate resistant to metronidazole. PLoS ONE 2014, 9, e82622. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.; Chong, P.; Zhang, J.; Hizon, R.; Du, T.; Graham, M.R.; Beniac, D.R.; Booth, T.F.; Kibsey, P.; Miller, M.; et al. Characterization of a stable, metronidazole-resistant Clostridium difficile clinical isolate. PLoS ONE 2013, 8, e53757. [Google Scholar] [CrossRef] [PubMed]
- Moura, I.; Monot, M.; Tani, C.; Spigaglia, P.; Barbanti, F.; Norais, N.; Dupuy, B.; Bouza, E.; Mastrantonio, P. Multidisciplinary analysis of a nontoxigenic Clostridium difficile strain with stable resistance to metronidazole. Antimicrob. Agents Chemother. 2014, 58, 4957–4960. [Google Scholar] [CrossRef] [PubMed]
- Ammam, F.; Meziane-Cherif, D.; Mengin-Lecreulx, D.; Blanot, D.; Patin, D.; Boneca, I.G.; Courvalin, P.; Lambert, T.; Candela, T. The functional vanGCd cluster of Clostridium difficile does not confer vancomycin resistance. Mol. Microbiol. 2013, 89, 612–625. [Google Scholar] [CrossRef] [PubMed]
- Ammam, F.; Marvaud, J.C.; Lambert, T. Distribution of the vanG-like gene cluster in Clostridium difficile clinical isolates. Can. J. Microbiol. 2012, 58, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Sebaihia, M.; Wren, B.W.; Mullany, P.; Fairweather, N.F.; Minton, N.; Stabler, R.; Thomson, N.R.; Roberts, A.P.; Cerdeno-Tarraga, A.M.; Wang, H.; et al. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat. Genet. 2006, 38, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Settle, C.D.; Wilcox, M.H.; Fawley, W.N.; Corrado, O.J.; Hawkey, P.M. Prospective study of the risk of Clostridium difficile diarrhoea in elderly patients following treatment with cefotaxime or piperacillin-tazobactam. Aliment. Pharmacol. Ther. 1998, 12, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, M.H.; Freeman, J.; Fawley, W.; MacKinlay, S.; Brown, A.; Donaldson, K.; Corrado, O. Long-term surveillance of cefotaxime and piperacillin-tazobactam prescribing and incidence of Clostridium difficile diarrhoea. J. Antimicrob. Chemother. 2004, 54, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Borriello, S.P.; Barclay, F.E. An in vitro model of colonisation resistance to Clostridium difficile infection. J. Med. Microbiol. 1986, 21, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Larson, H.E.; Borriello, S.P. Quantitative study of antibiotic-induced susceptibility to Clostridium difficile enterocecitis in hamsters. Antimicrob. Agents Chemother. 1990, 34, 1348–1353. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.; Baines, S.D.; Jabes, D.; Wilcox, M.H. Comparison of the efficacy of ramoplanin and vancomycin in both in vitro and in vivo models of clindamycin-induced Clostridium difficile infection. J. Antimicrob. Chemother. 2005, 56, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Baines, S.D.; Freeman, J.; Wilcox, M.H. Effects of piperacillin/tazobactam on Clostridium difficile growth and toxin production in a human gut model. J. Antimicrob. Chemother. 2005, 55, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Baines, S.D.; Saxton, K.; Freeman, J.; Wilcox, M.H. Tigecycline does not induce proliferation or cytotoxin production by epidemic Clostridium difficile strains in a human gut model. J. Antimicrob. Chemother. 2006, 58, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.; Baines, S.D.; Saxton, K.; Wilcox, M.H. Effect of metronidazole on growth and toxin production by epidemic Clostridium difficile PCR ribotypes 001 and 027 in a human gut model. J. Antimicrob. Chemother. 2007, 60, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Baines, S.D.; O’Connor, R.; Saxton, K.; Freeman, J.; Wilcox, M.H. Comparison of oritavancin versus vancomycin as treatments for clindamycin-induced Clostridium difficile PCR ribotype 027 infection in a human gut model. J. Antimicrob. Chemother. 2008, 62, 1078–1085. [Google Scholar] [CrossRef] [PubMed]
- Baines, S.D.; Freeman, J.; Wilcox, M.H. Tolevamer is not efficacious in the neutralization of cytotoxin in a human gut model of Clostridium difficile infection. Antimicrob. Agents Chemother. 2009, 53, 2202–2204. [Google Scholar] [CrossRef] [PubMed]
- Baines, S.D.; O’Connor, R.; Huscroft, G.; Saxton, K.; Freeman, J.; Wilcox, M.H. Mecillinam: A low-risk antimicrobial agent for induction of Clostridium difficile infection in an in vitro human gut model. J. Antimicrob. Chemother. 2009, 63, 838–839. [Google Scholar] [CrossRef] [PubMed]
- Baines, S.D.; O’Connor, R.; Saxton, K.; Freeman, J.; Wilcox, M.H. Activity of vancomycin against epidemic Clostridium difficile strains in a human gut model. J. Antimicrob. Chemother. 2009, 63, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Baines, S.D.; Crowther, G.S.; Todhunter, S.L.; Freeman, J.; Chilton, C.H.; Fawley, W.N.; Wilcox, M.H. Mixed infection by Clostridium difficile in an in vitro model of the human gut. J. Antimicrob. Chemother. 2013, 68, 1139–1143. [Google Scholar] [CrossRef] [PubMed]
- Crowther, G.S.; Baines, S.D.; Todhunter, S.L.; Freeman, J.; Chilton, C.H.; Wilcox, M.H. Evaluation of NVB302 versus vancomycin activity in an in vitro human gut model of Clostridium difficile infection. J. Antimicrob. Chemother. 2013, 68, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Baines, S.D.; Crowther, G.S.; Freeman, J.; Todhunter, S.; Vickers, R.; Wilcox, M.H. SMT19969 as a treatment for Clostridium difficile infection: An assessment of antimicrobial activity using conventional susceptibility testing and an in vitro gut model. J. Antimicrob. Chemother. 2014, 70, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Chilton, C.H.; Crowther, G.S.; Baines, S.D.; Todhunter, S.L.; Freeman, J.; Locher, H.H.; Athanasiou, A.; Wilcox, M.H. In vitro activity of cadazolid against clinically relevant Clostridium difficile isolates and in an in vitro gut model of C. difficile infection. J. Antimicrob. Chemother. 2014, 69, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Pultz, N.J.; Donskey, C.J. Effect of antibiotic treatment on growth of and toxin production by Clostridium difficile in the cecal contents of mice. Antimicrob. Agents Chemother. 2005, 49, 3529–3532. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.A.; Riggs, M.M.; Donskey, C.J. Effect of fluoroquinolone treatment on growth of and toxin production by epidemic and nonepidemic Clostridium difficile strains in the cecal contents of mice. Antimicrob. Agents Chemother. 2007, 51, 2674–2678. [Google Scholar] [CrossRef] [PubMed]
- Nerandzic, M.M.; Donskey, C.J. Effect of ceftobiprole treatment on growth of and toxin production by Clostridium difficile in cecal contents of mice. Antimicrob. Agents Chemother. 2011, 55, 2174–2177. [Google Scholar] [CrossRef] [PubMed]
- Jump, R.L.; Li, Y.; Pultz, M.J.; Kypriotakis, G.; Donskey, C.J. Tigecycline exhibits inhibitory activity against Clostridium difficile in the colon of mice and does not promote growth or toxin production. Antimicrob. Agents Chemother. 2011, 55, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Abujamel, T.; Cadnum, J.L.; Jury, L.A.; Sunkesula, V.C.; Kundrapu, S.; Jump, R.L.; Stintzi, A.C.; Donskey, C.J. Defining the vulnerable period for re-establishment of Clostridium difficile colonization after treatment of C. difficile infection with oral vancomycin or metronidazole. PLoS ONE 2013, 8, e76269. [Google Scholar] [CrossRef] [PubMed]
- Naaber, P.; Mikelsaar, R.H.; Salminen, S.; Mikelsaar, M. Bacterial translocation, intestinal microflora and morphological changes of intestinal mucosa in experimental models of Clostridium difficile infection. J. Med. Microbiol. 1998, 47, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Bourlioux, P. What is currently known about the molecular mechanisms of colonisation resistance. Anaerobe 1997, 3, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Larson, H.E.; Welch, A. In vitro and in vivo characterisation of resistance to colonisation with Clostridium difficile. J. Med. Microbiol. 1993, 38, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Boureau, H.; Decre, D.; Carlier, J.P.; Guichet, C.; Bourlioux, P. Identification of a Clostridium cocleatum strain involved in an anti-Clostridium difficile barrier effect and determination of its mucin-degrading enzymes. Res. Microbiol. 1993, 144, 405–410. [Google Scholar] [CrossRef]
- Lawley, T.D.; Clare, S.; Walker, A.W.; Stares, M.D.; Connor, T.R.; Raisen, C.; Goulding, D.; Rad, R.; Schreiber, F.; Brandt, C.; et al. Targeted restoration of the intestinal microbiota with a simple, defined bacteriotherapy resolves relapsing Clostridium difficile disease in mice. PLoS Pathog. 2012, 8, e1002995. [Google Scholar] [CrossRef] [PubMed]
- van Nood, E.; Speelman, P.; Nieuwdorp, M.; Keller, J. Fecal microbiota transplantation: Facts and controversies. Curr. Opin. Gastroenterol. 2014, 30, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Kager, L.; Liljeqvist, L.; Malmborg, A.S.; Nord, C.E. Effect of clindamycin prophylaxis on the colonic microflora in patients undergoing colorectal surgery. Antimicrob. Agents Chemother. 1981, 20, 736–740. [Google Scholar] [CrossRef] [PubMed]
- Brismar, B.; Edlund, C.; Malmborg, A.S.; Nord, C.E. Ciprofloxacin concentrations and impact of the colon microflora in patients undergoing colorectal surgery. Antimicrob. Agents Chemother. 1990, 34, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Chilton, C.H.; Freeman, J.; Crowther, G.S.; Todhunter, S.L.; Nicholson, S.; Wilcox, M.H. Co-amoxiclav induces proliferation and cytotoxin production of Clostridium difficile ribotype 027 in a human gut model. J. Antimicrob. Chemother. 2012, 67, 951–954. [Google Scholar] [CrossRef] [PubMed]
- Nord, C.E.; Brismar, B.; Kasholm-Tengve, B.; Tunevall, G. Effect of piperacillin/tazobactam treatment on human bowel microflora. J. Antimicrob. Chemother. 1993, 31, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Brismar, B.; Edlund, C.; Nord, C.E. Comparative effects of clarithromycin and erythromycin on the normal intestinal microflora. Scand. J. Infect. Dis. 1991, 23, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Manges, A.R.; Labbe, A.; Loo, V.G.; Atherton, J.K.; Behr, M.A.; Masson, L.; Tellis, P.A.; Brousseau, R. Comparative metagenomic study of alterations to the intestinal microbiota and risk of nosocomial Clostridum difficile-associated disease. J. Infect. Dis. 2010, 202, 1877–1884. [Google Scholar] [CrossRef] [PubMed]
- Antharam, V.C.; Li, E.C.; Ishmael, A.; Sharma, A.; Mai, V.; Rand, K.H.; Wang, G.P. Intestinal dysbiosis and depletion of butyrogenic bacteria in Clostridium difficile infection and nosocomial diarrhea. J. Clin. Microbiol. 2013, 51, 2884–2892. [Google Scholar] [CrossRef] [PubMed]
- Moubareck, C.; Gavini, F.; Vaugien, L.; Butel, M.J.; Doucet-Populaire, F. Antimicrobial susceptibility of bifidobacteria. J. Antimicrob. Chemother. 2005, 55, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Masco, L.; van Hoorde, K.; de Brandt, E.; Swings, J.; Huys, G. Antimicrobial susceptibility of Bifidobacterium strains from humans, animals and probiotic products. J. Antimicrob. Chemother. 2006, 58, 85–94. [Google Scholar] [CrossRef] [PubMed]
- van Hoek, A.H.; Mayrhofer, S.; Domig, K.J.; Aarts, H.J. Resistance determinant erm(X) is borne by transposon Tn5432 in Bifidobacterium thermophilum and Bifidobacterium animalis subsp. lactis. Int. J. Antimicrob. Agents 2008, 31, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Wexler, H.M.; Molitoris, E.; Finegold, S.M. In vitro activities of three of the newer quinolones against anaerobic bacteria. Antimicrob. Agents Chemother. 1992, 36, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Golan, Y.; McDermott, L.A.; Jacobus, N.V.; Goldstein, E.J.; Finegold, S.; Harrell, L.J.; Hecht, D.W.; Jenkins, S.G.; Pierson, C.; Venezia, R.; et al. Emergence of fluoroquinolone resistance among Bacteroides species. J. Antimicrob. Chemother. 2003, 52, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Stiefel, U.; Nerandzic, M.M.; Pultz, M.J.; Donskey, C.J. Gastrointestinal colonization with a cephalosporinase-producing bacteroides species preserves colonization resistance against vancomycin-resistant enterococcus and Clostridium difficile in cephalosporin-treated mice. Antimicrob. Agents Chemother. 2014, 58, 4535–4542. [Google Scholar] [CrossRef] [PubMed]
- Buffie, C.G.; Bucci, V.; Stein, R.R.; McKenney, P.T.; Ling, L.; Gobourne, A.; No, D.; Liu, H.; Kinnebrew, M.; Viale, A.; et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 2015, 517, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Hoverstad, T.; Carlstedt-Duke, B.; Lingaas, E.; Midtvedt, T.; Norin, K.E.; Saxerholt, H.; Steinbakk, M. Influence of ampicillin, clindamycin, and metronidazole on faecal excretion of short-chain fatty acids in healthy subjects. Scand. J. Gastroenterol. 1986, 21, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Bjorneklett, A.; Midtvedt, T. Influence of three antimicrobial agents—Penicillin, metronidazole, and doxycyclin—On the intestinal microflora of healthy humans. Scand. J. Gastroenterol. 1981, 16, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Chilton, C.H.; Crowther, G.S.; Freeman, J.; Todhunter, S.L.; Nicholson, S.; Longshaw, C.M.; Wilcox, M.H. Successful treatment of simulated Clostridium difficile infection in a human gut model by fidaxomicin first line and after vancomycin or metronidazole failure. J. Antimicrob. Chemother. 2014, 69, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Gerding, D.N. Metronidazole for Clostridium difficile-associated disease: Is it okay for Mom? Clin. Infect. Dis. 2005, 40, 1598–1600. [Google Scholar] [CrossRef] [PubMed]
- Bolton, R.P.; Culshaw, M.A. Faecal metronidazole concentrations during oral and intravenous therapy for antibiotic associated colitis due to Clostridium difficile. Gut 1986, 27, 1169–1172. [Google Scholar] [CrossRef] [PubMed]
- Arabi, Y.; Dimock, F.; Burdon, D.W.; Alexander-Williams, J.; Keighley, M.R. Influence of neomycin and metronidazole on colonic microflora of volunteers. J. Antimicrob. Chemother. 1979, 5, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Homann, S.R.; Bettin, K.M.; Quick, J.N.; Clabots, C.R.; Peterson, L.R.; Gerding, D.N. Treatment of asymptomatic Clostridium difficile carriers (fecal excretors) with vancomycin or metronidazole. A randomized, placebo-controlled trial. Ann. Intern. Med. 1992, 117, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Edlund, C.; Barkholt, L.; Olsson-Liljequist, B.; Nord, C.E. Effect of vancomycin on intestinal flora of patients who previously received antimicrobial therapy. Clin. Infect. Dis. 1997, 25, 729–732. [Google Scholar] [CrossRef] [PubMed]
- Onderdonk, A.B.; Louie, T.J.; Tally, F.P.; Bartlett, J.G. Activity of metronidazole against Escherichia coli in experimental intra-abdominal sepsis. J. Antimicrob. Chemother. 1979, 5, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, P.; Pensart, N.; Korten, V.; Murray, B.E.; Leclercq, R. Influence of oral glycopeptides on the fecal flora of human volunteers: Selection of highly glycopeptide-resistant enterococci. J. Infect. Dis. 1996, 173, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Sears, P.; Crook, D.W.; Louie, T.J.; Miller, M.A.; Weiss, K. Fidaxomicin attains high fecal concentrations with minimal plasma concentrations following oral administration in patients with Clostridium difficile infection. Clin. Infect. Dis. 2012, 55, S116–S120. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, E.J.; Babakhani, F.; Citron, D.M. Antimicrobial activities of fidaxomicin. Clin. Infect. Dis. 2012, 55, S143–S148. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, E.J.; Citron, D.M.; Tyrrell, K.L.; Merriam, C.V. Comparative in vitro activities of SMT19969, a new antimicrobial agent, against Clostridium difficile and 350 gram-positive and gram-negative aerobic and anaerobic intestinal flora isolates. Antimicrob. Agents Chemother. 2013, 57, 4872–4876. [Google Scholar] [CrossRef] [PubMed]
- Tannock, G.W.; Munro, K.; Taylor, C.; Lawley, B.; Young, W.; Byrne, B.; Emery, J.; Louie, T. A new macrocyclic antibiotic, fidaxomicin (OPT-80), causes less alteration to the bowel microbiota of Clostridium difficile-infected patients than does vancomycin. Microbiology 2010, 156, 3354–3359. [Google Scholar] [CrossRef] [PubMed]
- Chilton, C.H.; Freeman, J.; Baines, S.D.; Crowther, G.S.; Nicholson, S.; Wilcox, M.H. Evaluation of the effect of oritavancin on Clostridium difficile spore germination, outgrowth and recovery. J. Antimicrob. Chemother. 2013, 68, 2078–2082. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Kondo, T.; Toda, Y. Brief physical inactivity prolongs colonic transit time in elderly active men. Int. J. Sports Med. 1993, 14, 465–467. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.H.; Bingham, S.A.; Heaton, K.W.; Eastwood, M.A. Fecal weight, colon cancer risk, and dietary intake of non-starch polysaccharides (dietary fiber). Gastroenterology 1993, 103, 1783–1789. [Google Scholar]
- Donskey, C.; Louis Stokes Cleveland VA Medical Center, Cleveland, OH USA. Personal communication, 2015.
- Krook, A.; Lindstrom, B.; Kjellander, J.; Jarnerot, G.; Bodin, L. Relation between concentrations of metronidazole and Bacteroides spp. in faeces of patients with Crohn’s disease and healthy individuals. J. Clin. Pathol. 1981, 34, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.B.; Martyak, S.N.; Barza, M.; Curtis, L.; Weinstein, L. Penetration of clindamycin phosphate into the abnormal human biliary tract. Ann. Intern. Med. 1976, 84, 168–170. [Google Scholar] [CrossRef] [PubMed]
- Heimdahl, A.; Kager, L.; Nord, C.E. Changes in the oropharyngeal and colon microflora in relation to antimicrobial concentrations in saliva and faeces. Scand. J. Infect. Dis. 1985, 44, 52–58. [Google Scholar]
- Rashid, M.U.; Weintraub, A.; Nord, C.E. Development of antimicrobial resistance in the normal anaerobic microbiota during one year after administration of clindamycin or ciprofloxacin. Anaerobe 2015, 31, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Brumfitt, W.; Franklin, I.; Grady, D.; Hamilton-Miller, J.M.; Iliffe, A. Changes in the pharmacokinetics of ciprofloxacin and fecal flora during administration of a 7-day course to human volunteers. Antimicrob. Agents Chemother. 1984, 26, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Burkhardt, O.; Borner, K.; Stass, H.; Beyer, G.; Allewelt, M.; Nord, C.E.; Lode, H. Single- and multiple-dose pharmacokinetics of oral moxifloxacin and clarithromycin, and concentrations in serum, saliva and faeces. Scand. J. Infect. Dis. 2002, 34, 898–903. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.D.; Ke, S.; Palazzini, E.; Riopel, L.; Dupont, H. In vitro activity and fecal concentration of rifaximin after oral administration. Antimicrob. Agents Chemother. 2000, 44, 2205–2206. [Google Scholar] [CrossRef] [PubMed]
- Drummond, L.J.; Smith, D.G.; Poxton, I.R. Effects of sub-MIC concentrations of antibiotics on growth of and toxin production by Clostridium difficile. J. Med. Microbiol. 2003, 52, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Gerber, M.; Walch, C.; Loffler, B.; Tischendorf, K.; Reischl, U.; Ackermann, G. Effect of sub-MIC concentrations of metronidazole, vancomycin, clindamycin and linezolid on toxin gene transcription and production in Clostridium difficile. J. Med. Microbiol. 2008, 57, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Emerson, J.E.; Stabler, R.A.; Wren, B.W.; Fairweather, N.F. Microarray analysis of the transcriptional responses of Clostridium difficile to environmental and antibiotic stress. J. Med. Microbiol. 2008, 57, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Hennequin, C.; Collignon, A.; Karjalainen, T. Analysis of expression of GroEL (Hsp60) of Clostridium difficile in response to stress. Microb. Pathog. 2001, 31, 255–260. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baines, S.D.; Wilcox, M.H. Antimicrobial Resistance and Reduced Susceptibility in Clostridium difficile: Potential Consequences for Induction, Treatment, and Recurrence of C. difficile Infection. Antibiotics 2015, 4, 267-298. https://doi.org/10.3390/antibiotics4030267
Baines SD, Wilcox MH. Antimicrobial Resistance and Reduced Susceptibility in Clostridium difficile: Potential Consequences for Induction, Treatment, and Recurrence of C. difficile Infection. Antibiotics. 2015; 4(3):267-298. https://doi.org/10.3390/antibiotics4030267
Chicago/Turabian StyleBaines, Simon D., and Mark H. Wilcox. 2015. "Antimicrobial Resistance and Reduced Susceptibility in Clostridium difficile: Potential Consequences for Induction, Treatment, and Recurrence of C. difficile Infection" Antibiotics 4, no. 3: 267-298. https://doi.org/10.3390/antibiotics4030267