Strategies to Block HIV Transcription: Focus on Small Molecule Tat Inhibitors


Abstract
:1. Introduction
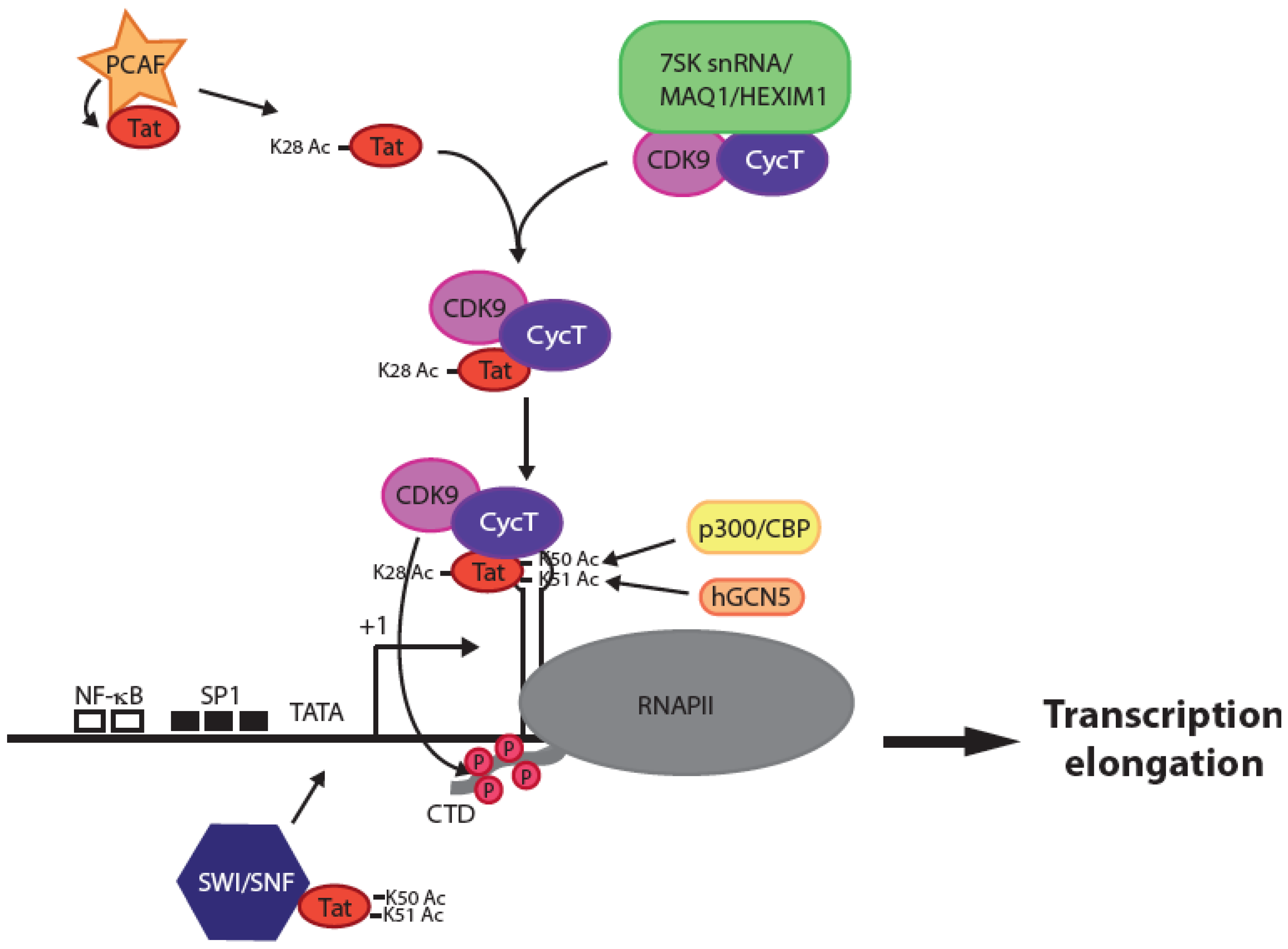
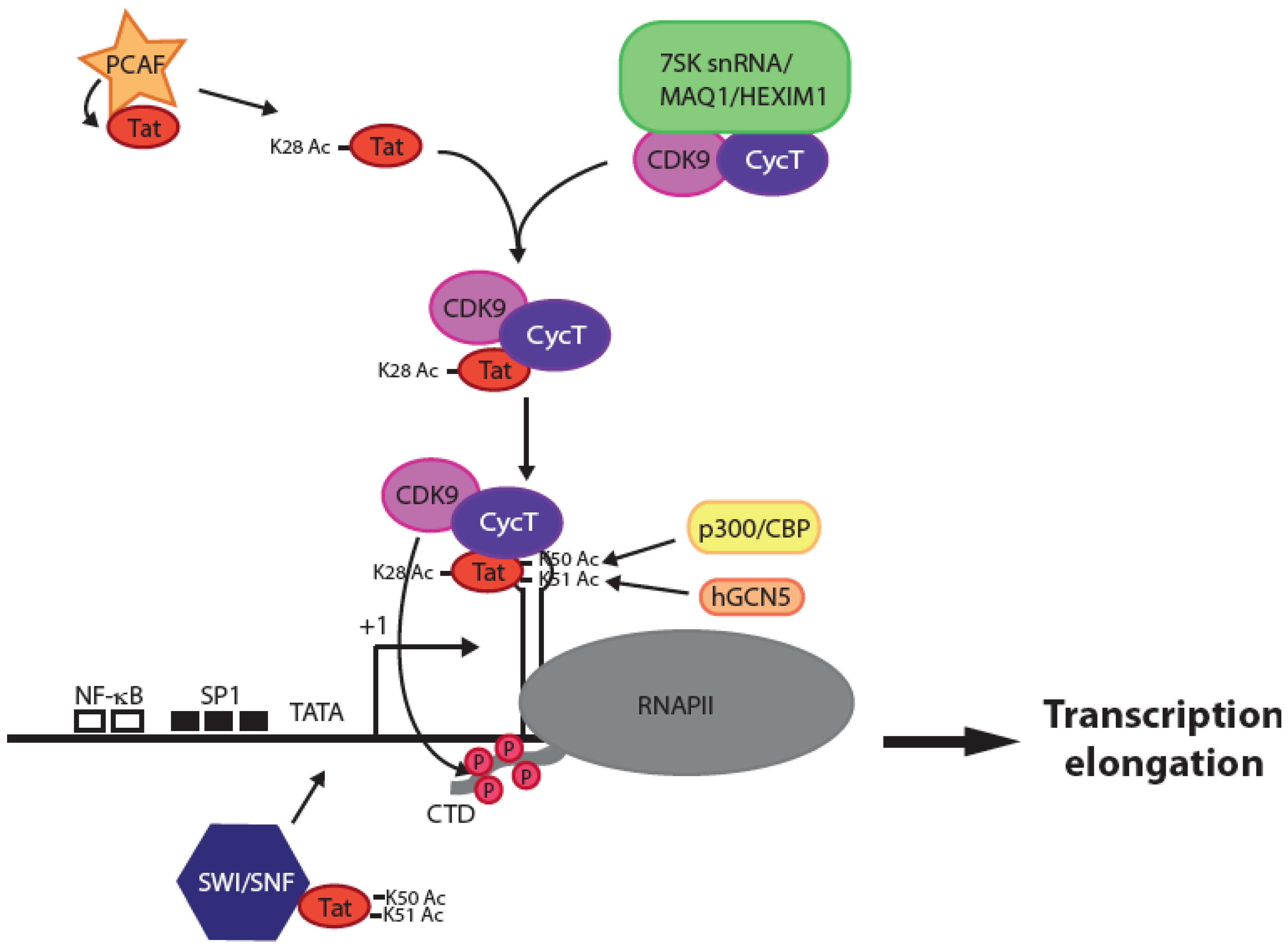
2. What Can We Expect from a Tat Inhibitor and What Features Make Such a Compound Different from Current ARVs?
3. Targets and Strategies: Approaches to Reduce HIV Transcription
3.1. Targeting Cellular Factors Involved in HIV Transcription
3.2. Non-Small Molecules Compounds Used to Inhibit Tat/TAR Mediated HIV Transcription
4. Small Molecules Inhibitors of HIV Transcription
4.1. Methods Used to Discover Small Molecule Inhibitors of Tat-Dependent Transcription
4.1.1. Biochemical Methods
4.1.2. LTR Reporter Activity
4.1.3. Alternative Methods
4.1.4. Docking Study
4.2. A Selection of Transcription Inhibitors with TI > 100
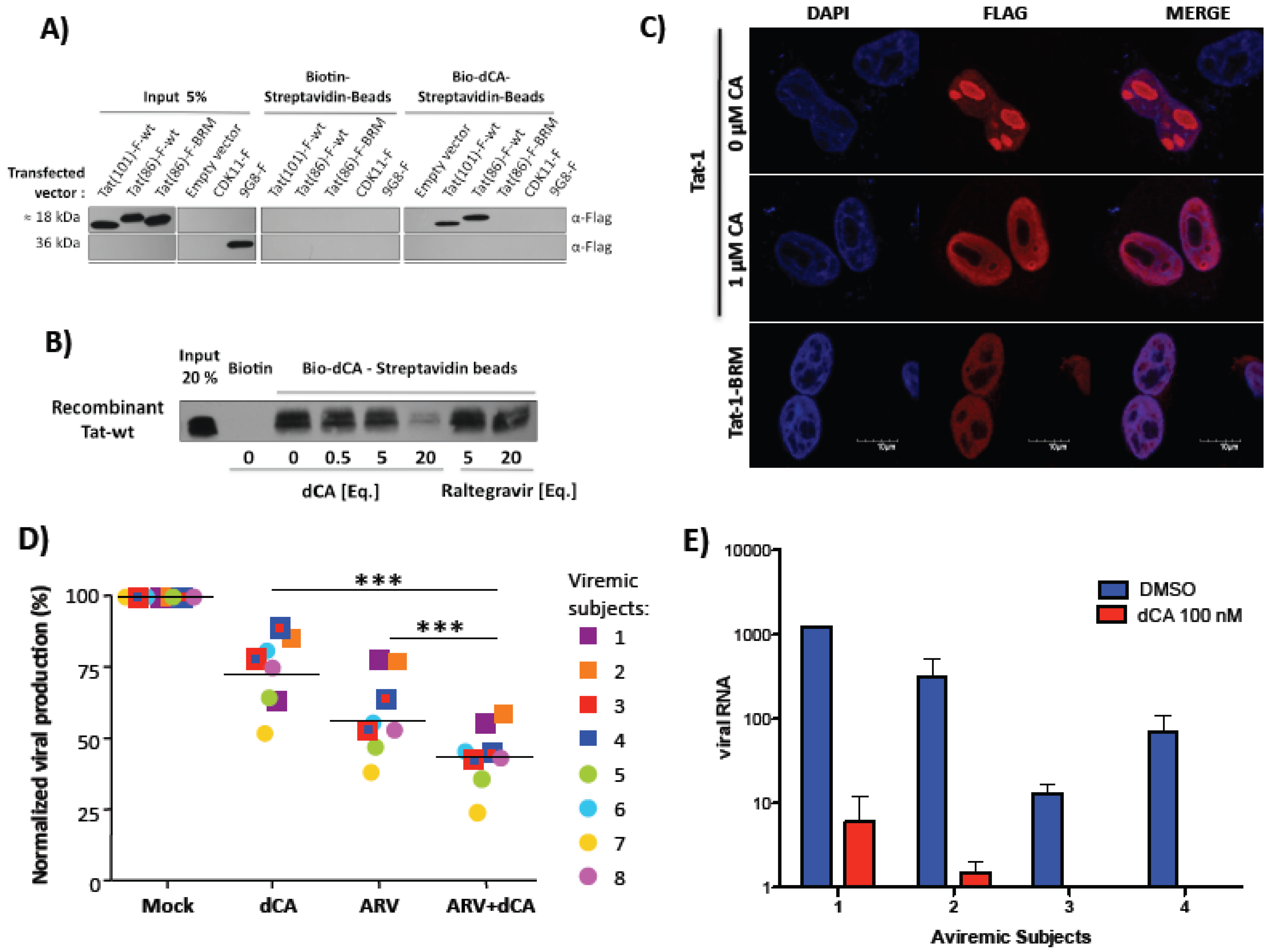
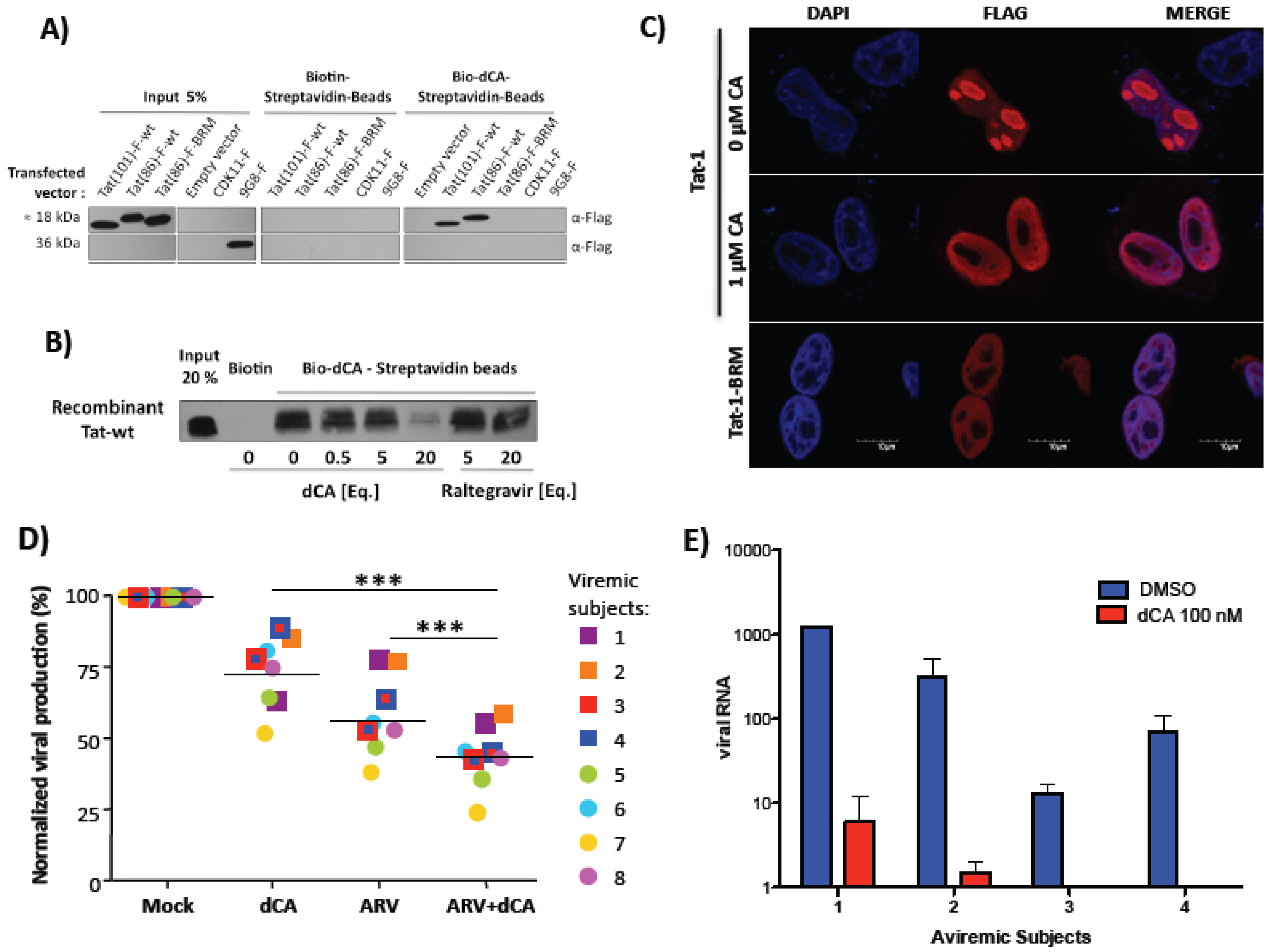
4.3. Didehydro-Cortistatin A (dCA)

{kind=link}
{kind=link}
| Compound | Structure | IC50 (nM) | CC50 (mM) | TI | Tested in | Cell types* | Screen | References |
|---|---|---|---|---|---|---|---|---|
| dCA |  | 0.0007-2.6 | 20 | >8000 | Acute, Chronic | Hela CD4+, T cell line, PBMCs, CD4+ T cells | N.A. | [146] |
| Durhamycin A |  | 4.8-11 | 5-25 | 431-5208 | Acute | Hela CD4+, T cell lines | LTR-reporter assay | [165] |
| WM5 |  | 30-850 | 2.21 to > 263 | 15->3333 | Acute, Chronic | Hela, T-cell lines, PBMCs | SAR | [187,206,207,211] |
| NM13 |  | 80 | ³296 | ³3707 | Acute | T-cell line | SAR | [91,208] |
| HM13N |  | 4-1200 | 1.5-26.33 | 17-1333 | Acute, Chronic, Latent | T-cell Lines, monocytic cell lines, PBMCs | SAR | [186] |
| Temacrazine |  | 0.1-72 | 1-10 | 21-2518 | Acute, Chronic, Latent | T cell line, monocytic cell lines | N.A. | [201] |
| NeoR |  | 1.7-5.3 | 275-500 | 33-250 | Acute, Chronic | T-cell line, promonocytic cell line, PBMCs | SAR | [197] |
5. Conclusions
Acknowledgments
References
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.R.; Ghattas, G.; Brenchley, J.M.; et al. Hiv reservoir size and persistence are driven by t cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef]
- Chun, T.W.; Stuyver, L.; Mizell, S.B.; Ehler, L.A.; Mican, J.A.; Baseler, M.; Lloyd, A.L.; Nowak, M.A.; Fauci, A.S. Presence of an inducible hiv-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 1997, 94, 13193–13197. [Google Scholar]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a reservoir for hiv-1 in patients on highly active antiretroviral therapy. Science 1997, 278, 1295–1300. [Google Scholar]
- Wong, J.K.; Hezareh, M.; Gunthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Spina, C.A.; Richman, D.D. Recovery of replication-competent hiv despite prolonged suppression of plasma viremia. Science 1997, 278, 1291–1295. [Google Scholar]
- Chun, T.W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of latent tissue reservoirs and total body viral load in hiv-1 infection. Nature 1997, 387, 183–188. [Google Scholar]
- Finzi, D.; Blankson, J.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.; Smith, K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent infection of cd4+ t cells provides a mechanism for lifelong persistence of hiv-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [CrossRef]
- Toohey, M.G.; Jones, K.A. In vitro formation of short rna polymerase ii transcripts that terminate within the hiv-1 and hiv-2 promoter-proximal downstream regions. Genes Dev. 1989, 3, 265–282. [Google Scholar] [CrossRef]
- Dingwall, C.; Ernberg, I.; Gait, M.J.; Green, S.M.; Heaphy, S.; Karn, J.; Lowe, A.D.; Singh, M.; Skinner, M.A. Hiv-1 tat protein stimulates transcription by binding to a u-rich bulge in the stem of the tar rna structure. EMBO J. 1990, 9, 4145–4153. [Google Scholar]
- Dingwall, C.; Ernberg, I.; Gait, M.J.; Green, S.M.; Heaphy, S.; Karn, J.; Lowe, A.D.; Singh, M.; Skinner, M.A.; Valerio, R. Human immunodeficiency virus 1 tat protein binds trans-activation-responsive region (tar) rna in vitro. Proc. Natl. Acad. Sci. USA 1989, 86, 6925–6929. [Google Scholar]
- Marshall, N.F.; Price, D.H. Control of formation of two distinct classes of rna polymerase ii elongation complexes. Mol. Cell. Biol. 1992, 12, 2078–2090. [Google Scholar]
- Marshall, N.F.; Price, D.H. Purification of p-tefb, a transcription factor required for the transition into productive elongation. J. Biol. Chem. 1995, 270, 12335–12338. [Google Scholar] [CrossRef]
- Michels, A.A.; Nguyen, V.T.; Fraldi, A.; Labas, V.; Edwards, M.; Bonnet, F.; Lania, L.; Bensaude, O. Maq1 and 7sk rna interact with cdk9/cyclin t complexes in a transcription-dependent manner. Mol. Cell. Biol. 2003, 23, 4859–4869. [Google Scholar] [CrossRef]
- Yik, J.H.; Chen, R.; Nishimura, R.; Jennings, J.L.; Link, A.J.; Zhou, Q. Inhibition of p-tefb (cdk9/cyclin t) kinase and rna polymerase ii transcription by the coordinated actions of hexim1 and 7sk snrna. Mol. Cell. 2003, 12, 971–982. [Google Scholar] [CrossRef]
- Peterlin, B.M.; Price, D.H. Controlling the elongation phase of transcription with p-tefb. Mol. Cell. 2006, 23, 297–305. [Google Scholar] [CrossRef]
- Van Lint, C. Role of chromatin in hiv-1 transcriptional regulation. Adv. Pharmacol. 2000, 48, 121–160. [Google Scholar] [CrossRef]
- Robison, A.J.; Nestler, E.J. Transcriptional and epigenetic mechanisms of addiction. Nat. Rev. Neurosci. 2011, 12, 623–637. [Google Scholar]
- Van Lint, C.; Emiliani, S.; Ott, M.; Verdin, E. Transcriptional activation and chromatin remodeling of the hiv-1 promoter in response to histone acetylation. EMBO J. 1996, 15, 1112–1120. [Google Scholar]
- Verdin, E. Dnase i-hypersensitive sites are associated with both long terminal repeats and with the intragenic enhancer of integrated human immunodeficiency virus type 1. J. Virol. 1991, 65, 6790–6799. [Google Scholar]
- Kiernan, R.E.; Vanhulle, C.; Schiltz, L.; Adam, E.; Xiao, H.; Maudoux, F.; Calomme, C.; Burny, A.; Nakatani, Y.; Jeang, K.T.; et al. Hiv-1 tat transcriptional activity is regulated by acetylation. EMBO J. 1999, 18, 6106–6118. [Google Scholar] [CrossRef]
- Ghose, R.; Liou, L.Y.; Herrmann, C.H.; Rice, A.P. Induction of tak (cyclin t1/p-tefb) in purified resting cd4(+) t lymphocytes by combination of cytokines. J. Virol. 2001, 75, 11336–11343. [Google Scholar] [CrossRef]
- Lassen, K.G.; Bailey, J.R.; Siliciano, R.F. Analysis of human immunodeficiency virus type 1 transcriptional elongation in resting cd4+ t cells in vivo. J. Virol. 2004, 78, 9105–9114. [Google Scholar] [CrossRef]
- Lin, X.; Irwin, D.; Kanazawa, S.; Huang, L.; Romeo, J.; Yen, T.S.; Peterlin, B.M. Transcriptional profiles of latent human immunodeficiency virus in infected individuals: Effects of tat on the host and reservoir. J. Virol. 2003, 77, 8227–8236. [Google Scholar] [CrossRef]
- He, G.; Margolis, D.M. Counterregulation of chromatin deacetylation and histone deacetylase occupancy at the integrated promoter of human immunodeficiency virus type 1 (hiv-1) by the hiv-1 repressor yy1 and hiv-1 activator tat. Mol. Cell. Biol. 2002, 22, 2965–2973. [Google Scholar] [CrossRef]
- Tyagi, M.; Karn, J. Cbf-1 promotes transcriptional silencing during the establishment of hiv-1 latency. EMBO J. 2007, 26, 4985–4995. [Google Scholar] [CrossRef]
- du Chene, I.; Basyuk, E.; Lin, Y.L.; Triboulet, R.; Knezevich, A.; Chable-Bessia, C.; Mettling, C.; Baillat, V.; Reynes, J.; Corbeau, P.; et al. Suv39h1 and hp1gamma are responsible for chromatin-mediated hiv-1 transcriptional silencing and post-integration latency. EMBO J. 2007, 26, 424–435. [Google Scholar] [CrossRef]
- Mahmoudi, T.; Parra, M.; Vries, R.G.; Kauder, S.E.; Verrijzer, C.P.; Ott, M.; Verdin, E. The swi/snf chromatin-remodeling complex is a cofactor for tat transactivation of the hiv promoter. J. Biol. Chem. 2006, 281, 19960–19968. [Google Scholar]
- Treand, C.; du Chene, I.; Bres, V.; Kiernan, R.; Benarous, R.; Benkirane, M.; Emiliani, S. Requirement for swi/snf chromatin-remodeling complex in tat-mediated activation of the hiv-1 promoter. EMBO J. 2006, 25, 1690–1699. [Google Scholar] [CrossRef]
- Gerritsen, M.E.; Williams, A.J.; Neish, A.S.; Moore, S.; Shi, Y.; Collins, T. Creb-binding protein/p300 are transcriptional coactivators of p65. Proc. Natl. Acad. Sci. USA 1997, 94, 2927–2932. [Google Scholar] [CrossRef]
- Agbottah, E.; Deng, L.; Dannenberg, L.O.; Pumfery, A.; Kashanchi, F. Effect of swi/snf chromatin remodeling complex on hiv-1 tat activated transcription. Retrovirology 2006, 3, 48. [Google Scholar] [CrossRef]
- Benkirane, M.; Chun, R.F.; Xiao, H.; Ogryzko, V.V.; Howard, B.H.; Nakatani, Y.; Jeang, K.T. Activation of integrated provirus requires histone acetyltransferase. P300 and p/caf are coactivators for hiv-1 tat. J. Biol. Chem. 1998, 273, 24898–24905. [Google Scholar]
- Col, E.; Caron, C.; Seigneurin-Berny, D.; Gracia, J.; Favier, A.; Khochbin, S. The histone acetyltransferase, hgcn5, interacts with and acetylates the hiv transactivator, tat. J. Biol. Chem. 2001, 276, 28179–28184. [Google Scholar]
- Hottiger, M.O.; Nabel, G.J. Interaction of human immunodeficiency virus type 1 tat with the transcriptional coactivators p300 and creb binding protein. J. Virol. 1998, 72, 8252–8256. [Google Scholar]
- Marzio, G.; Tyagi, M.; Gutierrez, M.I.; Giacca, M. Hiv-1 tat transactivator recruits p300 and creb-binding protein histone acetyltransferases to the viral promoter. Proc. Natl. Acad. Sci. USA 1998, 95, 13519–13524. [Google Scholar] [CrossRef]
- Nabel, G.; Baltimore, D. An inducible transcription factor activates expression of human immunodeficiency virus in t cells. Nature 1987, 326, 711–713. [Google Scholar] [CrossRef]
- Jones, K.A.; Kadonaga, J.T.; Luciw, P.A.; Tjian, R. Activation of the aids retrovirus promoter by the cellular transcription factor, sp1. Science 1986, 232, 755–759. [Google Scholar]
- Olsen, H.S.; Rosen, C.A. Contribution of the tata motif to tat-mediated transcriptional activation of human immunodeficiency virus gene expression. J. Virol. 1992, 66, 5594–5597. [Google Scholar]
- Verhoef, K.; Koper, M.; Berkhout, B. Determination of the minimal amount of tat activity required for human immunodeficiency virus type 1 replication. Virology 1997, 237, 228–236. [Google Scholar] [CrossRef]
- McCutchan, F.E.; Salminen, M.O.; Carr, J.K.; Burke, D.S. Hiv-1 genetic diversity. AIDS 1996, 10, S13–S20. [Google Scholar] [CrossRef]
- Simon, F.; Mauclere, P.; Roques, P.; Loussert-Ajaka, I.; Muller-Trutwin, M.C.; Saragosti, S.; Georges-Courbot, M.C.; Barre-Sinoussi, F.; Brun-Vezinet, F. Identification of a new human immunodeficiency virus type 1 distinct from group m and group o. Nat. Med. 1998, 4, 1032–1037. [Google Scholar] [CrossRef]
- Jeeninga, R.E.; Hoogenkamp, M.; Armand-Ugon, M.; de Baar, M.; Verhoef, K.; Berkhout, B. Functional differences between the long terminal repeat transcriptional promoters of human immunodeficiency virus type 1 subtypes a through g. J. Virol. 2000, 74, 3740–3751. [Google Scholar] [CrossRef]
- Montano, M.A.; Novitsky, V.A.; Blackard, J.T.; Cho, N.L.; Katzenstein, D.A.; Essex, M. Divergent transcriptional regulation among expanding human immunodeficiency virus type 1 subtypes. J. Virol. 1997, 71, 8657–8665. [Google Scholar]
- Irish, B.P.; Khan, Z.K.; Jain, P.; Nonnemacher, M.R.; Pirrone, V.; Rahman, S.; Rajagopalan, N.; Suchitra, J.B.; Mostoller, K.; Wigdahl, B. Molecular mechanisms of neurodegenerative diseases induced by human retroviruses: A review. Am. J. Infect. Dis. 2009, 5, 231–258. [Google Scholar] [CrossRef]
- Rappaport, J.; Joseph, J.; Croul, S.; Alexander, G.; Del Valle, L.; Amini, S.; Khalili, K. Molecular pathway involved in hiv-1-induced cns pathology: Role of viral regulatory protein, tat. J. Leukoc. Biol. 1999, 65, 458–465. [Google Scholar]
- Stevens, M.; de Clercq, E.; Balzarini, J. The regulation of hiv-1 transcription: Molecular targets for chemotherapeutic intervention. Med. Res. Rev. 2006, 26, 595–625. [Google Scholar] [CrossRef]
- Klebl, B.M.; Choidas, A. Cdk9/cyclin t1: A host cell target for antiretroviral therapy. Future Virol. 2006, 1, 317–330. [Google Scholar] [CrossRef]
- Wang, S.; Fischer, P.M. Cyclin-dependent kinase 9: A key transcriptional regulator and potential drug target in oncology, virology and cardiology. Trends Pharmacol. Sci. 2008, 29, 302–313. [Google Scholar] [CrossRef]
- Coley, W.; Kehn-Hall, K.; van Duyne, R.; Kashanchi, F. Novel hiv-1 therapeutics through targeting altered host cell pathways. Expert Opin. Biol. Ther. 2009, 9, 1369–1382. [Google Scholar] [CrossRef]
- Nemeth, G.; Varga, Z.; Greff, Z.; Bencze, G.; Sipos, A.; Szantai-Kis, C.; Baska, F.; Gyuris, A.; Kelemenics, K.; Szathmary, Z.; et al. Novel, selective cdk9 inhibitors for the treatment of hiv infection. Curr Med. Chem. 2011, 18, 342–358. [Google Scholar]
- Baumli, S.; Lolli, G.; Lowe, E.D.; Troiani, S.; Rusconi, L.; Bullock, A.N.; Debreczeni, J.E.; Knapp, S.; Johnson, L.N. The structure of p-tefb (cdk9/cyclin t1), its complex with flavopiridol and regulation by phosphorylation. EMBO J. 2008, 27, 1907–1918. [Google Scholar] [CrossRef]
- Chao, S.H.; Fujinaga, K.; Marion, J.E.; Taube, R.; Sausville, E.A.; Senderowicz, A.M.; Peterlin, B.M.; Price, D.H. Flavopiridol inhibits p-tefb and blocks hiv-1 replication. J. Biol. Chem. 2000, 275, 28345–28348. [Google Scholar]
- Ali, A.; Ghosh, A.; Nathans, R.S.; Sharova, N.; O'Brien, S.; Cao, H.; Stevenson, M.; Rana, T.M. Identification of flavopiridol analogues that selectively inhibit positive transcription elongation factor (p-tefb) and block hiv-1 replication. Chembiochem 2009, 10, 2072–2080. [Google Scholar] [CrossRef]
- Heredia, A.; Davis, C.; Bamba, D.; Le, N.; Gwarzo, M.Y.; Sadowska, M.; Gallo, R.C.; Redfield, R.R. Indirubin-3'-monoxime, a derivative of a chinese antileukemia medicine, inhibits p-tefb function and hiv-1 replication. AIDS 2005, 19, 2087–2095. [Google Scholar] [CrossRef]
- Toossi, Z.; Wu, M.; Hirsch, C.S.; Mayanja-Kizza, H.; Baseke, J.; Aung, H.; Canaday, D.H.; Fujinaga, K. Activation of p-tefb at sites of dual hiv/tb infection, and inhibition of mtb-induced hiv transcriptional activation by the inhibitor of cdk9, indirubin-3'-monoxime. AIDS Res. Hum. Retroviruses 2012, 28, 182–187. [Google Scholar] [CrossRef]
- Biglione, S.; Byers, S.A.; Price, J.P.; Nguyen, V.T.; Bensaude, O.; Price, D.H.; Maury, W. Inhibition of hiv-1 replication by p-tefb inhibitors drb, seliciclib and flavopiridol correlates with release of free p-tefb from the large, inactive form of the complex. Retrovirology 2007, 4, 47. [Google Scholar] [CrossRef]
- Agbottah, E.; de La Fuente, C.; Nekhai, S.; Barnett, A.; Gianella-Borradori, A.; Pumfery, A.; Kashanchi, F. Antiviral activity of cyc202 in hiv-1-infected cells. J. Biol. Chem. 2005, 280, 3029–3042. [Google Scholar]
- Debebe, Z.; Ammosova, T.; Breuer, D.; Lovejoy, D.B.; Kalinowski, D.S.; Kumar, K.; Jerebtsova, M.; Ray, P.; Kashanchi, F.; Gordeuk, V.R.; et al. Iron chelators of the di-2-pyridylketone thiosemicarbazone and 2-benzoylpyridine thiosemicarbazone series inhibit hiv-1 transcription: Identification of novel cellular targets--iron, cyclin-dependent kinase (cdk) 2, and cdk9. Mol. Pharmacol. 2011, 79, 185–196. [Google Scholar] [CrossRef]
- Bai, J.; Sui, J.; Zhu, R.Y.; Tallarico, A.S.; Gennari, F.; Zhang, D.; Marasco, W.A. Inhibition of tat-mediated transactivation and hiv-1 replication by human anti-hcyclint1 intrabodies. J. Biol. Chem. 2003, 278, 1433–1442. [Google Scholar]
- Sung, T.L.; Rice, A.P. Mir-198 inhibits hiv-1 gene expression and replication in monocytes and its mechanism of action appears to involve repression of cyclin t1. PLoS Pathog. 2009, 5, e1000263. [Google Scholar] [CrossRef]
- Jadlowsky, J.K.; Nojima, M.; Schulte, A.; Geyer, M.; Okamoto, T.; Fujinaga, K. Dominant negative mutant cyclin t1 proteins inhibit hiv transcription by specifically degrading tat. Retrovirology 2008, 5, 63. [Google Scholar] [CrossRef]
- Jadlowsky, J.K.; Nojima, M.; Okamoto, T.; Fujinaga, K. Dominant negative mutant cyclin t1 proteins that inhibit hiv transcription by forming a kinase inactive complex with tat. J. Gen. Virol. 2008, 89, 2783–2787. [Google Scholar] [CrossRef]
- Hoque, M.; Tian, B.; Mathews, M.B.; Pe'ery, T. Granulin and granulin repeats interact with the tat.P-tefb complex and inhibit tat transactivation. J. Biol. Chem. 2005, 280, 13648–13657. [Google Scholar]
- Hoque, M.; Young, T.M.; Lee, C.G.; Serrero, G.; Mathews, M.B.; Pe'ery, T. The growth factor granulin interacts with cyclin t1 and modulates p-tefb-dependent transcription. Mol. Cell. Biol. 2003, 23, 1688–1702. [Google Scholar]
- Fraldi, A.; Varrone, F.; Napolitano, G.; Michels, A.A.; Majello, B.; Bensaude, O.; Lania, L. Inhibition of tat activity by the hexim1 protein. Retrovirology 2005, 2, 42. [Google Scholar] [CrossRef]
- Young, T.M.; Wang, Q.; Pe'ery, T.; Mathews, M.B. The human i-mfa domain-containing protein, hic, interacts with cyclin t1 and modulates p-tefb-dependent transcription. Mol. Cell. Biol. 2003, 23, 6373–6384. [Google Scholar] [CrossRef]
- Deng, L.; Ammosova, T.; Pumfery, A.; Kashanchi, F.; Nekhai, S. Hiv-1 tat interaction with rna polymerase ii c-terminal domain (ctd) and a dynamic association with cdk2 induce ctd phosphorylation and transcription from hiv-1 promoter. J. Biol. Chem. 2002, 277, 33922–33929. [Google Scholar]
- Nekhai, S.; Zhou, M.; Fernandez, A.; Lane, W.S.; Lamb, N.J.; Brady, J.; Kumar, A. Hiv-1 tat-associated rna polymerase c-terminal domain kinase, cdk2, phosphorylates cdk7 and stimulates tat-mediated transcription. Biochem. J. 2002, 364, 649–657. [Google Scholar] [CrossRef]
- Pumfery, A.; de la Fuente, C.; Berro, R.; Nekhai, S.; Kashanchi, F.; Chao, S.H. Potential use of pharmacological cyclin-dependent kinase inhibitors as anti-hiv therapeutics. Curr. Pharm. Des. 2006, 12, 1949–1961. [Google Scholar] [CrossRef]
- Guendel, I.; Agbottah, E.T.; Kehn-Hall, K.; Kashanchi, F. Inhibition of human immunodeficiency virus type-1 by cdk inhibitors. AIDS Res. Ther. 2010, 7, 7. [Google Scholar] [CrossRef]
- Ammosova, T.; Berro, R.; Kashanchi, F.; Nekhai, S. Rna interference directed to cdk2 inhibits hiv-1 transcription. Virology 2005, 341, 171–178. [Google Scholar] [CrossRef]
- Agbottah, E.; Zhang, N.; Dadgar, S.; Pumfery, A.; Wade, J.D.; Zeng, C.; Kashanchi, F. Inhibition of hiv-1 virus replication using small soluble tat peptides. Virology 2006, 345, 373–389. [Google Scholar] [CrossRef]
- Van Duyne, R.; Cardenas, J.; Easley, R.; Wu, W.; Kehn-Hall, K.; Klase, Z.; Mendez, S.; Zeng, C.; Chen, H.; Saifuddin, M.; et al. Effect of transcription peptide inhibitors on hiv-1 replication. Virology 2008, 376, 308–322. [Google Scholar] [CrossRef]
- Stevens, M.; Balzarini, J.; Lagoja, I.M.; Noppen, B.; Francois, K.; van Aerschot, A.; Herdewijn, P.; de Clercq, E.; Pannecouque, C. Inhibition of human immunodeficiency virus type 1 transcription by n-aminoimidazole derivatives. Virology 2007, 365, 220–237. [Google Scholar] [CrossRef]
- Karn, J. Tackling tat. J. Mol. Biol. 1999, 293, 235–254. [Google Scholar] [CrossRef]
- Baba, M. Recent status of hiv-1 gene expression inhibitors. Antiviral Res. 2006, 71, 301–306. [Google Scholar] [CrossRef]
- Gnabre, J.N.; Brady, J.N.; Clanton, D.J.; Ito, Y.; Dittmer, J.; Bates, R.B.; Huang, R.C. Inhibition of human immunodeficiency virus type 1 transcription and replication by DNA sequence-selective plant lignans. Proc. Natl. Acad. Sci. USA 1995, 92, 11239–11243. [Google Scholar]
- Huang, R.C.; Li, Y.; Giza, P.E.; Gnabre, J.N.; Abd-Elazem, I.S.; King, K.Y.; Hwu, J.R. Novel antiviral agent tetraglycylated nordihydroguaiaretic acid hydrochloride as a host-dependent viral inhibitor. Antiviral Res. 2003, 58, 57–64. [Google Scholar] [CrossRef]
- Hwu, J.R.; Tseng, W.N.; Gnabre, J.; Giza, P.; Huang, R.C. Antiviral activities of methylated nordihydroguaiaretic acids. 1. Synthesis, structure identification, and inhibition of tat-regulated hiv transactivation. J. Med. Chem. 1998, 41, 2994–3000. [Google Scholar] [CrossRef]
- Bedoya, L.M.; Abad, M.J.; Calonge, E.; Saavedra, L.A.; Gutierrez, C.M.; Kouznetsov, V.V.; Alcami, J.; Bermejo, P. Quinoline-based compounds as modulators of hiv transcription through nf-kappab and sp1 inhibition. Antiviral Res. 2010, 87, 338–344. [Google Scholar] [CrossRef]
- Osorio, A.A.; Munoz, A.; Torres-Romero, D.; Bedoya, L.M.; Perestelo, N.R.; Jimenez, I.A.; Alcami, J.; Bazzocchi, I.L. Olean-18-ene triterpenoids from celastraceae species inhibit hiv replication targeting nf-kb and sp1 dependent transcription. Eur J. Med. Chem. 2012, 52, 295–303. [Google Scholar] [CrossRef]
- Takada, N.; Sanda, T.; Okamoto, H.; Yang, J.P.; Asamitsu, K.; Sarol, L.; Kimura, G.; Uranishi, H.; Tetsuka, T.; Okamoto, T. Rela-associated inhibitor blocks transcription of human immunodeficiency virus type 1 by inhibiting nf-kappab and sp1 actions. J. Virol. 2002, 76, 8019–8030. [Google Scholar]
- Pande, V.; Ramos, M.J. Nuclear factor kappa b: A potential target for anti-hiv chemotherapy. Curr. Med. Chem. 2003, 10, 1603–1615. [Google Scholar] [CrossRef]
- Fujiwara, N.; Nakajima, T.; Ueda, Y.; Fujita, H.; Kawakami, H. Novel piperidinylpyrimidine derivatives as inhibitors of hiv-1 ltr activation. Bioorg. Med. Chem. 2008, 16, 9804–9816. [Google Scholar] [CrossRef]
- Haraguchi, S.; Day, N.K.; Kamchaisatian, W.; Beigier-Pompadre, M.; Stenger, S.; Tangsinmankong, N.; Sleasman, J.W.; Pizzo, S.V.; Cianciolo, G.J. Lmp-420, a small-molecule inhibitor of tnf-alpha, reduces replication of hiv-1 and mycobacterium tuberculosis in human cells. AIDS Res. Ther. 2006, 3, 8. [Google Scholar] [CrossRef]
- Biswas, D.K.; Dezube, B.J.; Ahlers, C.M.; Pardee, A.B. Pentoxifylline inhibits hiv-1 ltr-driven gene expression by blocking nf-kappa b action. J. Acquir. Immune. Defic. Syndr. 1993, 6, 778–786. [Google Scholar]
- Fazely, F.; Dezube, B.J.; Allen-Ryan, J.; Pardee, A.B.; Ruprecht, R.M. Pentoxifylline (trental) decreases the replication of the human immunodeficiency virus type 1 in human peripheral blood mononuclear cells and in cultured t cells. Blood 1991, 77, 1653–1656. [Google Scholar]
- Navarro, J.; Punzon, M.C.; Pizarro, A.; Fernandez-Cruz, E.; Fresno, M.; Munoz-Fernandez, M.A. Pentoxifylline inhibits acute hiv-1 replication in human t cells by a mechanism not involving inhibition of tumour necrosis factor synthesis or nuclear factor-kappa b activation. AIDS 1996, 10, 469–475. [Google Scholar] [CrossRef]
- Smith, J.A.; Nunnari, G.; Preuss, M.; Pomerantz, R.J.; Daniel, R. Pentoxifylline suppresses transduction by hiv-1-based vectors. Intervirology 2007, 50, 377–386. [Google Scholar] [CrossRef]
- Asamitsu, K.; Yamaguchi, T.; Nakata, K.; Hibi, Y.; Victoriano, A.F.; Imai, K.; Onozaki, K.; Kitade, Y.; Okamoto, T. Inhibition of human immunodeficiency virus type 1 replication by blocking ikappab kinase with noraristeromycin. J. Biochem. 2008, 144, 581–589. [Google Scholar] [CrossRef]
- Balasubramanyam, K.; Varier, R.A.; Altaf, M.; Swaminathan, V.; Siddappa, N.B.; Ranga, U.; Kundu, T.K. Curcumin, a novel p300/creb-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J. Biol. Chem. 2004, 279, 51163–51171. [Google Scholar]
- Mantelingu, K.; Reddy, B.A.; Swaminathan, V.; Kishore, A.H.; Siddappa, N.B.; Kumar, G.V.; Nagashankar, G.; Natesh, N.; Roy, S.; Sadhale, P.P.; et al. Specific inhibition of p300-hat alters global gene expression and represses hiv replication. Chem. Biol. 2007, 14, 645–657. [Google Scholar] [CrossRef]
- Sarli, V.; Giannis, A. Selective inhibition of cbp/p300 hat. Chem. Biol. 2007, 14, 605–606. [Google Scholar] [CrossRef]
- Dorr, A.; Kiermer, V.; Pedal, A.; Rackwitz, H.R.; Henklein, P.; Schubert, U.; Zhou, M.M.; Verdin, E.; Ott, M. Transcriptional synergy between tat and pcaf is dependent on the binding of acetylated tat to the pcaf bromodomain. EMBO J. 2002, 21, 2715–2723. [Google Scholar] [CrossRef]
- Zeng, L.; Li, J.; Muller, M.; Yan, S.; Mujtaba, S.; Pan, C.; Wang, Z.; Zhou, M.M. Selective small molecules blocking hiv-1 tat and coactivator pcaf association. J. Am. Chem. Soc. 2005, 127, 2376–2377. [Google Scholar]
- Pan, C.; Mezei, M.; Mujtaba, S.; Muller, M.; Zeng, L.; Li, J.; Wang, Z.; Zhou, M.M. Structure-guided optimization of small molecules inhibiting human immunodeficiency virus 1 tat association with the human coactivator p300/creb binding protein-associated factor. J. Med. Chem. 2007, 50, 2285–2288. [Google Scholar] [CrossRef]
- Zhang, H.S.; Sang, W.W.; Ruan, Z.; Wang, Y.O. Akt/nox2/nf-kappab signaling pathway is involved in tat-induced hiv-1 long terminal repeat (ltr) transactivation. Arch. Biochem. Biophys. 2011, 505, 266–272. [Google Scholar] [CrossRef]
- Zhang, H.S.; Wu, T.C.; Sang, W.W.; Ruan, Z. Egcg inhibits tat-induced ltr transactivation: Role of nrf2, akt, ampk signaling pathway. Life Sci. 2012, 90, 747–754. [Google Scholar] [CrossRef]
- Lin, P.H.; Ke, Y.Y.; Su, C.T.; Shiao, H.Y.; Hsieh, H.P.; Chao, Y.K.; Lee, C.N.; Kao, C.L.; Chao, Y.S.; Chang, S.Y. Inhibition of hiv-1 tat-mediated transcription by a coumarin derivative, bprhiv001, through the akt pathway. J. Virol. 2011, 85, 9114–9126. [Google Scholar] [CrossRef]
- Guendel, I.; Carpio, L.; Easley, R.; van Duyne, R.; Coley, W.; Agbottah, E.; Dowd, C.; Kashanchi, F.; Kehn-Hall, K. 9-aminoacridine inhibition of hiv-1 tat dependent transcription. Virol. J. 2009, 6, 114. [Google Scholar] [CrossRef]
- Ammosova, T.; Jerebtsova, M.; Beullens, M.; Voloshin, Y.; Ray, P.E.; Kumar, A.; Bollen, M.; Nekhai, S. Nuclear protein phosphatase-1 regulates hiv-1 transcription. J. Biol. Chem. 2003, 278, 32189–32194. [Google Scholar]
- Ammosova, T.; Yedavalli, V.R.; Niu, X.; Jerebtsova, M.; van Eynde, A.; Beullens, M.; Bollen, M.; Jeang, K.T.; Nekhai, S. Expression of a protein phosphatase 1 inhibitor, cdnipp1, increases cdk9 threonine 186 phosphorylation and inhibits hiv-1 transcription. J. Biol. Chem. 2011, 286, 3798–3804. [Google Scholar]
- Ammosova, T.; Platonov, M.; Yedavalli, V.R.; Obukhov, Y.; Gordeuk, V.R.; Jeang, K.T.; Kovalskyy, D.; Nekhai, S. Small molecules targeted to a non-catalytic "rvxf" binding site of protein phosphatase-1 inhibit hiv-1. PLoS One 2012, 7, e39481. [Google Scholar]
- Campagna, M.; Rivas, C. Antiviral activity of resveratrol. Biochem. Soc. Trans. 2010, 38, 50–53. [Google Scholar] [CrossRef]
- Lee, E.O.; Kim, S.E.; Park, H.K.; Kang, J.L.; Chong, Y.H. Extracellular hiv-1 tat upregulates tnf-alpha dependent mcp-1/ccl2 production via activation of erk1/2 pathway in rat hippocampal slice cultures: Inhibition by resveratrol, a polyphenolic phytostilbene. Exp. Neurol. 2011, 229, 399–408. [Google Scholar] [CrossRef]
- Zhang, H.S.; Zhou, Y.; Wu, M.R.; Zhou, H.S.; Xu, F. Resveratrol inhibited tat-induced hiv-1 ltr transactivation via nad(+)-dependent sirt1 activity. Life Sci. 2009, 85, 484–489. [Google Scholar] [CrossRef]
- Pagans, S.; Pedal, A.; North, B.J.; Kaehlcke, K.; Marshall, B.L.; Dorr, A.; Hetzer-Egger, C.; Henklein, P.; Frye, R.; McBurney, M.W.; et al. Sirt1 regulates hiv transcription via tat deacetylation. PLoS Biol. 2005, 3, e41. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.S.; Brent, M.M.; Getachew, R.; Jayakumar, P.; Chen, L.F.; Schnolzer, M.; McBurney, M.W.; Marmorstein, R.; Greene, W.C.; Ott, M. Human immunodeficiency virus type 1 tat protein inhibits the sirt1 deacetylase and induces t cell hyperactivation. Cell. Host Microbe 2008, 3, 158–167. [Google Scholar] [CrossRef]
- Richter, S.N.; Palu, G. Inhibitors of hiv-1 tat-mediated transactivation. Curr. Med. Chem. 2006, 13, 1305–1315. [Google Scholar] [CrossRef]
- Turner, J.J.; Fabani, M.; Arzumanov, A.A.; Ivanova, G.; Gait, M.J. Targeting the hiv-1 rna leader sequence with synthetic oligonucleotides and sirna: Chemistry and cell delivery. Biochim. Biophys. Acta 2006, 1758, 290–300. [Google Scholar]
- Burnett, J.C.; Rossi, J.J. Rna-based therapeutics: Current progress and future prospects. Chem. Biol. 2012, 19, 60–71. [Google Scholar] [CrossRef]
- Burnett, J.C.; Rossi, J.J. Stem cells, ribozymes and hiv. Gene Ther. 2009, 16, 1178–1179. [Google Scholar] [CrossRef]
- Mitsuyasu, R.T.; Merigan, T.C.; Carr, A.; Zack, J.A.; Winters, M.A.; Workman, C.; Bloch, M.; Lalezari, J.; Becker, S.; Thornton, L.; et al. Phase 2 gene therapy trial of an anti-hiv ribozyme in autologous cd34+ cells. Nat. Med. 2009, 15, 285–292. [Google Scholar] [CrossRef]
- Mulhbacher, J.; St-Pierre, P.; Lafontaine, D.A. Therapeutic applications of ribozymes and riboswitches. Curr. Opin. Pharmacol. 2010, 10, 551–556. [Google Scholar] [CrossRef]
- Zeller, S.J.; Kumar, P. Rna-based gene therapy for the treatment and prevention of hiv: From bench to bedside. Yale J. Biol. Med. 2011, 84, 301–309. [Google Scholar]
- Eekels, J.J.; Berkhout, B. Toward a durable treatment of hiv-1 infection using rna interference. Prog. Mol. Biol. Transl. Sci. 2011, 102, 141–163. [Google Scholar] [CrossRef]
- Zhou, J.; Rossi, J.J. Current progress in the development of rnai-based therapeutics for hiv-1. Gene Ther. 2011, 18, 1134–1138. [Google Scholar] [CrossRef]
- Scherer, L.; Rossi, J.J.; Weinberg, M.S. Progress and prospects: Rna-based therapies for treatment of hiv infection. Gene Ther. 2007, 14, 1057–1064. [Google Scholar] [CrossRef]
- Li, M.J.; Kim, J.; Li, S.; Zaia, J.; Yee, J.K.; Anderson, J.; Akkina, R.; Rossi, J.J. Long-term inhibition of hiv-1 infection in primary hematopoietic cells by lentiviral vector delivery of a triple combination of anti-hiv shrna, anti-ccr5 ribozyme, and a nucleolar-localizing tar decoy. Mol. Ther. 2005, 12, 900–909. [Google Scholar] [CrossRef]
- Anderson, J.; Li, M.J.; Palmer, B.; Remling, L.; Li, S.; Yam, P.; Yee, J.K.; Rossi, J.; Zaia, J.; Akkina, R. Safety and efficacy of a lentiviral vector containing three anti-hiv genes—ccr5 ribozyme, tat-rev sirna, and tar decoy—in scid-hu mouse-derived t cells. Mol. Ther. 2007, 15, 1182–1188. [Google Scholar]
- DiGiusto, D.L.; Krishnan, A.; Li, L.; Li, H.; Li, S.; Rao, A.; Mi, S.; Yam, P.; Stinson, S.; Kalos, M.; et al. Rna-based gene therapy for hiv with lentiviral vector-modified cd34(+) cells in patients undergoing transplantation for aids-related lymphoma. Sci. Transl. Med. 2010, 2, 36–ra43. [Google Scholar] [CrossRef]
- Kiem, H.P.; Wu, R.A.; Sun, G.; von Laer, D.; Rossi, J.J.; Trobridge, G.D. Foamy combinatorial anti-hiv vectors with mgmtp140k potently inhibit hiv-1 and shiv replication and mediate selection in vivo. Gene Ther. 2010, 17, 37–49. [Google Scholar] [CrossRef]
- Hamy, F.; Felder, E.R.; Heizmann, G.; Lazdins, J.; Aboul-ela, F.; Varani, G.; Karn, J.; Klimkait, T. An inhibitor of the tat/tar rna interaction that effectively suppresses hiv-1 replication. Proc. Natl. Acad. Sci. USA 1997, 94, 3548–3553. [Google Scholar]
- Davidson, A.; Leeper, T.C.; Athanassiou, Z.; Patora-Komisarska, K.; Karn, J.; Robinson, J.A.; Varani, G. Simultaneous recognition of hiv-1 tar rna bulge and loop sequences by cyclic peptide mimics of tat protein. Proc. Natl. Acad. Sci. USA 2009, 106, 11931–11936. [Google Scholar]
- Davidson, A.; Patora-Komisarska, K.; Robinson, J.A.; Varani, G. Essential structural requirements for specific recognition of hiv tar rna by peptide mimetics of tat protein. Nucleic Acids Res. 2011, 39, 248–256. [Google Scholar] [CrossRef]
- Lalonde, M.S.; Lobritz, M.A.; Ratcliff, A.; Chamanian, M.; Athanassiou, Z.; Tyagi, M.; Wong, J.; Robinson, J.A.; Karn, J.; Varani, G.; et al. Inhibition of both hiv-1 reverse transcription and gene expression by a cyclic peptide that binds the tat-transactivating response element (tar) rna. PLoS Pathog. 2011, 7, e1002038. [Google Scholar] [CrossRef] [Green Version]
- D’Orso, I.; Grunwell, J.R.; Nakamura, R.L.; Das, C.; Frankel, A.D. Targeting tat inhibitors in the assembly of human immunodeficiency virus type 1 transcription complexes. J. Virol. 2008, 82, 9492–9504. [Google Scholar] [CrossRef]
- Campbell, G.R.; Loret, E.P. What does the structure-function relationship of the hiv-1 tat protein teach us about developing an aids vaccine? Retrovirology 2009, 6, 50. [Google Scholar] [CrossRef]
- Goldstein, G. Hiv-1 tat protein as a potential aids vaccine. Nat. Med. 1996, 2, 960–964. [Google Scholar] [CrossRef]
- Bellino, S.; Francavilla, V.; Longo, O.; Tripiciano, A.; Paniccia, G.; Arancio, A.; Fiorelli, V.; Scoglio, A.; Collacchi, B.; Campagna, M.; et al. Parallel conduction of the phase i preventive and therapeutic trials based on the tat vaccine candidate. Rev. Recent Clin. Trials 2009, 4, 195–204. [Google Scholar] [CrossRef]
- Ensoli, B.; Bellino, S.; Tripiciano, A.; Longo, O.; Francavilla, V.; Marcotullio, S.; Cafaro, A.; Picconi, O.; Paniccia, G.; Scoglio, A.; et al. Therapeutic immunization with hiv-1 tat reduces immune activation and loss of regulatory t-cells and improves immune function in subjects on haart. PLoS One 2010, 5, e13540. [Google Scholar]
- Ensoli, B.; Fiorelli, V.; Ensoli, F.; Lazzarin, A.; Visintini, R.; Narciso, P.; di Carlo, A.; Monini, P.; Magnani, M.; Garaci, E. The therapeutic phase i trial of the recombinant native hiv-1 tat protein. AIDS 2008, 22, 2207–2209. [Google Scholar] [CrossRef]
- Gavioli, R.; Cellini, S.; Castaldello, A.; Voltan, R.; Gallerani, E.; Gagliardoni, F.; Fortini, C.; Cofano, E.B.; Triulzi, C.; Cafaro, A.; et al. The tat protein broadens t cell responses directed to the hiv-1 antigens gag and env: Implications for the design of new vaccination strategies against aids. Vaccine 2008, 26, 727–737. [Google Scholar] [CrossRef]
- Longo, O.; Tripiciano, A.; Fiorelli, V.; Bellino, S.; Scoglio, A.; Collacchi, B.; Alvarez, M.J.; Francavilla, V.; Arancio, A.; Paniccia, G.; et al. Phase i therapeutic trial of the hiv-1 tat protein and long term follow-up. Vaccine 2009, 27, 3306–3312. [Google Scholar] [CrossRef]
- Goldstein, G.; Chicca, J. Exploratory clinical studies of a synthetic hiv-1 tat epitope vaccine in asymptomatic treatment-naive and antiretroviral-controlled hiv-1 infected subjects plus healthy uninfected subjects. Hum. Vaccin Immunother. 2012, 8, 479–485. [Google Scholar]
- Goldstein, G.; Chicca, J.J., 2nd. A universal anti-hiv-1 tat epitope vaccine that is fully synthetic and self-adjuvanting. Vaccine 2010, 28, 1008–1014. [Google Scholar] [CrossRef]
- Allard, S.D.; de Keersmaecker, B.; de Goede, A.L.; Verschuren, E.J.; Koetsveld, J.; Reedijk, M.L.; Wylock, C.; de Bel, A.V.; Vandeloo, J.; Pistoor, F.; et al. A phase i/iia immunotherapy trial of hiv-1-infected patients with tat, rev and nef expressing dendritic cells followed by treatment interruption. Clin. Immunol. 2012, 142, 252–268. [Google Scholar] [CrossRef]
- Mediouni, S.; Watkins, J.D.; Pierres, M.; Bole, A.; Loret, E.P.; Baillat, G. A monoclonal antibody directed against a conformational epitope of the hiv-1 trans-activator (tat) protein neutralizes cross-clade. J. Biol. Chem. 2012, 287, 11942–11950. [Google Scholar]
- Kutsch, O.; Levy, D.N.; Bates, P.J.; Decker, J.; Kosloff, B.R.; Shaw, G.M.; Priebe, W.; Benveniste, E.N. Bis-anthracycline antibiotics inhibit human immunodeficiency virus type 1 transcription. Antimicrob. Agents Chemother. 2004, 48, 1652–1663. [Google Scholar] [CrossRef]
- Mischiati, C.; Jeang, K.T.; Feriotto, G.; Breda, L.; Borgatti, M.; Bianchi, N.; Gambari, R. Aromatic polyamidines inhibiting the tat-induced hiv-1 transcription recognize structured tar-rna. Antisense Nucleic Acid Drug Dev. 2001, 11, 209–217. [Google Scholar] [CrossRef]
- Yu, X.; Lin, W.; Li, J.; Yang, M. Synthesis and biological evaluation of novel beta-carboline derivatives as tat-tar interaction inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 3127–3130. [Google Scholar]
- Yu, X.; Lin, W.; Pang, R.; Yang, M. Design, synthesis and bioactivities of tar rna targeting beta-carboline derivatives based on tat-tar interaction. Eur. J. Med. Chem. 2005, 40, 831–839. [Google Scholar] [CrossRef]
- Ankel, H.; Turriziani, O.; Antonelli, G. Prostaglandin a inhibits replication of human immunodeficiency virus during acute infection. J. Gen. Virol. 1991, 72, 2797–2800. [Google Scholar] [CrossRef]
- Hughes-Fulford, M.; McGrath, M.S.; Hanks, D.; Erickson, S.; Pulliam, L. Effects of dimethyl prostaglandin a1 on herpes simplex virus and human immunodeficiency virus replication. Antimicrob. Agents Chemother. 1992, 36, 2253–2258. [Google Scholar] [CrossRef]
- Rozera, C.; Carattoli, A.; de Marco, A.; Amici, C.; Giorgi, C.; Santoro, M.G. Inhibition of hiv-1 replication by cyclopentenone prostaglandins in acutely infected human cells. Evidence for a transcriptional block. J. Clin. Invest. 1996, 97, 1795–1803. [Google Scholar] [CrossRef]
- Li, C.J.; Zhang, L.J.; Dezube, B.J.; Crumpacker, C.S.; Pardee, A.B. Three inhibitors of type 1 human immunodeficiency virus long terminal repeat-directed gene expression and virus replication. Proc. Natl. Acad. Sci. USA 1993, 90, 1839–1842. [Google Scholar] [CrossRef]
- Valente, S.T.; Gilmartin, G.M.; Venkatarama, K.; Arriagada, G.; Goff, S.P. Hiv-1 mrna 3' end processing is distinctively regulated by eif3f, cdk11, and splice factor 9g8. Mol. Cell. 2009, 36, 279–289. [Google Scholar] [CrossRef]
- Mousseau, G.; Clementz, M.A.; Bakeman, W.N.; Nagarsheth, N.; Cameron, M.; Shi, J.; Baran, P.; Fromentin, R.; Chomont, N.; Valente, S.T. An analog of the natural steroidal alkaloid cortistatin a potently suppresses tat-dependent hiv transcription. Cell. Host Microbe 2012, 12, 97–108. [Google Scholar] [CrossRef]
- Mei, H.Y.; Mack, D.P.; Galan, A.A.; Halim, N.S.; Heldsinger, A.; Loo, J.A.; Moreland, D.W.; Sannes-Lowery, K.A.; Sharmeen, L.; Truong, H.N.; et al. Discovery of selective, small-molecule inhibitors of rna complexes—i. The tat protein/tar rna complexes required for hiv-1 transcription. Bioorg. Med. Chem. 1997, 5, 1173–1184. [Google Scholar] [CrossRef]
- Xavier, K.A.; Eder, P.S.; Giordano, T. Rna as a drug target: Methods for biophysical characterization and screening. Trends Biotechnol. 2000, 18, 349–356. [Google Scholar] [CrossRef]
- Gelus, N.; Bailly, C.; Hamy, F.; Klimkait, T.; Wilson, W.D.; Boykin, D.W. Inhibition of hiv-1 tat-tar interaction by diphenylfuran derivatives: Effects of the terminal basic side chains. Bioorg. Med. Chem. 1999, 7, 1089–1096. [Google Scholar] [CrossRef]
- Hamy, F.; Brondani, V.; Florsheimer, A.; Stark, W.; Blommers, M.J.; Klimkait, T. A new class of hiv-1 tat antagonist acting through tat-tar inhibition. Biochemistry 1998, 37, 5086–5095. [Google Scholar]
- Mei, H.-Y.; Galan, A.A.; Halim, N.S.; Mack, D.P.; Moreland, D.W.; Sanders, K.B.; Hoa, N.T.; Czarnik, A.W. Inhibition of an hiv-1 tat-derived peptide binding to tar rna by aminoglycoside antibiotics. Bioorg. Med. Chem. Lett. 1995, 5, 2755–2760. [Google Scholar] [CrossRef]
- Hsu, M.C.; Schutt, A.D.; Holly, M.; Slice, L.W.; Sherman, M.I.; Richman, D.D.; Potash, M.J.; Volsky, D.J. Inhibition of hiv replication in acute and chronic infections In vitro by a tat antagonist. Science 1991, 254, 1799–1802. [Google Scholar]
- Hsu, M.C.; Dhingra, U.; Earley, J.V.; Holly, M.; Keith, D.; Nalin, C.M.; Richou, A.R.; Schutt, A.D.; Tam, S.Y.; Potash, M.J.; et al. Inhibition of type 1 human immunodeficiency virus replication by a tat antagonist to which the virus remains sensitive after prolonged exposure In vitro. Proc. Natl. Acad. Sci. USA 1993, 90, 6395–6399. [Google Scholar]
- Dunne, A.L.; Siregar, H.; Mills, J.; Crowe, S.M. Hiv replication in chronically infected macrophages is not inhibited by the tat inhibitors ro-5-3335 and ro-24-7429. J. Leukoc. Biol. 1994, 56, 369–373. [Google Scholar]
- Witvrouw, M.; Pauwels, R.; Vandamme, A.M.; Schols, D.; Reymen, D.; Yamamoto, N.; Desmyter, J.; de Clercq, E. Cell type-specific anti-human immunodeficiency virus type 1 activity of the transactivation inhibitor ro5-3335. Antimicrob. Agents Chemother. 1992, 36, 2628–2633. [Google Scholar] [CrossRef]
- Braddock, M.; Cannon, P.; Muckenthaler, M.; Kingsman, A.J.; Kingsman, S.M. Inhibition of human immunodeficiency virus type 1 tat-dependent activation of translation in xenopus oocytes by the benzodiazepine ro24-7429 requires trans-activation response element loop sequences. J. Virol. 1994, 68, 25–33. [Google Scholar]
- Michne, W.F.; Schroeder, J.D.; Bailey, T.R.; Young, D.C.; Hughes, J.V.; Dutko, F.J. Keto/enol epoxy steroids: A new structural class of hiv-1 tat inhibitors. J. Med. Chem. 1993, 36, 2701–2702. [Google Scholar] [CrossRef]
- Pang, R.; Zhang, C.; Yuan, D.; Yang, M. Design and sar of new substituted purines bearing aryl groups at n9 position as hiv-1 tat-tar interaction inhibitors. Bioorg. Med. Chem. 2008, 16, 8178–8186. [Google Scholar] [CrossRef]
- Yuan, D.; He, M.; Pang, R.; Lin, S.S.; Li, Z.; Yang, M. The design, synthesis, and biological evaluation of novel substituted purines as hiv-1 tat-tar inhibitors. Bioorg. Med. Chem. 2007, 15, 265–272. [Google Scholar] [CrossRef]
- Uchiumi, F.; Maruta, H.; Inoue, J.; Yamamoto, T.; Tanuma, S. Inhibitory effect of tannic acid on human immunodeficiency virus promoter activity induced by 12-o-tetra decanoylphorbol-13-acetate in jurkat t-cells. Biochem. Biophys. Res. Commun. 1996, 220, 411–417. [Google Scholar] [CrossRef]
- Chandra, A.; Demirhan, I.; Arya, S.K.; Chandra, P. D-penicillamine inhibits transactivation of human immunodeficiency virus type-1 (hiv-1) ltr by transactivator protein. FEBS Lett. 1988, 236, 282–286. [Google Scholar] [CrossRef]
- Chandra, P.; Sarin, P.S. Selective inhibition of replication of the aids-associated virus htlv-iii/lav by synthetic d-penicillamine. Arzneimittelforschung 1986, 36, 184–186. [Google Scholar]
- Kalebic, T.; Schein, P.S. Organic thiophosphate wr-151327 suppresses expression of hiv in chronically infected cells. AIDS Res. Hum. Retroviruses 1994, 10, 727–733. [Google Scholar] [CrossRef]
- Li, C.J.; Wang, C.; Pardee, A.B. Camptothecin inhibits tat-mediated transactivation of type 1 human immunodeficiency virus. J. Biol.Chem. 1994, 269, 7051–7054. [Google Scholar]
- Jayasuriya, H.; Lingham, R.B.; Graham, P.; Quamina, D.; Herranz, L.; Genilloud, O.; Gagliardi, M.; Danzeisen, R.; Tomassini, J.E.; Zink, D.L.; et al. Durhamycin a, a potent inhibitor of hiv tat transactivation. J. Nat. Prod. 2002, 65, 1091–1095. [Google Scholar] [CrossRef]
- Jayasuriya, H.; Zink, D.L.; Polishook, J.D.; Bills, G.F.; Dombrowski, A.W.; Genilloud, O.; Pelaez, F.F.; Herranz, L.; Quamina, D.; Lingham, R.B.; et al. Identification of diverse microbial metabolites as potent inhibitors of hiv-1 tat transactivation. Chem. Biodivers. 2005, 2, 112–122. [Google Scholar] [CrossRef]
- Uchiumi, F.; Hatano, T.; Ito, H.; Yoshida, T.; Tanuma, S. Transcriptional suppression of the hiv promoter by natural compounds. Antiviral Res. 2003, 58, 89–98. [Google Scholar] [CrossRef]
- Bedoya, L.M.; del Olmo, E.; Sancho, R.; Barboza, B.; Beltran, M.; Garcia-Cadenas, A.E.; Sanchez-Palomino, S.; Lopez-Perez, J.L.; Munoz, E.; San Feliciano, A.; et al. Anti-hiv activity of stilbene-related heterocyclic compounds. Bioorg. Med. Chem. Lett. 2006, 16, 4075–4079. [Google Scholar]
- Bedoya, L.M.; Beltran, M.; Sancho, R.; Olmedo, D.A.; Sanchez-Palomino, S.; del Olmo, E.; Lopez-Perez, J.L.; Munoz, E.; San Feliciano, A.; Alcami, J. 4-phenylcoumarins as hiv transcription inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 4447–4450. [Google Scholar] [CrossRef]
- Barthelemy, S.; Vergnes, L.; Moynier, M.; Guyot, D.; Labidalle, S.; Bahraoui, E. Curcumin and curcumin derivatives inhibit tat-mediated transactivation of type 1 human immunodeficiency virus long terminal repeat. Res. Virol. 1998, 149, 43–52. [Google Scholar] [CrossRef]
- Kalantari, P.; Narayan, V.; Henderson, A.J.; Prabhu, K.S. 15-deoxy-delta12,14-prostaglandin j2 inhibits hiv-1 transactivating protein, tat, through covalent modification. FASEB J. 2009, 23, 2366–2373. [Google Scholar] [CrossRef]
- Kim, S.E.; Lee, E.O.; Yang, J.H.; Kang, J.H.; Suh, Y.H.; Chong, Y.H. 15-deoxy-delta12,14-prostaglandin j2 inhibits human immunodeficiency virus-1 tat-induced monocyte chemoattractant protein-1/ccl2 production by blocking the extracellular signal-regulated kinase-1/2 signaling pathway independently of peroxisome proliferator-activated receptor-gamma and heme oxygenase-1 in rat hippocampal slices. J. Neurosci. Res. 2012, 90, 1732–1742. [Google Scholar] [CrossRef]
- Narayan, V.; Ravindra, K.C.; Chiaro, C.; Cary, D.; Aggarwal, B.B.; Henderson, A.J.; Prabhu, K.S. Celastrol inhibits tat-mediated human immunodeficiency virus (hiv) transcription and replication. J. Mol. Biol. 2011, 410, 972–983. [Google Scholar] [CrossRef]
- Baba, M.; Okamoto, M.; Takeuchi, H. Inhibition of human immunodeficiency virus type 1 replication in acutely and chronically infected cells by em2487, a novel substance produced by a streptomyces species. Antimicrob. Agents Chemother. 1999, 43, 2350–2355. [Google Scholar]
- Shoji, S.; Furuishi, K.; Misumi, S.; Miyazaki, T.; Kino, M.; Yamataka, K. Thiamine disulfide as a potent inhibitor of human immunodeficiency virus (type-1) production. Biochem. Biophys. Res. Commun. 1994, 205, 967–975. [Google Scholar] [CrossRef]
- Kira, T.; Hashimoto, K.; Baba, M.; Okamoto, T.; Shigeta, S. 2-glycineamide-5-chlorophenyl 2-pyrryl ketone, a non-benzodiazepin tat antagonist, is effective against acute and chronic hiv-1 infections in vitro. Antiviral Res. 1996, 32, 55–62. [Google Scholar] [CrossRef]
- Chande, A.G.; Baba, M.; Mukhopadhyaya, R. Short communication: A single step assay for rapid evaluation of inhibitors targeting hiv type 1 tat-mediated long terminal repeat transactivation. AIDS Res. Hum. Retroviruses 2012, 28, 902–906. [Google Scholar] [CrossRef]
- Wang, X.; Yamataka, K.; Okamoto, M.; Ikeda, S.; Baba, M. Potent and selective inhibition of tat-dependent hiv-1 replication in chronically infected cells by a novel naphthalene derivative jtk-101. Antivir. Chem. Chemother. 2007, 18, 201–211. [Google Scholar]
- Davidson, A.; Begley, D.W.; Lau, C.; Varani, G. A small-molecule probe induces a conformation in hiv tar rna capable of binding drug-like fragments. J. Mol. Biol. 2011, 410, 984–996. [Google Scholar] [CrossRef]
- Murchie, A.I.; Davis, B.; Isel, C.; Afshar, M.; Drysdale, M.J.; Bower, J.; Potter, A.J.; Starkey, I.D.; Swarbrick, T.M.; Mirza, S.; et al. Structure-based drug design targeting an inactive rna conformation: Exploiting the flexibility of hiv-1 tar rna. J. Mol. Biol. 2004, 336, 625–638. [Google Scholar] [CrossRef]
- Hwang, S.; Tamilarasu, N.; Kibler, K.; Cao, H.; Ali, A.; Ping, Y.H.; Jeang, K.T.; Rana, T.M. Discovery of a small molecule tat-trans-activation-responsive rna antagonist that potently inhibits human immunodeficiency virus-1 replication. J. Biol. Chem. 2003, 278, 39092–39103. [Google Scholar]
- Hamy, F.; Felder, E.; Lipson, K.; Klimkait, T. Merged screening for human immunodeficiency virus tat and rev inhibitors. J. Biomol. Screen. 2001, 6, 179–187. [Google Scholar] [CrossRef]
- Hamy, F.; Gelus, N.; Zeller, M.; Lazdins, J.L.; Bailly, C.; Klimkait, T. Blocking hiv replication by targeting tat protein. Chem. Biol. 2000, 7, 669–676. [Google Scholar] [CrossRef]
- Hamasaki, K.; Ueno, A. Aminoglycoside antibiotics, neamine and its derivatives as potent inhibitors for the rna-protein interactions derived from hiv-1 activators. Bioorg. Med. Chem. Lett. 2001, 11, 591–594. [Google Scholar] [CrossRef]
- Yajima, S.; Shionoya, H.; Akagi, T.; Hamasaki, K. Neamine derivatives having a nucleobase with a lysine or an arginine as a linker, their synthesis and evaluation as potential inhibitors for hiv tar-tat. Bioorg. Med. Chem. 2006, 14, 2799–2809. [Google Scholar] [CrossRef]
- Massari, S.; Daelemans, D.; Barreca, M.L.; Knezevich, A.; Sabatini, S.; Cecchetti, V.; Marcello, A.; Pannecouque, C.; Tabarrini, O. A 1,8-naphthyridone derivative targets the hiv-1 tat-mediated transcription and potently inhibits the hiv-1 replication. J. Med. Chem. 2010, 53, 641–648. [Google Scholar] [CrossRef]
- Parolin, C.; Gatto, B.; Del Vecchio, C.; Pecere, T.; Tramontano, E.; Cecchetti, V.; Fravolini, A.; Masiero, S.; Palumbo, M.; Palu, G. New anti-human immunodeficiency virus type 1 6-aminoquinolones: Mechanism of action. Antimicrob. Agents Chemother. 2003, 47, 889–896. [Google Scholar] [CrossRef]
- Lind, K.E.; Du, Z.; Fujinaga, K.; Peterlin, B.M.; James, T.L. Structure-based computational database screening, In vitro assay, and nmr assessment of compounds that target tar rna. Chem. Biol. 2002, 9, 185–193. [Google Scholar] [CrossRef]
- Mayer, M.; James, T.L. Nmr-based characterization of phenothiazines as a rna binding scaffold. J. Am. Chem. Soc. 2004, 126, 4453–4460. [Google Scholar] [CrossRef]
- Renner, S.; Ludwig, V.; Boden, O.; Scheffer, U.; Gobel, M.; Schneider, G. New inhibitors of the tat-tar rna interaction found with a “fuzzy” pharmacophore model. Chembiochem 2005, 6, 1119–1125. [Google Scholar] [CrossRef]
- Filikov, A.V.; Mohan, V.; Vickers, T.A.; Griffey, R.H.; Cook, P.D.; Abagyan, R.A.; James, T.L. Identification of ligands for rna targets via structure-based virtual screening: Hiv-1 tar. J. Comput. Aided Mol. Des. 2000, 14, 593–610. [Google Scholar] [CrossRef]
- Schuller, A.; Suhartono, M.; Fechner, U.; Tanrikulu, Y.; Breitung, S.; Scheffer, U.; Gobel, M.W.; Schneider, G. The concept of template-based de novo design from drug-derived molecular fragments and its application to tar rna. J. Comput. Aided Mol. Des. 2008, 22, 59–68. [Google Scholar] [CrossRef]
- Stelzer, A.C.; Frank, A.T.; Kratz, J.D.; Swanson, M.D.; Gonzalez-Hernandez, M.J.; Lee, J.; Andricioaei, I.; Markovitz, D.M.; Al-Hashimi, H.M. Discovery of selective bioactive small molecules by targeting an rna dynamic ensemble. Nat. Chem. Biol. 2011, 7, 553–559. [Google Scholar] [CrossRef]
- Lapidot, A.; Berchanski, A.; Borkow, G. Insight into the mechanisms of aminoglycoside derivatives interaction with hiv-1 entry steps and viral gene transcription. FEBS J. 2008, 275, 5236–5257. [Google Scholar] [CrossRef]
- Litovchick, A.; Evdokimov, A.G.; Lapidot, A. Arginine-aminoglycoside conjugates that bind to hiv transactivation responsive element rna in vitro. FEBS Lett. 1999, 445, 73–79. [Google Scholar] [CrossRef]
- Litovchick, A.; Evdokimov, A.G.; Lapidot, A. Aminoglycoside-arginine conjugates that bind tar rna: Synthesis, characterization, and antiviral activity. Biochemistry 2000, 39, 2838–2852. [Google Scholar] [CrossRef]
- Litovchick, A.; Lapidot, A.; Eisenstein, M.; Kalinkovich, A.; Borkow, G. Neomycin b-arginine conjugate, a novel hiv-1 tat antagonist: Synthesis and anti-hiv activities. Biochemistry 2001, 40, 15612–15623. [Google Scholar] [CrossRef]
- Cabrera, C.; Gutierrez, A.; Blanco, J.; Barretina, J.; Litovchick, A.; Lapidot, A.; Evdokimov, A.G.; Clotet, B.; Este, J.A. Anti-human immunodeficiency virus activity of novel aminoglycoside-arginine conjugates at early stages of infection. AIDS Res. Hum. Retroviruses 2000, 16, 627–634. [Google Scholar] [CrossRef]
- Catani, M.V.; Corasaniti, M.T.; Ranalli, M.; Amantea, D.; Litovchick, A.; Lapidot, A.; Melino, G. The tat antagonist neomycin b hexa-arginine conjugate inhibits gp-120-induced death of human neuroblastoma cells. J. Neurochem. 2003, 84, 1237–1245. [Google Scholar] [CrossRef]
- Carriere, M.; Vijayabaskar, V.; Applefield, D.; Harvey, I.; Garneau, P.; Lorsch, J.; Lapidot, A.; Pelletier, J. Inhibition of protein synthesis by aminoglycoside-arginine conjugates. RNA 2002, 8, 1267–1279. [Google Scholar] [CrossRef]
- Turpin, J.A.; Buckheit, R.W., Jr.; Derse, D.; Hollingshead, M.; Williamson, K.; Palamone, C.; Osterling, M.C.; Hill, S.A.; Graham, L.; Schaeffer, C.A.; et al. Inhibition of acute-, latent-, and chronic-phase human immunodeficiency virus type 1 (hiv-1) replication by a bistriazoloacridone analog that selectively inhibits hiv-1 transcription. Antimicrob. Agents Chemother. 1998, 42, 487–494. [Google Scholar]
- Tabarrini, O.; Massari, S.; Cecchetti, V. 6-desfluoroquinolones as hiv-1 tat-mediated transcription inhibitors. Future Med. Chem. 2010, 2, 1161–1180. [Google Scholar] [CrossRef]
- Baba, M.; Okamoto, M.; Makino, M.; Kimura, Y.; Ikeuchi, T.; Sakaguchi, T.; Okamoto, T. Potent and selective inhibition of human immunodeficiency virus type 1 transcription by piperazinyloxoquinoline derivatives. Antimicrob. Agents Chemother. 1997, 41, 1250–1255. [Google Scholar]
- Witvrouw, M.; Daelemans, D.; Pannecouque, C.; Neyts, J.; Andrei, G.; Snoeck, R.; Vandamme, A.M.; Balzarini, J.; Desmyter, J.; Baba, M.; et al. Broad-spectrum antiviral activity and mechanism of antiviral action of the fluoroquinolone derivative k-12. Antivir. Chem. Chemother. 1998, 9, 403–411. [Google Scholar]
- Okamoto, M.; Okamoto, T.; Baba, M. Inhibition of human immunodeficiency virus type 1 replication by combination of transcription inhibitor k-12 and other antiretroviral agents in acutely and chronically infected cells. Antimicrob. Agents Chemother. 1999, 43, 492–497. [Google Scholar]
- Cecchetti, V.; Parolin, C.; Moro, S.; Pecere, T.; Filipponi, E.; Calistri, A.; Tabarrini, O.; Gatto, B.; Palumbo, M.; Fravolini, A.; Palu, G. 6-aminoquinolones as new potential anti-hiv agents. J. Med. Chem. 2000, 43, 3799–3802. [Google Scholar] [CrossRef]
- Richter, S.; Parolin, C.; Gatto, B.; Del Vecchio, C.; Brocca-Cofano, E.; Fravolini, A.; Palu, G.; Palumbo, M. Inhibition of human immunodeficiency virus type 1 tat-trans-activation-responsive region interaction by an antiviral quinolone derivative. Antimicrob. Agents Chemother. 2004, 48, 1895–1899. [Google Scholar]
- Tabarrini, O.; Massari, S.; Sancineto, L.; Daelemans, D.; Sabatini, S.; Manfroni, G.; Cecchetti, V.; Pannecouque, C. Structural investigation of the naphthyridone scaffold: Identification of a 1,6-naphthyridone derivative with potent and selective anti-hiv activity. ChemMedChem 2011, 6, 1249–1257. [Google Scholar] [CrossRef]
- Stevens, M.; Pollicita, M.; Pannecouque, C.; Verbeken, E.; Tabarrini, O.; Cecchetti, V.; Aquaro, S.; Perno, C.F.; Fravolini, A.; de Clercq, E.; Schols, D.; et al. Novel in vivo model for the study of human immunodeficiency virus type 1 transcription inhibitors: Evaluation of new 6-desfluoroquinolone derivatives. Antimicrob. Agents Chemother. 2007, 51, 1407–1413. [Google Scholar] [CrossRef]
- Tabarrini, O.; Massari, S.; Daelemans, D.; Stevens, M.; Manfroni, G.; Sabatini, S.; Balzarini, J.; Cecchetti, V.; Pannecouque, C.; Fravolini, A. Structure-activity relationship study on anti-hiv 6-desfluoroquinolones. J. Med. Chem. 2008, 51, 5454–5458. [Google Scholar]
- Tabarrini, O.; Stevens, M.; Cecchetti, V.; Sabatini, S.; Dell'Uomo, M.; Manfroni, G.; Palumbo, M.; Pannecouque, C.; de Clercq, E.; Fravolini, A. Structure modifications of 6-aminoquinolones with potent anti-hiv activity. J. Med. Chem. 2004, 47, 5567–5578. [Google Scholar] [CrossRef]
- Palmer, S.; Maldarelli, F.; Wiegand, A.; Bernstein, B.; Hanna, G.J.; Brun, S.C.; Kempf, D.J.; Mellors, J.W.; Coffin, J.M.; King, M.S. Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2008, 105, 3879–3884. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mousseau, G.; Valente, S. Strategies to Block HIV Transcription: Focus on Small Molecule Tat Inhibitors. Biology 2012, 1, 668-697. https://doi.org/10.3390/biology1030668
Mousseau G, Valente S. Strategies to Block HIV Transcription: Focus on Small Molecule Tat Inhibitors. Biology. 2012; 1(3):668-697. https://doi.org/10.3390/biology1030668
Chicago/Turabian StyleMousseau, Guillaume, and Susana Valente. 2012. "Strategies to Block HIV Transcription: Focus on Small Molecule Tat Inhibitors" Biology 1, no. 3: 668-697. https://doi.org/10.3390/biology1030668