
In Silico Prediction of the Phosphorylation of NS3 as an Essential Mechanism for Dengue Virus Replication and the Antiviral Activity of Quercetin



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Predicting Phosphorylation
2.2. System Preparation for Molecular Simulation
2.2.1. Initial Structure
2.2.2. Phosphorylated Structure
2.2.3. Solvation
2.2.4. Simulations Steps
2.2.5. Minimization
2.2.6. Heating
2.2.7. Equilibration
2.2.8. MD Simulation
2.2.9. Trajectories Analysis
2.2.10. Docking
2.2.11. Binding Pocket Calculation
3. Results
3.1. Phosphorylation Sites Prediction
3.2. Effects of NS3 Phosphorylation and Conformational Change in Protein Structure
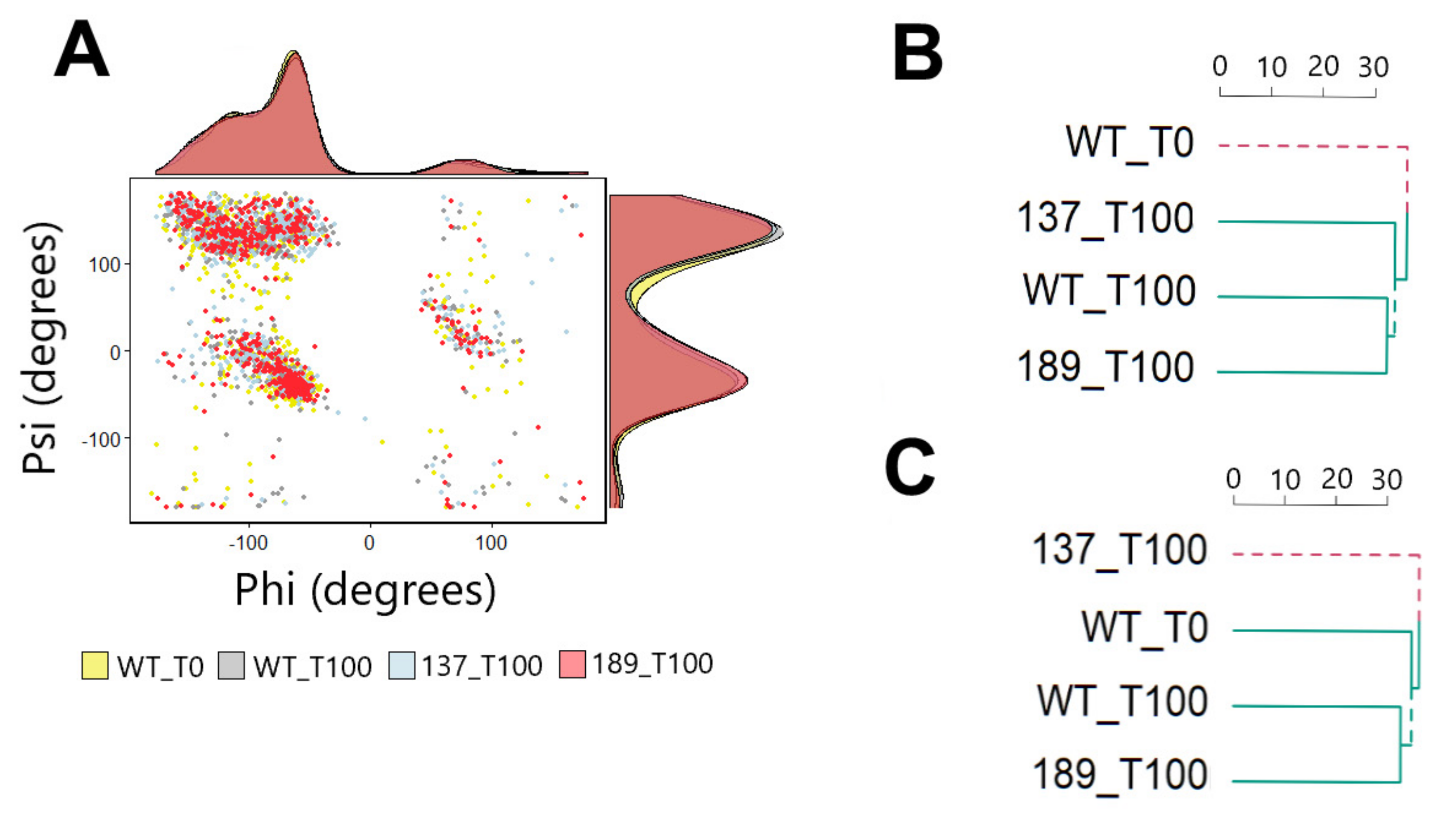
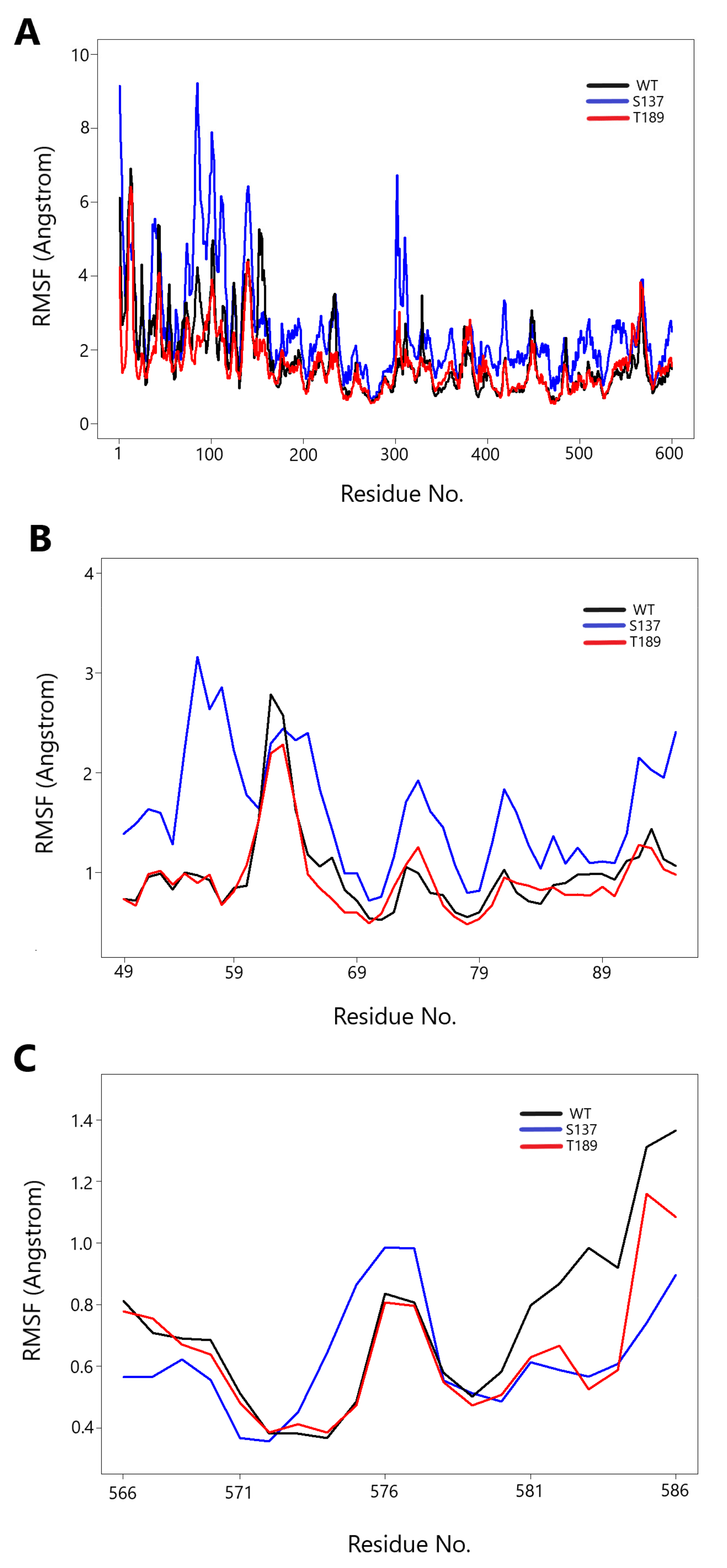
3.3. Protein Movement Changes with Phosphorylation
3.4. Hydrophobicity of the NS3–NS5 Contact Site Increases with Phosphorylation
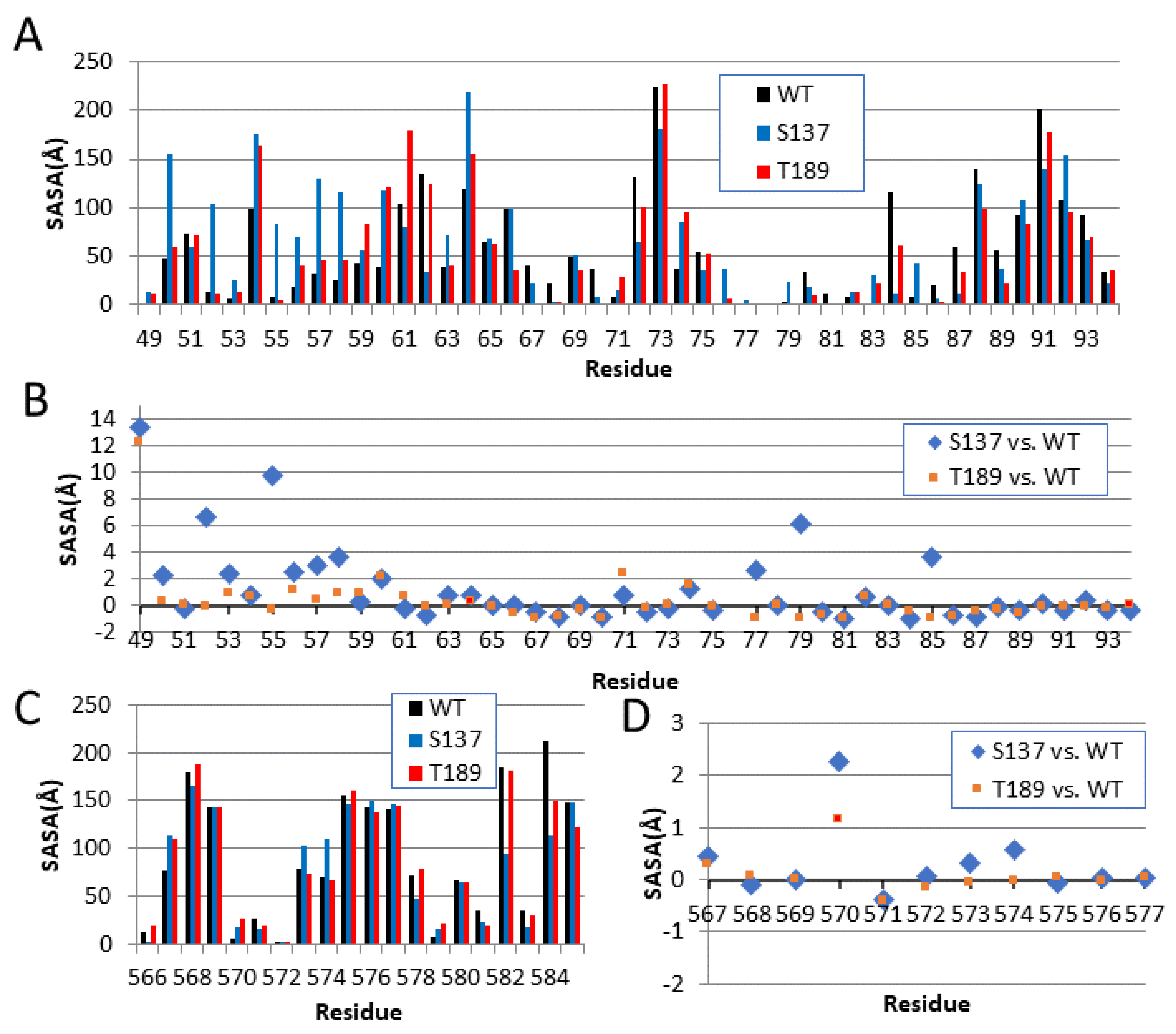
3.5. Solvent Accessibility in the NS3-NS2B Interaction and the Catalytic Domain
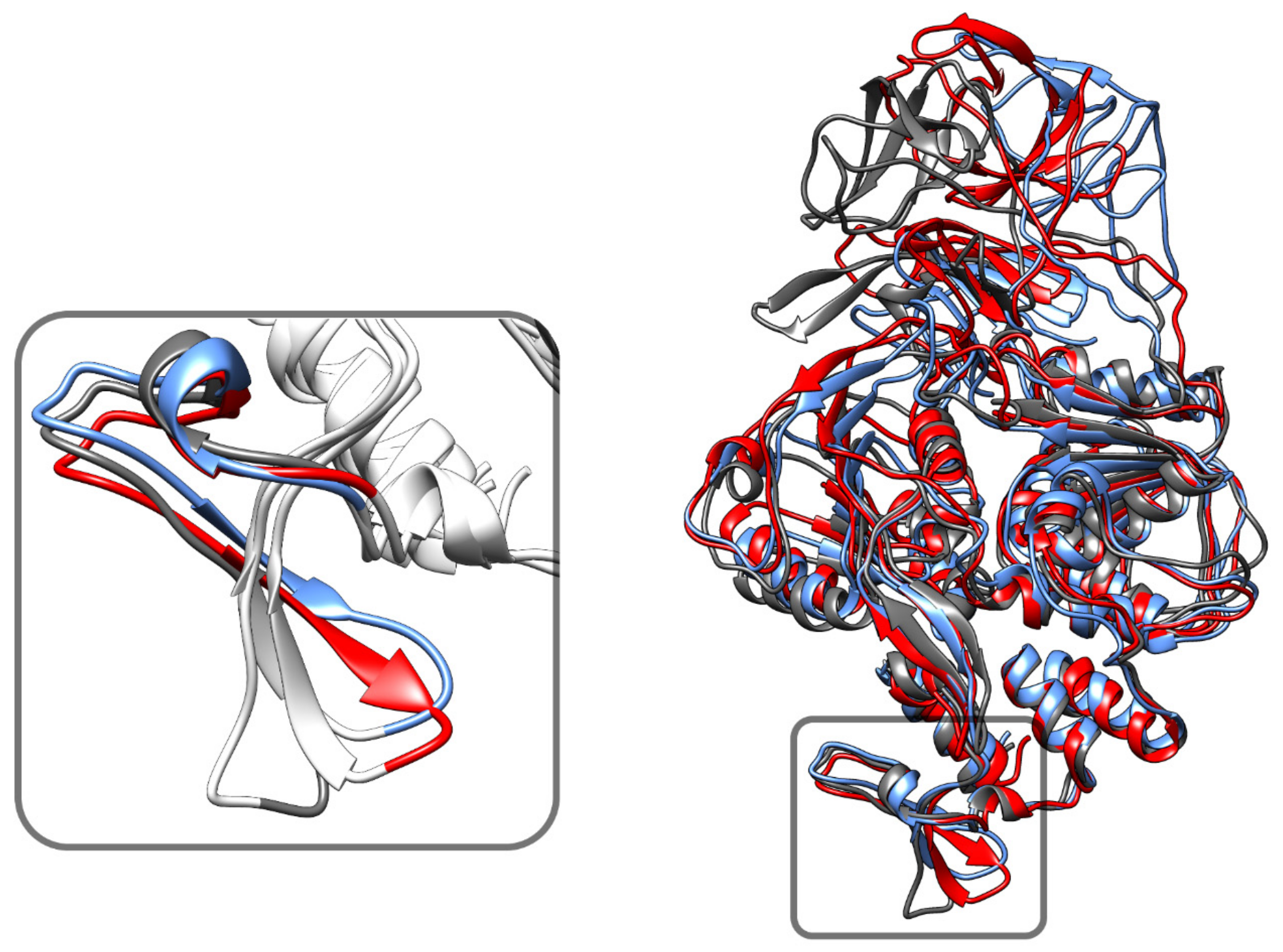
3.6. Phosphorylation Induced Site-Specific Structural Changes
3.7. Docking Analysis of NS3–NS5 Interaction
3.8. Interactions of NS3 with Quercetin
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GPS | Netphos | ScanSite | ||||||
|---|---|---|---|---|---|---|---|---|
| Kinase | Position | Score | Kinase | Position | Score | Kinase | Position | Score |
| MAPK | 9 | 23.1 | cdk5 | 9 | 0.59 | CDK5 | 9 | 0.64 |
| SRC | 23 | 10.1 | SRC | 23 | 0.49 | |||
| PIKK | 34 | 9.57 | ||||||
| PKC | 45 | 9.56 | PRKCZ(PKC) | 45 | 0.52 | |||
| PKC | 122 | 0.55 | PRKCE(PKC) | 68 | 0.49 | |||
| PIKK | 127 | 9.45 | ||||||
| MAPK | 131 | 28 | cdk5 | 131 | 0.68 | GSK3A | 131 | 0.56 |
| MAPK | 134 | 11.3 | MAPK3 | 135 | 0.61 | |||
| MAPK | 137 | 28.8 | GSK3 | 137 | 0.49 | CDK1 | 137 | 0.6 |
| PKC | 163 | 0.53 | ||||||
| PKC | 168 | 0.68 | ||||||
| PRKDC(PIKK) | 171 | 0.58 | ||||||
| AKT | 189 | 10.9 | PKB(AKT) | 189 | 0.73 | CAMK2G | 189 | 0.53 |
| PKC | 200 | 0.73 | ||||||
| PKC | 218 | 0.71 | ||||||
| MAPK | 244 | 25.7 | cdk5 | 244 | 0.65 | CDK1 | 244 | 0.63 |
| PKC | 266 | 0.85 | ||||||
| MAPK | 271 | 31.4 | cdk5 | 271 | 0.65 | AKT1 | 271 | 0.69 |
| PKC | 293 | 0.53 | PRKCD(PKC) | 293 | 0.49 | |||
| PKC | 301 | 0.67 | ||||||
| MAPK | 317 | 28.3 | cdk5 | 317 | 0.65 | MAPK3 | 317 | 0.63 |
| MAPK | 317 | 28.3 | cdk5 | 317 | 0.65 | MAPK3 | 317 | |
| PKC | 352 | 0.6 | GSK3A | 321 | ||||
| PKC | 358 | 0.63 | CAMK2G | 358 | ||||
| PKC | 364 | 9.68 | PRKCA(PKC) | 364 | ||||
| PKC | 386 | 0.61 | CAMK2G | 386 | ||||
| PKC | 389 | 10.7 | PRKACG(PKC) | 389 | ||||
| PKC | 453 | 9.95 | ||||||
| SRC | 472 | 11.9 | ||||||
| MAPK | 500 | 29.9 | MAPK3 | 500 | ||||
| PKC | 507 | 9.59 | ||||||
| SRC | 523 | 10.3 | ||||||
| AKT1 | 602 | |||||||
| MAPK | 317 | 28.3 | cdk5 | 317 | 0.65 | MAPK3 | 317 | |
| PKC | 352 | 0.6 | GSK3A | 321 | ||||
| PKC | 358 | 0.63 | CAMK2G | 358 | ||||
| PKC | 364 | 9.68 | PRKCA(PKC) | 364 | 0.53 | |||
| PKC | 386 | 0.61 | CAMK2G | 386 | 0.56 | |||
| PKC | 389 | 10.7 | PRKACG(PKC) | 389 | 0.6 | |||
| PKC | 453 | 9.95 | ||||||
| SRC | 472 | 11.9 | ||||||
| MAPK | 500 | 29.9 | MAPK3 | 500 | 0.59 | |||
| PKC | 507 | 9.59 | ||||||
| SRC | 523 | 10.3 | ||||||
| AKT1 | 602 | 0.71 | ||||||
| PKC | 364 | 9.68 | PRKCA(PKC) | 364 | 0.53 | |||
| PKC | 386 | 0.61 | CAMK2G | 386 | 0.56 | |||
| PKC | 389 | 10.7 | PRKACG(PKC) | 389 | 0.6 | |||
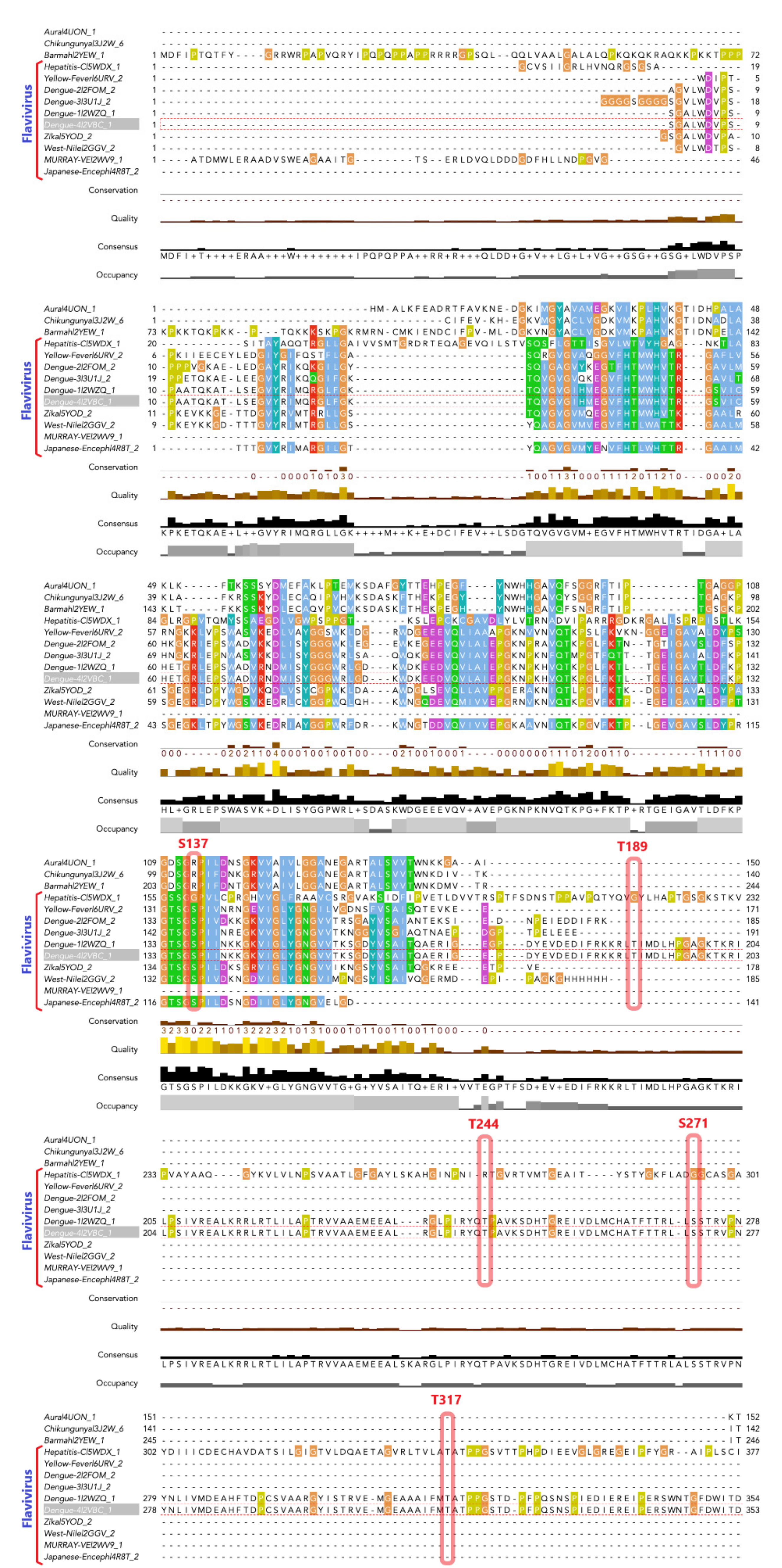
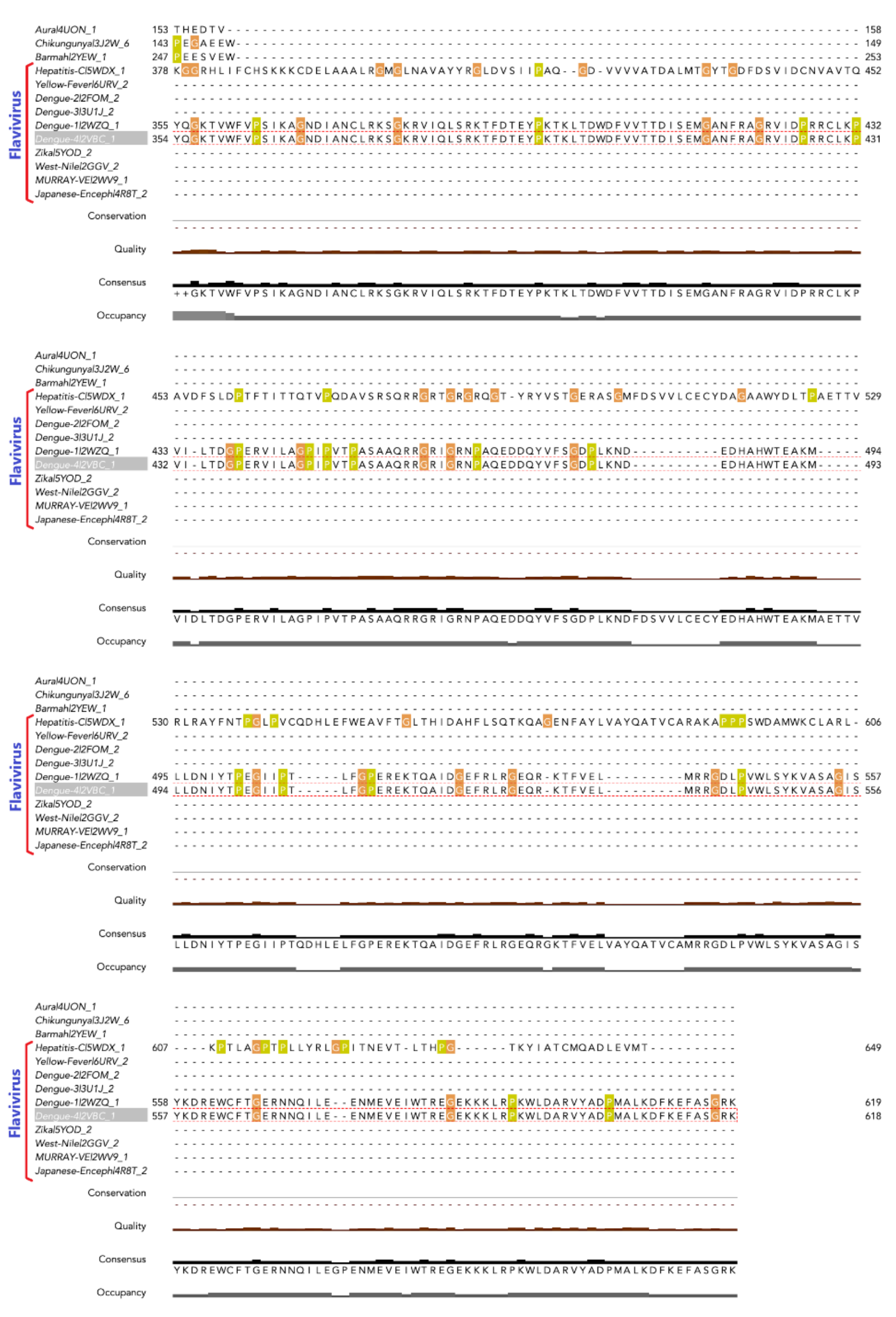
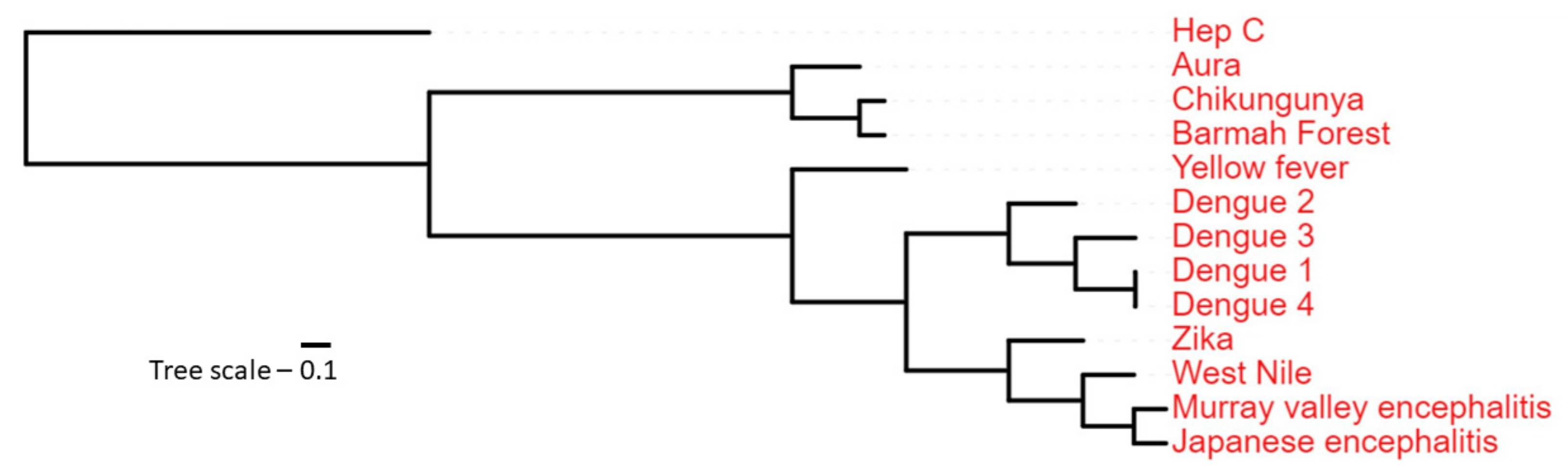

References
- Friedrich, M.J. Asymptomatic People May Contribute to Dengue TransmissionAsymptomatic People May Contribute to Dengue TransmissionGlobal Health. JAMA 2016, 315, 242. [Google Scholar] [CrossRef]
- Beesetti, H.; Khanna, N.; Swaminathan, S. Drugs for dengue: A patent review (2010–2014). Expert Opin. Ther. Pat. 2014, 24, 1171–1184. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.W.K.; Watanabe, S.; Jin, J.Y.; Pompon, J.; Teng, D.; Alonso, S.; Vijaykrishna, D.; Halstead, S.B.; Marzinek, J.K.; Bond, P.J.; et al. A T164S mutation in the dengue virus NS1 protein is associated with greater disease severity in mice. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Murphy, B.R.; Whitehead, S.S. Immune response to dengue virus and prospects for a vaccine. Annu. Rev. Immunol. 2011, 29, 587–619. [Google Scholar] [CrossRef] [PubMed]
- Brady, O.J.; Gething, P.W.; Bhatt, S.; Messina, J.P.; Brownstein, J.S.; Hoen, A.G.; Moyes, C.L.; Farlow, A.W.; Scott, T.W.; Hay, S.I. Refining the global spatial limits of dengue virus transmission by evidence-based consensus. PLoS Negl. Trop Dis. 2012, 6, e1760. [Google Scholar] [CrossRef]
- Gubler, D.J. Dengue, Urbanization and Globalization: The Unholy Trinity of the 21(st) Century. Trop Med. Health 2011, 39, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Bidet, K.; Ho, V.; Chu, C.W.; Naim, A.N.H.; Thazin, K.; Chan, K.R.; Low, J.G.H.; Choy, M.M.; Wong, L.H.; Florez de Sessions, P.; et al. Mimicking immune signatures of flavivirus infection with targeted adjuvants improves dengue subunit vaccine immunogenicity. NPJ Vaccines 2019, 4, 3. [Google Scholar] [CrossRef] [Green Version]
- Messina, J.P.; Brady, O.J.; Golding, N.; Kraemer, M.U.G.; Wint, G.R.W.; Ray, S.E.; Pigott, D.M.; Shearer, F.M.; Johnson, K.; Earl, L.; et al. The current and future global distribution and population at risk of dengue. Nat. Microbiol. 2019. [Google Scholar] [CrossRef]
- Feinberg, M.B.; Ahmed, R. Advancing dengue vaccine development. Science 2017, 358, 865–866. [Google Scholar] [CrossRef]
- Friedrich, M.J. Global Temperature Affects DengueGlobal Temperature Affects DengueGlobal Health. JAMA 2018, 320, 227. [Google Scholar] [CrossRef]
- Freedman, D.; LH, C.; Kozarsky, P. Medical Considerations before International Travel. N. Engl. J. Med. 2016, 375, 247–260. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.L.; Durbin, A.P. Dengue vaccines: Implications for dengue control. Curr. Opin. Infect. Dis. 2017, 30, 449–454. [Google Scholar] [CrossRef]
- Senthilvel, P.; Lavanya, P.; Kumar, K.M.; Swetha, R.; Anitha, P.; Bag, S.; Sarveswari, S.; Vijayakumar, V.; Ramaiah, S.; Anbarasu, A. Flavonoid from Carica papaya inhibits NS2B-NS3 protease and prevents Dengue 2 viral assembly. Bioinformation 2013, 9, 889–895. [Google Scholar] [CrossRef]
- de Sousa, L.R.; Wu, H.; Nebo, L.; Fernandes, J.B.; da Silva, M.F.; Kiefer, W.; Kanitz, M.; Bodem, J.; Diederich, W.E.; Schirmeister, T.; et al. Flavonoids as noncompetitive inhibitors of Dengue virus NS2B-NS3 protease: Inhibition kinetics and docking studies. Bioorg. Med. Chem. 2015, 23, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Bartholomeusz, A.I.; Wright, P.J. Synthesis of dengue virus RNA in vitro: Initiation and the involvement of proteins NS3 and NS5. Arch. Virol. 1993, 128, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Kuhn, R.J.; Rossmann, M.G. A structural perspective of the flavivirus life cycle. Nat. Rev. Microbiol. 2005, 3, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Limjindaporn, T.; Wongwiwat, W.; Noisakran, S.; Srisawat, C.; Netsawang, J.; Puttikhunt, C.; Kasinrerk, W.; Avirutnan, P.; Thiemmeca, S.; Sriburi, R.; et al. Interaction of dengue virus envelope protein with endoplasmic reticulum-resident chaperones facilitates dengue virus production. Biochem. Biophys. Res. Commun. 2009, 379, 196–200. [Google Scholar] [CrossRef]
- Reid, D.W.; Campos, R.K.; Child, J.R.; Zheng, T.; Chan, K.W.K.; Bradrick, S.S.; Vasudevan, S.G.; Garcia-Blanco, M.A.; Nicchitta, C.V. Dengue Virus Selectively Annexes Endoplasmic Reticulum-Associated Translation Machinery as a Strategy for Co-opting Host Cell Protein Synthesis. J. Virol. 2018, 92, e01766-17. [Google Scholar] [CrossRef] [Green Version]
- Neufeldt, C.J.; Cortese, M.; Acosta, E.G.; Bartenschlager, R. Rewiring cellular networks by members of the Flaviviridae family. Nat. Rev. Microbiol. 2018, 16, 125. [Google Scholar] [CrossRef]
- Kuno, G.; Chang, G.J.; Tsuchiya, K.R.; Karabatsos, N.; Cropp, C.B. Phylogeny of the genus Flavivirus. J. Virol. 1998, 72, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Bera, A.K.; Kuhn, R.J.; Smith, J.L. Structure of the Flavivirus helicase: Implications for catalytic activity, protein interactions, and proteolytic processing. J. Virol. 2005, 79, 10268–10277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, D.; Vasudevan, S.G.; Lescar, J. The flavivirus NS2B-NS3 protease-helicase as a target for antiviral drug development. Antivir. Res. 2015, 118, 148–158. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, J.; Li, H. Chapter 7—Flavivirus NS2B/NS3 Protease: Structure, Function, and Inhibition. In Viral Proteases and Their Inhibitors; Gupta, S., Ed.; Academic Press: New York, NY, USA, 2017; pp. 163–188. [Google Scholar]
- Brinkworth, R.I.; Fairlie, D.P.; Leung, D.; Young, P.R. Homology model of the dengue 2 virus NS3 protease: Putative interactions with both substrate and NS2B cofactor. J. Gen. Virol. 1999, 80 Pt 5, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, L.; Loukatou, S.; Sofia, K.; Maroulis, D.; Vlachakis, D. An updated evolutionary study of Flaviviridae NS3 helicase and NS5 RNA-dependent RNA polymerase reveals novel invariable motifs as potential pharmacological targets. Mol. Biosyst. 2016, 12, 2080–2093. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Zhang, L.; Ramachandra, M.; Kusukawa, J.; Ebner, K.E.; Padmanabhan, R. Association between NS3 and NS5 proteins of dengue virus type 2 in the putative RNA replicase is linked to differential phosphorylation of NS5. J. Biol. Chem. 1995, 270, 19100–19106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natarajan, S. NS3 protease from flavivirus as a target for designing antiviral inhibitors against dengue virus. Genet. Mol. Biol. 2010, 33, 214–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alomair, L. Combining Protein Interactions and Functionality Classification in NS3 to Determine Specific Antiviral Targets in Dengue; George Mason University: Fairfax, VA, USA, 2017. [Google Scholar]
- Xue, Y.; Liu, Z.; Cao, J.; Ma, Q.; Gao, X.; Wang, Q.; Jin, C.; Zhou, Y.; Wen, L.; Ren, J. GPS 2.1: Enhanced prediction of kinase-specific phosphorylation sites with an algorithm of motif length selection. Protein Eng. Des. Sel. 2011, 24, 255–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blom, N.; Sicheritz-Ponten, T.; Gupta, R.; Gammeltoft, S.; Brunak, S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 2004, 4, 1633–1649. [Google Scholar] [CrossRef]
- Obenauer, J.C.; Cantley, L.C.; Yaffe, M.B. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003, 31, 3635–3641. [Google Scholar] [CrossRef] [Green Version]
- Vlachakis, D.; Bencurova, E.; Papageorgiou, L.; Bhide, M.; Kossida, S. Protein phosphorylation prediction: Limitations, merits and pitfalls. J. Mol. Biochem. 2015, 4, 36–41. [Google Scholar]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Luo, D.; Xu, T.; Hunke, C.; Gruber, G.; Vasudevan, S.G.; Lescar, J. Crystal structure of the NS3 protease-helicase from dengue virus. J. Virol. 2008, 82, 173–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Harrach, M.F.; Drossel, B. Structure and dynamics of TIP3P, TIP4P, and TIP5P water near smooth and atomistic walls of different hydroaffinity. J. Chem. Phys. 2014, 140, 174501. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; Mackerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Grant, B.J.; Rodrigues, A.P.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skjaerven, L.; Yao, X.Q.; Scarabelli, G.; Grant, B.J. Integrating protein structural dynamics and evolutionary analysis with Bio3D. BMC Bioinform. 2014, 15, 399. [Google Scholar] [CrossRef] [Green Version]
- Haynes, W. Bonferroni Correction. In Encyclopedia of Systems Biology; Dubitzky, W., Wolkenhauer, O., Cho, K.-H., Yokota, H., Eds.; Springer: New York, NY, USA, 2013; p. 154. [Google Scholar] [CrossRef]
- Vajda, S.; Yueh, C.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Kozakov, D. New additions to the ClusPro server motivated by CAPRI. Proteins 2017, 85, 435–444. [Google Scholar] [CrossRef] [Green Version]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Kozakov, D.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Vajda, S. How good is automated protein docking? Proteins 2013, 81, 2159–2166. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alder, B.J.; Wainwright, T.E. Studies in Molecular Dynamics. I. General Method. J. Chem. Phys. 1959, 31, 459–466. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zheng, Q.C.; Kong, C.P.; Tian, Y.; Zhan, J.; Zhang, J.L.; Zhang, H.X. Heparin makes differences: A molecular dynamics simulation study on the human betaII-tryptase monomer. Mol. Biosyst. 2015, 11, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, S.M.; Serber, Z.; Ferrell, J.E., Jr. A mechanism for the evolution of phosphorylation sites. Cell 2011, 147, 934–946. [Google Scholar] [CrossRef] [Green Version]
- Boekhorst, J.; van Breukelen, B.; Heck, A., Jr.; Snel, B. Comparative phosphoproteomics reveals evolutionary and functional conservation of phosphorylation across eukaryotes. Genome Biol. 2008, 9, R144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macek, B.; Gnad, F.; Soufi, B.; Kumar, C.; Olsen, J.V.; Mijakovic, I.; Mann, M. Phosphoproteome analysis of E. coli reveals evolutionary conservation of bacterial Ser/Thr/Tyr phosphorylation. Mol. Cell Proteom. 2008, 7, 299–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, H.; Takahashi, C.; Moreland, N.J.; Chang, Y.T.; Sawasaki, T.; Ryo, A.; Vasudevan, S.G.; Suzuki, Y.; Yamamoto, N. Establishment of a robust dengue virus NS3-NS5 binding assay for identification of protein-protein interaction inhibitors. Antiviral Res. 2012, 96, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Hu, H.S.; Wu, R.H.; Wu, S.H.; Lee, S.J.; Jiaang, W.T.; Chern, J.H.; Huang, Z.S.; Wu, H.N.; Chang, C.M.; et al. A novel dengue virus inhibitor, BP13944, discovered by high-throughput screening with dengue virus replicon cells selects for resistance in the viral NS2B/NS3 protease. Antimicrob. Agents Chemother. 2014, 58, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noble, C.G.; Seh, C.C.; Chao, A.T.; Shi, P.Y. Ligand-bound structures of the dengue virus protease reveal the active conformation. J. Virol. 2012, 86, 438–446. [Google Scholar] [CrossRef] [Green Version]
- Silva, E.M.; Conde, J.N.; Allonso, D.; Ventura, G.T.; Coelho, D.R.; Carneiro, P.H.; Silva, M.L.; Paes, M.V.; Rabelo, K.; Weissmuller, G.; et al. Dengue virus nonstructural 3 protein interacts directly with human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and reduces its glycolytic activity. Sci. Rep. 2019, 9, 2651. [Google Scholar] [CrossRef]
- Salaemae, W.; Junaid, M.; Angsuthanasombat, C.; Katzenmeier, G. Structure-guided mutagenesis of active site residues in the dengue virus two-component protease NS2B-NS3. J. Biomed. Sci. 2010, 17, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tay, M.Y.; Saw, W.G.; Zhao, Y.; Chan, K.W.; Singh, D.; Chong, Y.; Forwood, J.K.; Ooi, E.E.; Gruber, G.; Lescar, J.; et al. The C-terminal 50 amino acid residues of dengue NS3 protein are important for NS3-NS5 interaction and viral replication. J. Biol. Chem. 2015, 290, 2379–2394. [Google Scholar] [CrossRef] [Green Version]
- Brand, C.; Bisaillon, M.; Geiss, B.J. Organization of the Flavivirus RNA replicase complex. Wiley Interdiscip. Rev. RNA 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Swarbrick, C.M.D.; Basavannacharya, C.; Chan, K.W.K.; Chan, S.A.; Singh, D.; Wei, N.; Phoo, W.W.; Luo, D.; Lescar, J.; Vasudevan, S.G. NS3 helicase from dengue virus specifically recognizes viral RNA sequence to ensure optimal replication. Nucleic Acids Res. 2017, 45, 12904–12920. [Google Scholar] [CrossRef] [Green Version]
- Zandi, K.; Teoh, B.T.; Sam, S.S.; Wong, P.F.; Mustafa, M.R.; Abubakar, S. Antiviral activity of four types of bioflavonoid against dengue virus type-2. Virol. J. 2011, 8, 560. [Google Scholar] [CrossRef] [Green Version]
- Ceballos-Olvera, I.; Chavez-Salinas, S.; Medina, F.; Ludert, J.E.; del Angel, R.M. JNK phosphorylation, induced during dengue virus infection, is important for viral infection and requires the presence of cholesterol. Virology 2010, 396, 30–36. [Google Scholar] [CrossRef] [Green Version]
- Sreekanth, G.P.; Chuncharunee, A.; Cheunsuchon, B.; Noisakran, S.; Yenchitsomanus, P.T.; Limjindaporn, T. JNK1/2 inhibitor reduces dengue virus-induced liver injury. Antivir. Res. 2017, 141, 7–18. [Google Scholar] [CrossRef]
- Zou, G.; Chen, Y.L.; Dong, H.; Lim, C.C.; Yap, L.J.; Yau, Y.H.; Shochat, S.G.; Lescar, J.; Shi, P.Y. Functional analysis of two cavities in flavivirus NS5 polymerase. J. Biol. Chem. 2011, 286, 14362–14372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teramoto, T.; Balasubramanian, A.; Choi, K.H.; Padmanabhan, R. Serotype-specific interactions among functional domains of dengue virus 2 nonstructural proteins (NS) 5 and NS3 are crucial for viral RNA replication. J. Biol. Chem. 2017, 292, 9465–9479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, T.J.; Weir, R.C.; Grakoui, A.; McCourt, D.W.; Bazan, J.F.; Fletterick, R.J.; Rice, C.M. Evidence that the N-terminal domain of nonstructural protein NS3 from yellow fever virus is a serine protease responsible for site-specific cleavages in the viral polyprotein. Proc. Natl. Acad. Sci. USA 1990, 87, 8898–8902. [Google Scholar] [CrossRef] [Green Version]
- Agnihotri, S.; Narula, R.; Joshi, K.; Rana, S.; Singh, M. In silico modeling of ligand molecule for non structural 3 (NS3) protein target of flaviviruses. Bioinformation 2012, 8, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Mukhametov, A.; Newhouse, E.I.; Aziz, N.A.; Saito, J.A.; Alam, M. Allosteric pocket of the dengue virus (serotype 2) NS2B/NS3 protease: In silico ligand screening and molecular dynamics studies of inhibition. J. Mol. Graph. Model. 2014, 52, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jiang, H.; Li, F.; Hu, B.; Wang, Y.; Wang, M.; Wang, J.; Cheng, M. Computational insight into dengue virus NS2B-NS3 protease inhibition: A combined ligand- and structure-based approach. Comput. Biol. Chem. 2018, 77, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Velmurugan, D.; Mythily, U.; Rao, K. Design and docking studies of peptide inhibitors as potential antiviral drugs for dengue virus ns2b/ns3 protease. Protein Pept. Lett. 2014, 21, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Niyomrattanakit, P.; Winoyanuwattikun, P.; Chanprapaph, S.; Angsuthanasombat, C.; Panyim, S.; Katzenmeier, G. Identification of Residues in the Dengue Virus Type 2 NS2B Cofactor That Are Critical for NS3 Protease Activation. J. Virol. 2004, 78, 13708–13716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez, J.L.; Hegemann, B.; Hutchins, J.R.; Peters, J.M.; Durbin, R. A systematic comparative and structural analysis of protein phosphorylation sites based on the mtcPTM database. Genome Biol. 2007, 8, R90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ubersax, J.A.; Ferrell Jr, J.E. Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Henriques, J.; Lindorff-Larsen, K. Protein Dynamics Enables Phosphorylation of Buried Residues in Cdk2/Cyclin-A-Bound p27. Biophys. J. 2020, 119, 2010–2018. [Google Scholar] [CrossRef]
- Somavarapu, A.K.; Balakrishnan, S.; Gautam, A.K.; Palmer, D.S.; Venkatraman, P. Structural interrogation of phosphoproteome identified by mass spectrometry reveals allowed and disallowed regions of phosphoconformation. BMC Struct Biol. 2014, 14, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trost, B.; Kusalik, A. Computational prediction of eukaryotic phosphorylation sites. Bioinformatics 2011, 27, 2927–2935. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Xu, H.; Lin, S.; Deng, W.; Zhou, J.; Zhang, Y.; Shi, Y.; Peng, D.; Xue, Y. GPS 5.0: An Update on the Prediction of Kinase-specific Phosphorylation Sites in Proteins. Genom. Proteom. Bioinform. 2020, 18, 72–80. [Google Scholar] [CrossRef]
- Perera, R.; Kuhn, R.J. Structural proteomics of dengue virus. Curr. Opin. Microbiol. 2008, 11, 369–377. [Google Scholar] [CrossRef] [Green Version]
- Murthy, H.M.; Judge, K.; DeLucas, L.; Padmanabhan, R. Crystal structure of Dengue virus NS3 protease in complex with a Bowman-Birk inhibitor: Implications for flaviviral polyprotein processing and drug design. J. Mol. Biol. 2000, 301, 759–767. [Google Scholar] [CrossRef]
- Lin, K.H.; Nalivaika, E.A.; Prachanronarong, K.L.; Yilmaz, N.K.; Schiffer, C.A. Dengue Protease Substrate Recognition: Binding of the Prime Side. ACS Infect. Dis. 2016, 2, 734–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiryaev, S.A.; Kozlov, I.A.; Ratnikov, B.I.; Smith, J.W.; Lebl, M.; Strongin, A.Y. Cleavage preference distinguishes the two-component NS2B-NS3 serine proteinases of Dengue and West Nile viruses. Biochem. J. 2007, 401, 743–752. [Google Scholar] [CrossRef]
- Bera, A.K.; Kuhn, R.J.; Smith, J.L. Functional characterization of cis and trans activity of the Flavivirus NS2B-NS3 protease. J. Biol. Chem. 2007, 282, 12883–12892. [Google Scholar] [CrossRef] [Green Version]
- Niyomrattanakit, P.; Yahorava, S.; Mutule, I.; Mutulis, F.; Petrovska, R.; Prusis, P.; Katzenmeier, G.; Wikberg, J.E. Probing the substrate specificity of the dengue virus type 2 NS3 serine protease by using internally quenched fluorescent peptides. Biochem. J. 2006, 397, 203–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valle, R.P.; Falgout, B. Mutagenesis of the NS3 protease of dengue virus type 2. J. Virol. 1998, 72, 624–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshizumi, M.; Tsuchiya, K.; Kirima, K.; Kyaw, M.; Suzaki, Y.; Tamaki, T. Quercetin inhibits Shc- and phosphatidylinositol 3-kinase-mediated c-Jun N-terminal kinase activation by angiotensin II in cultured rat aortic smooth muscle cells. Mol. Pharmacol. 2001, 60, 656–665. [Google Scholar] [PubMed]
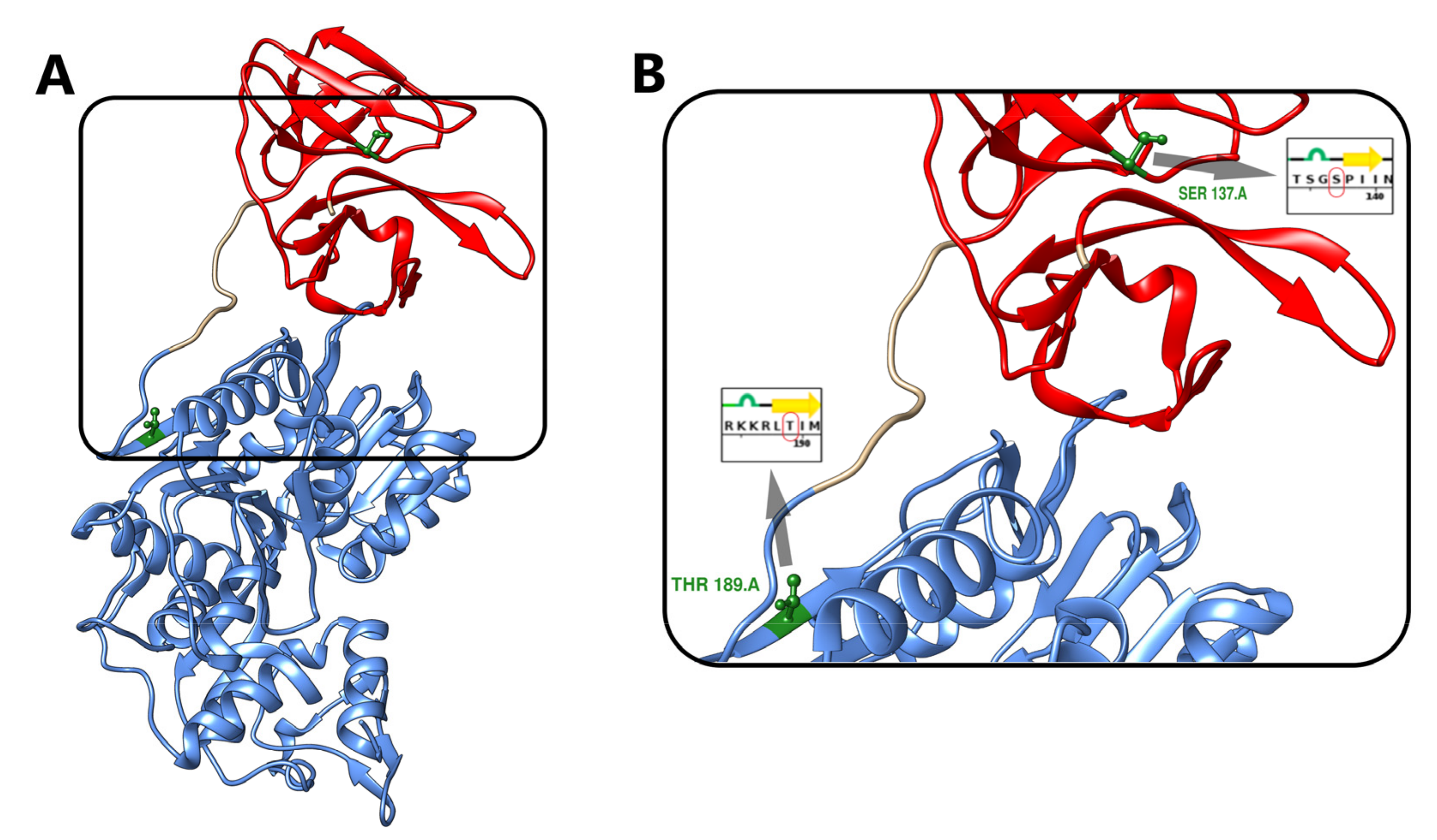




| Effect | DFn | DFd | F | ges | p |
|---|---|---|---|---|---|
| Time Step | 4795 | 9590 | 4.443 | 0.69 | <0.0001 |
| Category | 2 | 9590 | 59671.861 | 0.926 | <0.0001 |
| RMSD Emmean Comparisons 1 | ||||||
|---|---|---|---|---|---|---|
| Category | Emmean | SE | DF | S137 | T189 | WT |
| S137 | 7.785 | 0.012 | 14385 | NA | <0.0001 | <0.0001 |
| T189 | 3.871 | 0.012 | 14385 | <0.0001 | NA | <0.0001 |
| WT | 5.396 | 0.012 | 14385 | <0.0001 | <0.0001 | NA |
| Effect | DFn | DFd | F | ges | p |
|---|---|---|---|---|---|
| Time Step | 4795 | 9590 | 0.843 | 0.297 | 1 |
| Category | 2 | 9590 | 1296396.55 | 0.996 | <0.0001 |
| ID | N-Lobe Catalytic Domain 120–1168 | Linker 169–179 | NS3 Loop 566–585 | C-Lobe Helicase 180–618 |
|---|---|---|---|---|
| WT-S137 | 28.97 Å | 8.82 Å | 3.66 Å | 2.82 Å |
| WT-T189 | 16.20 Å | 9.78 Å | 3.38 Å | 3.05 Å |
| RMSD Emmean Comparisons 1 | ||||||
|---|---|---|---|---|---|---|
| Category | Emmean | SE | DF | S137 | T189 | WT |
| S137 | 11.002 | 0.0045 | 14385 | NA | <0.0001 | <0.0001 |
| T189 | 1.908 | 0.0045 | 14385 | <0.0001 | NA | <0.0001 |
| WT | 1.733 | 0.0045 | 14385 | <0.0001 | <0.0001 | NA |
| ID | Cluster Members | Weighted Score Lowest Energy a |
|---|---|---|
| WT-NS5 | 29 | −963.8 Kcal/mol |
| T189-NS5 | 28 | −949.2 Kcal/mol |
| S137-NS5 | 49 | −1141.8 Kcal/mol |
| Residue | WT | S137 | T189 |
|---|---|---|---|
| 75 | 54.42 | 35.07 | 52.11 |
| 177 | 100.35 | 22.45 | 122.17 |
| 179 | 56.54 | 100.64 | 82.61 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alomair, L.; Almsned, F.; Ullah, A.; Jafri, M.S. In Silico Prediction of the Phosphorylation of NS3 as an Essential Mechanism for Dengue Virus Replication and the Antiviral Activity of Quercetin. Biology 2021, 10, 1067. https://doi.org/10.3390/biology10101067
Alomair L, Almsned F, Ullah A, Jafri MS. In Silico Prediction of the Phosphorylation of NS3 as an Essential Mechanism for Dengue Virus Replication and the Antiviral Activity of Quercetin. Biology. 2021; 10(10):1067. https://doi.org/10.3390/biology10101067
Chicago/Turabian StyleAlomair, Lamya, Fahad Almsned, Aman Ullah, and Mohsin S. Jafri. 2021. "In Silico Prediction of the Phosphorylation of NS3 as an Essential Mechanism for Dengue Virus Replication and the Antiviral Activity of Quercetin" Biology 10, no. 10: 1067. https://doi.org/10.3390/biology10101067