
De novo Assembly and Analysis of Tissue-Specific Transcriptomes of the Edible Red Sea Urchin Loxechinus albus Using RNA-Seq

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Design and Sampling
2.2. Isolation of RNA and Sequencing
2.3. Processing of Raw Data, De novo Assembly, and Validation of Assembly
2.4. Functional Annotation and Analysis of Differentially Expressed Transcripts
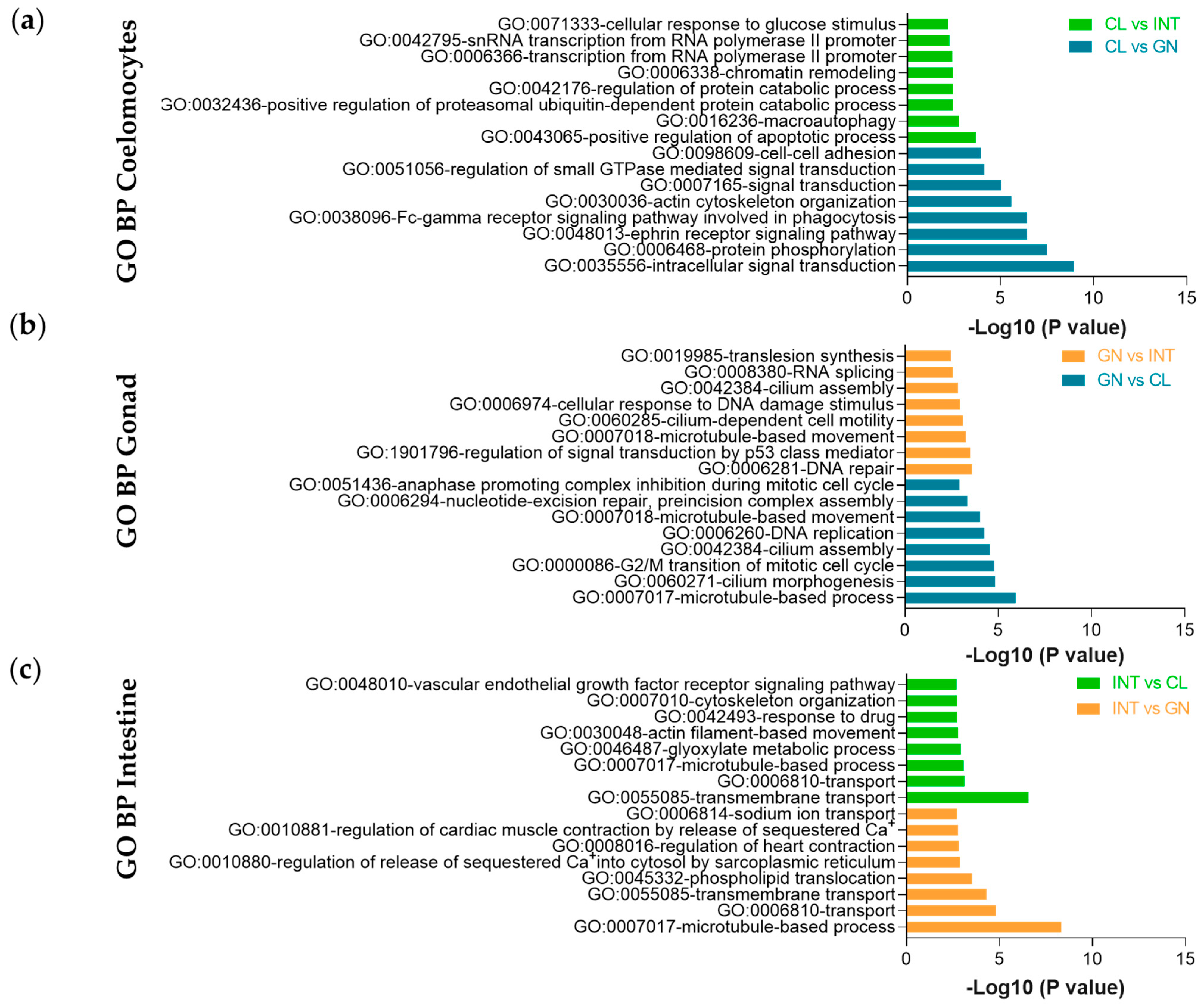
2.5. Gene Ontology and KEGG Enrichment Analysis
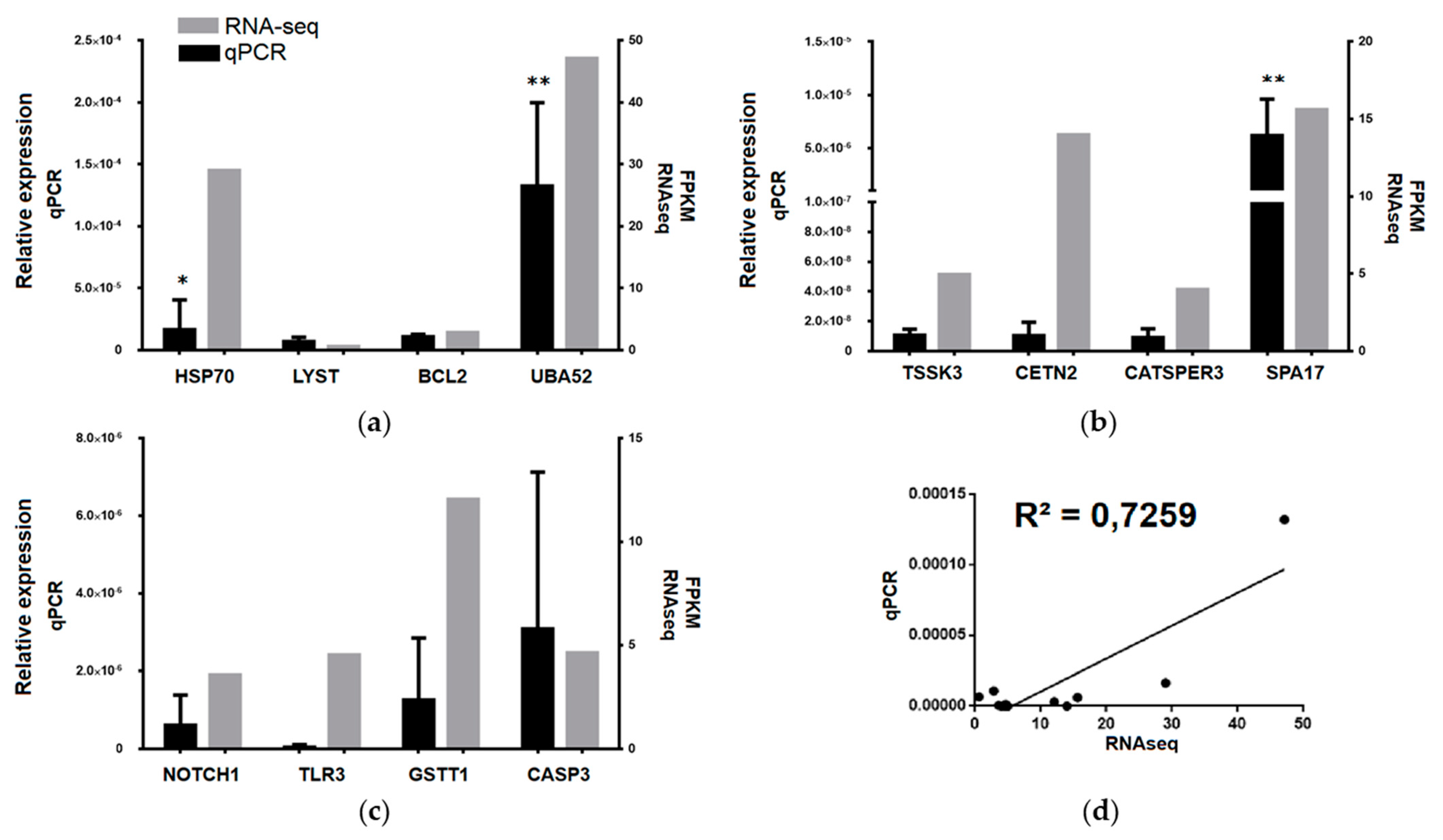
2.6. Validation of RNA-Seq by Real-Time qPCR
2.7. Statistical Analysis
3. Results
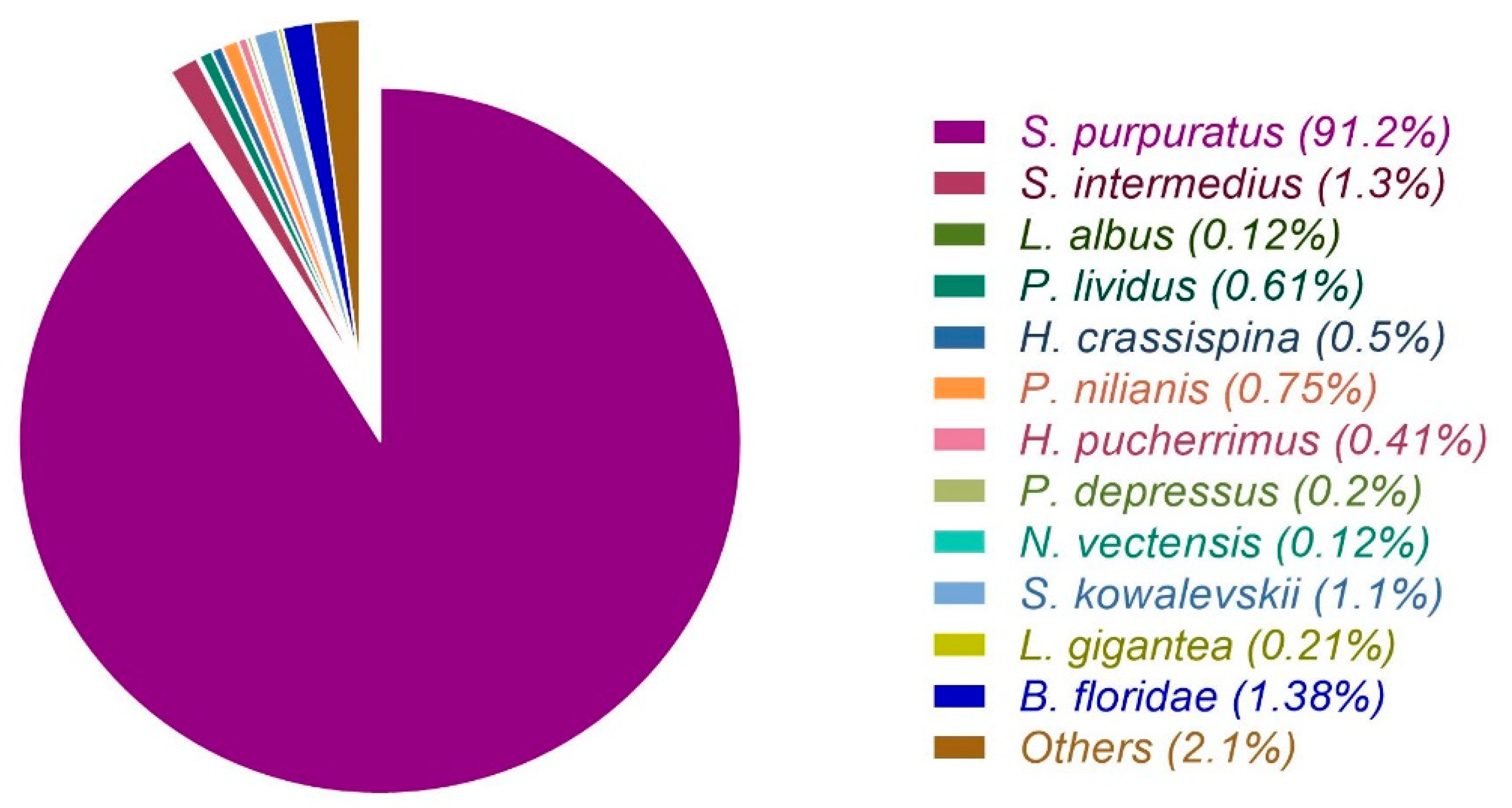
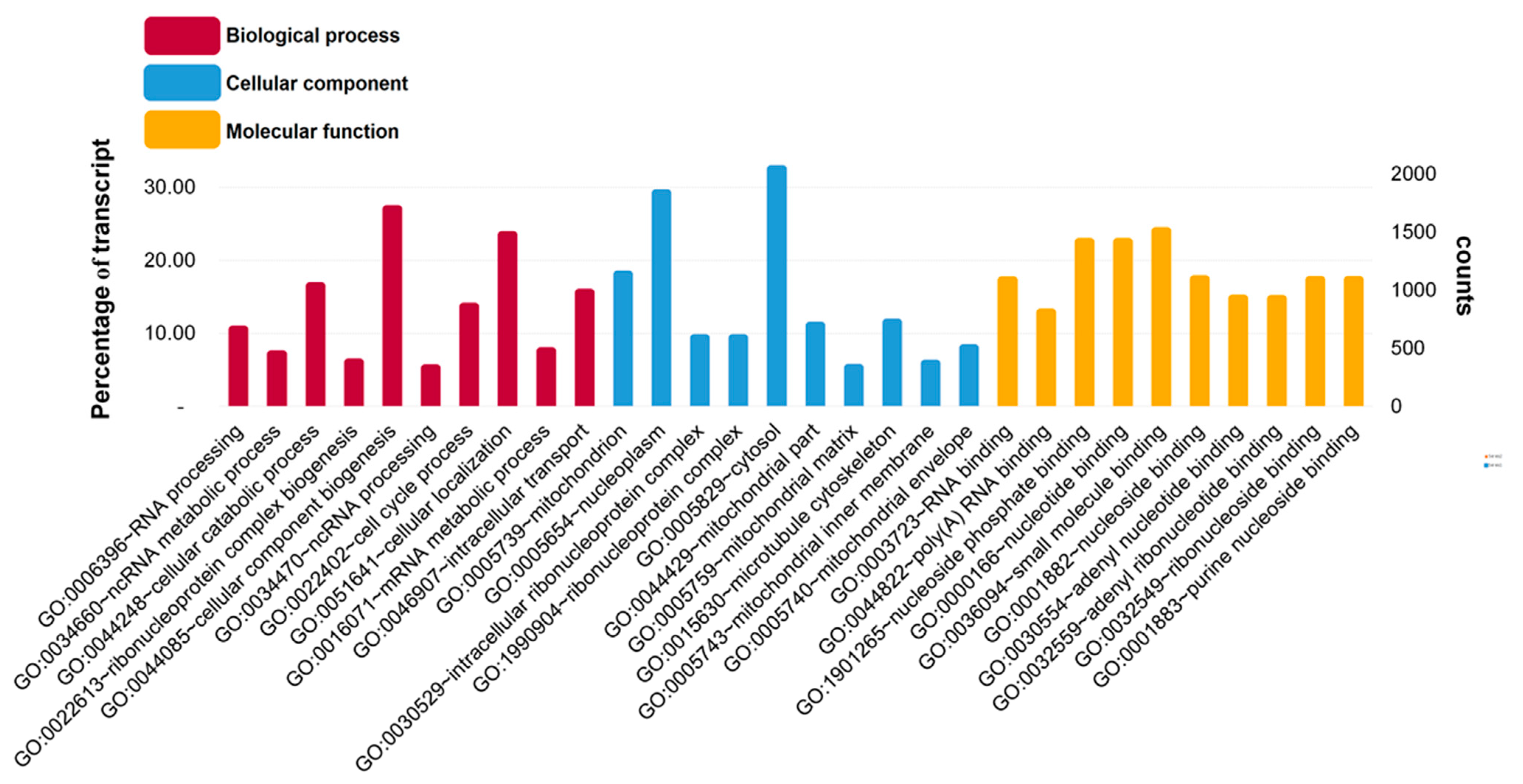
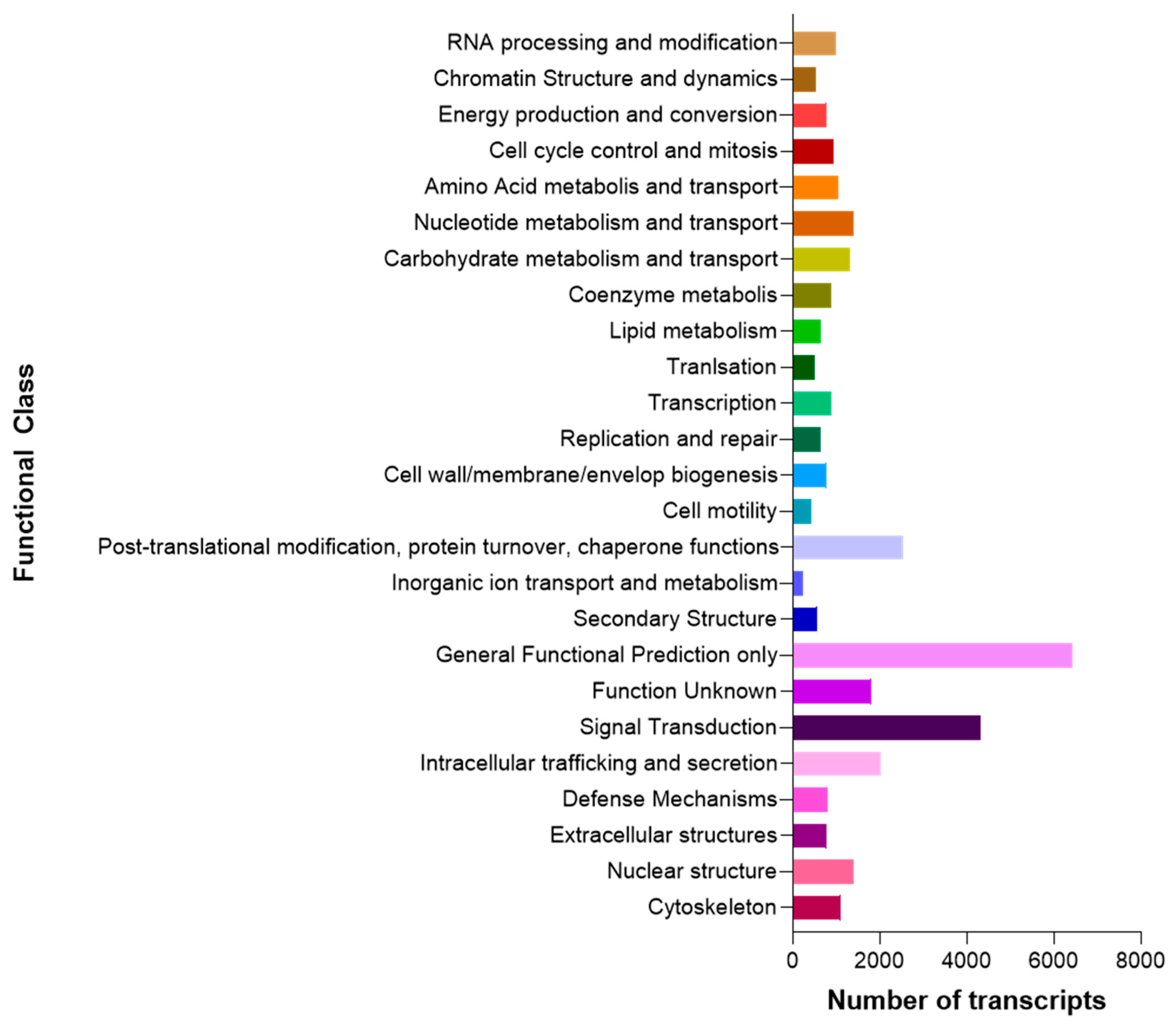
3.1. Raw Data Sequencing, De novo Assembly Transcriptome and Functional Annotation
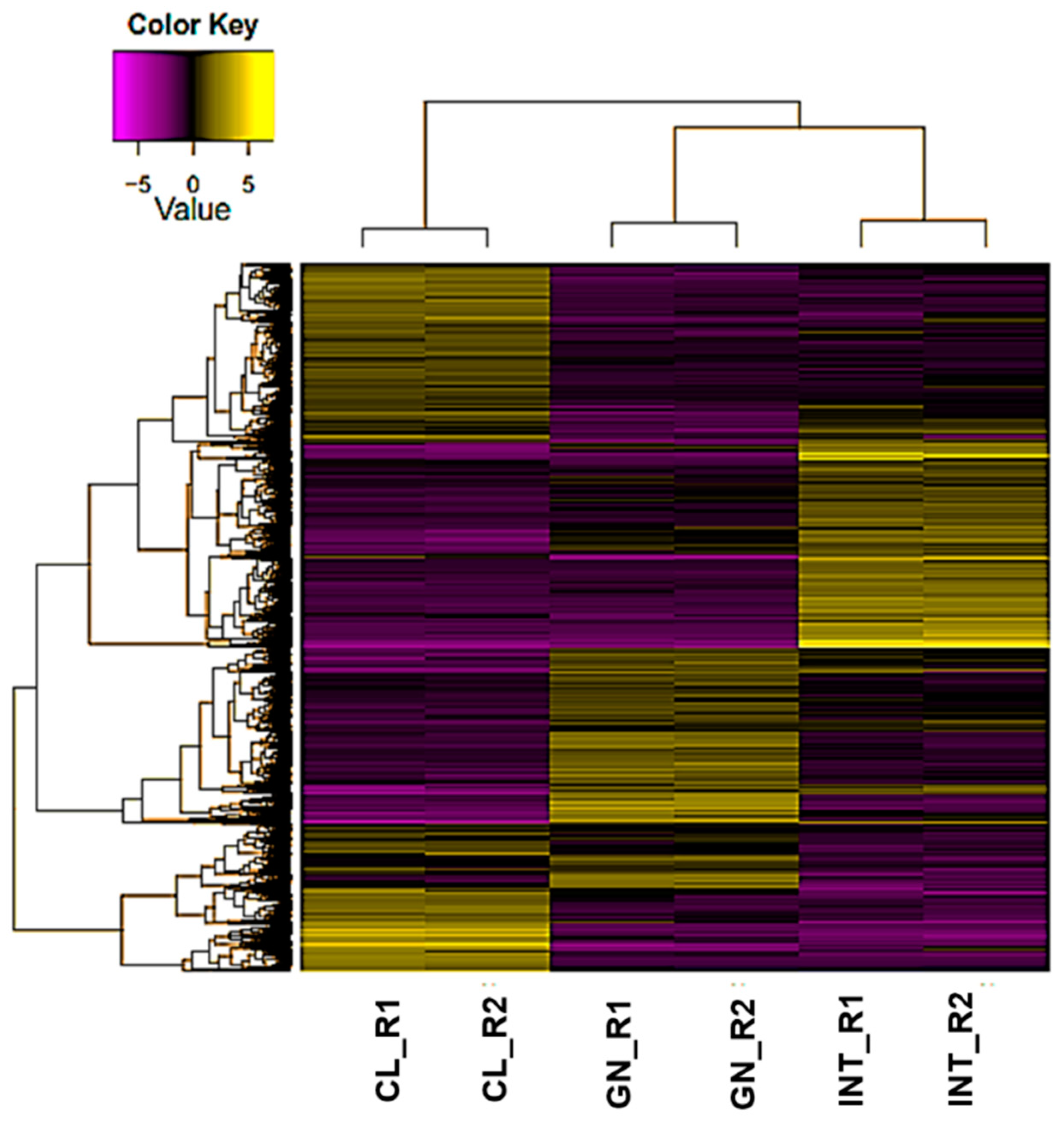
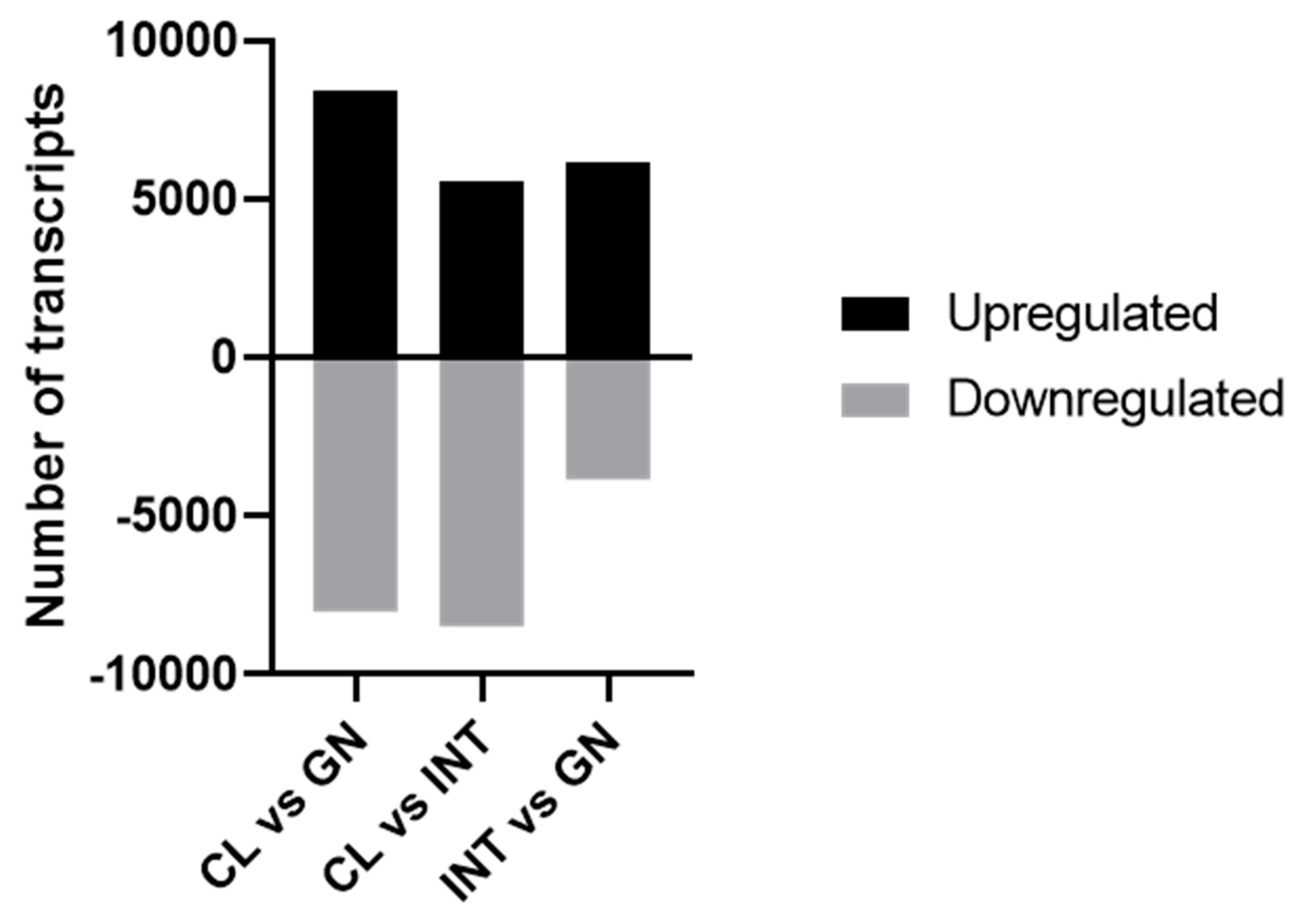
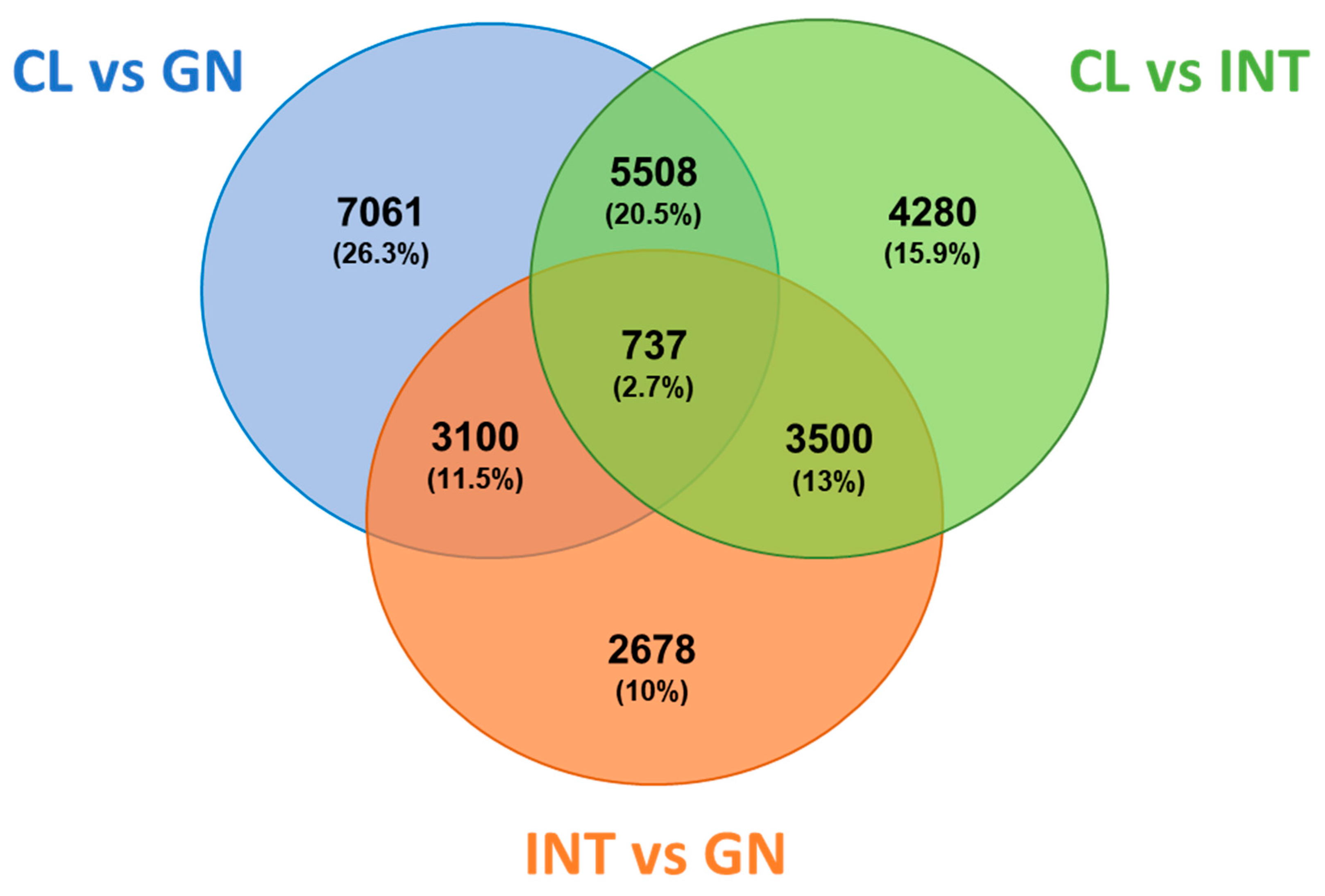
3.2. Assessment of Differentially Expressed Transcripts
3.3. Transcriptomic Data Validation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Oyarzún, S.T.; Marín, S.L.; Valladares, C.; Iriarte, J.L. Reproductive Cycle of Loxechinus albus (Echinodermata: Echinoidea) in Two Areas of the Magellan Region (53oS, 70-72oW), Chile. Sci. Mar. 1999, 63, 439–449. [Google Scholar] [CrossRef]
- Vásquez, J.A.; Donoso, G.A. Loxechinus albus. In Developments in Aquaculture and Fisheries Science; Elsevier: Amsterdam, The Netherlands, 2013; Volume 38, pp. 285–296. ISBN 978-0-12-396491-5. [Google Scholar]
- Vásquez, J.A. Loxechinus albus. In Developments in Aquaculture and Fisheries Science; Elsevier: Amsterdam, The Netherlands, 2020; Volume 43, pp. 431–445. ISBN 978-0-12-819570-3. [Google Scholar]
- Olave, S.; Bustos, E.; Lawrence, J.M.; Carcamo, P. The Effect of Size and Diet on Gonad Production by the Chilean Sea Urchin Loxechinus albus. JWAS 2001, 32, 210–214. [Google Scholar] [CrossRef]
- Cea, G.; Gaitán-Espitia, J.D.; Cárdenas, L. Complete Mitogenome of the Edible Sea Urchin Loxechinus albus: Genetic Structure and Comparative Genomics within Echinozoa. Mol. Biol. Rep. 2015, 42, 1081–1089. [Google Scholar] [CrossRef]
- Gaitán-Espitia, J.D.; Sánchez, R.; Bruning, P.; Cárdenas, L. Functional Insights into the Testis Transcriptome of the Edible Sea Urchin Loxechinus albus. Sci. Rep. 2016, 6, 36516. [Google Scholar] [CrossRef] [Green Version]
- Chandhini, S.; Rejish Kumar, V.J. Transcriptomics in Aquaculture: Current Status and Applications. Rev. Aquac. 2018, 11, 1379–1397. [Google Scholar] [CrossRef]
- Inbakandan, D. Transcriptomics in Aquaculture. In Encyclopedia of Marine Biotechnology; Kim, S., Ed.; Wiley: Hoboken, NJ, USA, 2020; pp. 1919–1936. ISBN 978-1-119-14377-2. [Google Scholar]
- Jung, G.; Lee, Y.-H. Complete Mitochondrial Genome of Chilean Sea Urchin: Loxechinus albus (Camarodonta, Parechinidae). Mitochondrial DNA 2015, 26, 883–884. [Google Scholar] [CrossRef]
- Oulas, A.; Pavloudi, C.; Polymenakou, P.; Pavlopoulos, G.A.; Papanikolaou, N.; Kotoulas, G.; Arvanitidis, C.; Iliopoulos, L. Metagenomics: Tools and Insights for Analyzing Next-Generation Sequencing Data Derived from Biodiversity Studies. Bioinform. Biol. Insights 2015, 9, 75–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergara-Amado, J.; Silva, A.X.; Manzi, C.; Nespolo, R.F.; Cárdenas, L. Differential Expression of Stress Candidate Genes for Thermal Tolerance in the Sea Urchin Loxechinus albus. J. Therm. Biol. 2017, 68, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.M.; Johnson, K.M.; Kelly, M.W.; Hofmann, G.E. Transcriptomics Reveal Transgenerational Effects in Purple Sea Urchin Embryos: Adult Acclimation to Upwelling Conditions Alters the Response of Their Progeny to Differential pCO2 Levels. Mol. Ecol. 2018, 27, 1120–1137. [Google Scholar] [CrossRef]
- Zhan, Y.; Li, J.; Sun, J.; Zhang, W.; Li, Y.; Cui, D.; Hu, W.; Chang, Y. The Impact of Chronic Heat Stress on the Growth, Survival, Feeding, and Differential Gene Expression in the Sea Urchin Strongylocentrotus Intermedius. Front. Genet. 2019, 10, 301. [Google Scholar] [CrossRef] [Green Version]
- Malanga, G.; Perez, A.; Calvo, J.; Puntarulo, S. The Effect of Seasonality on Oxidative Metabolism in the Sea Urchin Loxechinus albus. Mar. Biol. 2009, 156, 763–770. [Google Scholar] [CrossRef]
- Flores, L.; Ernst, B.; Parma, A.M. Growth Pattern of the Sea Urchin, Loxechinus albus (Molina, 1782) in Southern Chile: Evaluation of Growth Models. Mar. Biol. 2010, 157, 967–977. [Google Scholar] [CrossRef]
- Cárcamo, P.F. Effects of Food Type and Feeding Frequency on the Performance of Early Juveniles of the Sea Urchin Loxechinus albus (Echinodermata: Echinoidea): Implications for Aquaculture and Restocking. Aquaculture 2015, 436, 172–178. [Google Scholar] [CrossRef]
- Dupré, E.; Carvajal, J. Cryopreservation of Embryos and Larvae of the Edible Sea Urchin Loxechinus albus (Molina, 1782). Cryobiology. 2019, 86, 84–88. [Google Scholar] [CrossRef]
- Dettleff, P.; Villagra, M.; González, J.; Fuentes, M.; Estrada, J.M.; Valenzuela, C.; Molina, A.; Valdés, J.A. Effect of Bacterial LPS, Poly I:C and Temperature on the Immune Response of Coelomocytes in Short Term Cultures of Red Sea Urchin (Loxechinus albus). Fish Shellfish Immunol. 2020, S1050464820306513. [Google Scholar] [CrossRef]
- Arévalo, A.; Bruning, P.; Sanchez, R.; Cárdenas, L. Development and Characterization of EST-Microsatellites for the Edible Sea Urchin Loxechinus albus Using next Generation Sequencing. Conserv. Genet. Resour. 2014, 6, 433–435. [Google Scholar] [CrossRef]
- Dodt, M.; Roehr, J.; Ahmed, R.; Dieterich, C. FLEXBAR—Flexible Barcode and Adapter Processing for Next-Generation Sequencing Platforms. Biology. 2012, 1, 895–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-Length Transcriptome Assembly from RNA-Seq Data without a Reference Genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, N.M.; Oshlack, A. Corset: Enabling Differential Gene Expression Analysis for de Novoassembled Transcriptomes. Genome Biol. 2014, 15, 410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinformatics. 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A Universal Tool for Annotation, Visualization and Analysis in Functional Genomics Research. Bioinformatics. 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Zhang, Y.; Cui, H.; Liu, J.; Wu, Y.; Cheng, Y.; Xu, H.; Huang, X.; Li, S.; Zhou, A.; et al. WEGO 2.0: A Web Tool for Analyzing and Plotting GO Annotations, 2018 Update. Nucleic Acids Res. 2018, 46, W71–W75. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and Memory-Efficient Alignment of Short DNA Sequences to the Human Genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, P. Q-Gene: Processing Quantitative Real-Time RT-PCR Data. Bioinformatics 2003, 19, 1439–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillard, G.B.; Garama, D.J.; Brown, C.M. The Transcriptome of the NZ Endemic Sea Urchin Kina (Evechinus chloroticus). BMC Genom. 2014, 15, 45. [Google Scholar] [CrossRef] [Green Version]
- Dilly, G.F.; Gaitán-Espitia, J.D.; Hofmann, G.E. Characterization of the Antarctic Sea Urchin (Sterechinus neumayeri) Transcriptome and Mitogenome: A Molecular Resource for Phylogenetics, Ecophysiology and Global Change Biology. Mol. Ecol. Resour. 2015, 15, 425–436. [Google Scholar] [CrossRef]
- Wang, H.; Ding, J.; Ding, S.; Chang, Y. Transcriptome Analysis to Characterize the Genes Related to Gonad Growth and Fatty Acid Metabolism in the Sea Urchin Strongylocentrotus intermedius. Genes Genom. 2019, 41, 1397–1415. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Liu, X.; Zhou, Z.; Xu, J. De Novo Transcriptomic Analysis of Gonad of Strongylocentrotus nudus and Gene Discovery for Biosynthesis of Polyunsaturated Fatty Acids. Genes Genom. 2019, 41, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, J.R.M.C. Immunology in sea urchins. In Developments in Aquaculture and Fisheries Science; Elsevier: Amsterdam, The Netherlands, 2020; Volume 43, pp. 227–236. ISBN 978-0-12-819570-3. [Google Scholar]
- Smith, L.C.; Hawley, T.S.; Henson, J.H.; Majeske, A.J.; Oren, M.; Rosental, B. Methods for Collection, Handling, and Analysis of Sea Urchin Coelomocytes. Methods Cell. Biol. 2019, 150, 357–389. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chang, Y.; Wang, X.; Qiu, X.; Liu, Y. De Novo Assembly and Analysis of Tissue-Specific Transcriptomes Revealed the Tissue-Specific Genes and Profile of Immunity from Strongylocentrotus intermedius. Fish Shellfish Immunol. 2015, 46, 723–736. [Google Scholar] [CrossRef]
- Pérez-Portela, R.; Turon, X.; Riesgo, A. Characterization of the Transcriptome and Gene Expression of Four Different Tissues in the Ecologically Relevant Sea Urchin Arbacia lixula Using RNA-Seq. Mol. Ecol. Resour. 2016, 16, 794–808. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lv, Z.; Li, C.; Sun, Y.; Jiang, H.; Zhao, M.; Zhao, X.; Shao, Y.; Chang, Y. Transcriptome Profiling Reveals Key Roles of Phagosome and NOD-like Receptor Pathway in Spotting Diseased Strongylocentrotus intermedius. Fish Shellfish Immunol. 2019, 84, 521–531. [Google Scholar] [CrossRef]
- Lubkowska, A.; Pluta, W.; Strońska, A.; Lalko, A. Role of Heat Shock Proteins (HSP70 and HSP90) in Viral Infection. IJMS 2021, 22, 9366. [Google Scholar] [CrossRef]
- Chiaramonte, M.; Inguglia, L.; Vazzana, M.; Deidun, A.; Arizza, V. Stress and Immune Response to Bacterial LPS in the Sea Urchin Paracentrotus lividus (Lamarck, 1816). Fish Shellfish Immunol. 2019, 92, 384–394. [Google Scholar] [CrossRef]
- Sepulveda, F.E.; Burgess, A.; Heiligenstein, X.; Goudin, N.; Ménager, M.M.; Romao, M.; Côte, M.; Mahlaoui, N.; Fischer, A.; Raposo, G.; et al. LYST Controls the Biogenesis of the Endosomal Compartment Required for Secretory Lysosome Function: LYST Controls the Biogenesis of Secretory Lysosome. Traffic 2015, 16, 191–203. [Google Scholar] [CrossRef]
- Westphal, A.; Cheng, W.; Yu, J.; Grassl, G.; Krautkrämer, M.; Holst, O.; Föger, N.; Lee, K.-H. Lysosomal Trafficking Regulator Lyst Links Membrane Trafficking to Toll-like Receptor–Mediated Inflammatory Responses. J. Exp. Med. 2017, 214, 227–244. [Google Scholar] [CrossRef] [Green Version]
- Banjara, S.; Suraweera, C.D.; Hinds, M.G.; Kvansakul, M. The Bcl-2 Family: Ancient Origins, Conserved Structures, and Divergent Mechanisms. Biomolecules 2020, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.; Chen, K.; Lv, Z.; Shao, Y.; Zhang, W.; Zhao, X.; Li, C. Bcl-2 Mediates Coelomocytes Apoptosis by Suppressing Cytochrome c Release in Vibrio splendidus Challenged Apostichopus Japonicus. Dev. Comp. Immunol. 2020, 103, 103533. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Oshima, S.; Maeyashiki, C.; Nibe, Y.; Otsubo, K.; Matsuzawa, Y.; Nemoto, Y.; Nagaishi, T.; Okamoto, R.; Tsuchiya, K.; et al. The Ubiquitin Hybrid Gene UBA52 Regulates Ubiquitination of Ribosome and Sustains Embryonic Development. Sci. Rep. 2016, 6, 36780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebner, P.; Versteeg, G.A.; Ikeda, F. Ubiquitin Enzymes in the Regulation of Immune Responses. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 425–460. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.W.; Unuma, T.; Lesser, M.P. Chapter 2 Gametogenesis and reproduction of sea urchins. In Edible Sea Urchins: Biology and Ecology; Lawrence, J.M., Ed.; Developments in Aquaculture and Fisheries Science; Elsevier: Amsterdam, The Netherlands, 2007; Volume 37, pp. 11–33. [Google Scholar]
- Walker, C.W.; Harrington, L.M.; Lesser, M.P.; Fagerberg, W.R. Nutritive Phagocyte Incubation Chambers Provide a Structural and Nutritive Microenvironment for Germ Cells of Strongylocentrotus droebachiensis, the Green Sea Urchin. Biol. Bull. 2005, 209, 31–48. [Google Scholar] [CrossRef]
- Jia, Z.; Wang, Q.; Wu, K.; Wei, Z.; Zhou, Z.; Liu, X. De Novo Transcriptome Sequencing and Comparative Analysis to Discover Genes Involved in Ovarian Maturity in Strongylocentrotus nudus. Comp. Biochem. Physiol. D Genom. Proteom. 2017, 23, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.-H.; Zhang, J.; Zhang, W.-J.; Chang, Y.-Q. Gonadal Transcriptomic Analysis and Identification of Candidate Sex-Related Genes in Mesocentrotus nudus. Gene 2019, 698, 72–81. [Google Scholar] [CrossRef]
- Wang, H.; Ding, J.; Ding, S.; Chang, Y. Integrated Metabolomic and Transcriptomic Analyses Identify Critical Genes in Eicosapentaenoic Acid Biosynthesis and Metabolism in the Sea Urchin Strongylocentrotus intermedius. Sci. Rep. 2020, 10, 1697. [Google Scholar] [CrossRef]
- Salicioni, A.M.; Gervasi, M.G.; Sosnik, J.; Tourzani, D.A.; Nayyab, S.; Caraballo, D.A.; Visconti, P.E. Testis-Specific Serine Kinase Protein Family in Male Fertility and as Targets for Non-Hormonal Male Contraception. Biol. Reprod. 2020, 103, 264–274. [Google Scholar] [CrossRef]
- Xue, X.; Zhang, L.; Li, Y.; Wei, H.; Wu, S.; Liu, T.; Liu, L.; Xing, Q.; Wang, S.; Bao, Z. Expression of the Testis-Specific Serine/Threonine Kinases Suggests Their Role in Spermiogenesis of Bay Scallop Argopecten irradians. Front. Physiol. 2021, 12, 657559. [Google Scholar] [CrossRef]
- Mullee, L.I.; Morrison, C.G. Centrosomes in the DNA Damage Response—The Hub Outside the Centre. Chromosome Res. 2016, 24, 35–51. [Google Scholar] [CrossRef]
- Singh, A.P.; Rajender, S. CatSper Channel, Sperm Function and Male Fertility. Reprod. Biomed. Online 2015, 30, 28–38. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.-L.; O’Doherty, A.M.; Wang, S.; Zheng, H.; Sanders, K.M.; Yan, W. Catsper3 and Catsper4 Encode Two Cation Channel-Like Proteins Exclusively Expressed in the Testis. Biol. Reprod. 2005, 73, 1235–1242. [Google Scholar] [CrossRef] [Green Version]
- Chiriva-Internati, M. Sperm Protein 17: Clinical Relevance of a Cancer/Testis Antigen, from Contraception to Cancer Immunotherapy, and Beyond. Int. Rev. Immunol. 2011, 30, 138–149. [Google Scholar] [CrossRef]
- Santella, L.; Limatola, N.; Vasilev, F.; Chun, J.T. Maturation and Fertilization of Echinoderm Eggs: Role of Actin Cytoskeleton Dynamics. Biochem. Biophys. Res. Commun. 2018, 506, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Holland, N.D. Digestive system in regular sea urchins. In Developments in Aquaculture and Fisheries Science; Elsevier: Amsterdam, The Netherlands, 2020; Volume 43, pp. 147–163. ISBN 978-0-12-819570-3. [Google Scholar]
- Ramírez-Gómez, F.; Ortíz-Pineda, P.A.; Rojas-Cartagena, C.; Suárez-Castillo, E.C.; García-Arrarás, J.E.; García-Ararrás, J.E. Immune-Related Genes Associated with Intestinal Tissue in the Sea Cucumber Holothuria glaberrima. Immunogenetics 2008, 60, 57–71. [Google Scholar] [CrossRef]
- Yang, A.-F.; Zhou, Z.-C.; He, C.-B.; Hu, J.-J.; Chen, Z.; Gao, X.-G.; Dong, Y.; Jiang, B.; Liu, W.-D.; Guan, X.-Y.; et al. Analysis of Expressed Sequence Tags from Body Wall, Intestine and Respiratory Tree of Sea Cucumber (Apostichopus japonicus). Aquaculture 2009, 296, 193–199. [Google Scholar] [CrossRef]
- Hibino, T.; Loza-Coll, M.; Messier, C.; Majeske, A.J.; Cohen, A.H.; Terwilliger, D.P.; Buckley, K.M.; Brockton, V.; Nair, S.V.; Berney, K.; et al. The Immune Gene Repertoire Encoded in the Purple Sea Urchin Genome. Dev. Biol. 2006, 300, 349–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garis, M.; Garrett-Sinha, L.A. Notch Signaling in B Cell Immune Responses. Front. Immunol. 2021, 11, 609324. [Google Scholar] [CrossRef] [PubMed]
- Dunkin, D.; Iuga, A.C.; Mimouna, S.; Harris, C.L.; Haure-Mirande, J.-V.; Bozec, D.; Yeretssian, G.; Dahan, S. Intestinal Epithelial Notch-1 Protects from Colorectal Mucinous Adenocarcinoma. Oncotarget 2018, 9, 33536–33548. [Google Scholar] [CrossRef] [PubMed]
- Habib, Y.J.; Zhang, Z. The Involvement of Crustaceans Toll-like Receptors in Pathogen Recognition. Fish Shellfish Immunol. 2020, 102, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Vaish, S.; Gupta, D.; Mehrotra, R.; Mehrotra, S.; Basantani, M.K. Glutathione S-Transferase: A Versatile Protein Family. 3 Biotech. 2020, 10, 321. [Google Scholar] [CrossRef] [PubMed]
- Cunha, I.; García, L.M.; Guilhermino, L. Sea-Urchin (Paracentrotus lividus) Glutathione S-Transferases and Cholinesterase Activities as Biomarkers of Environmental Contamination. J. Environ. Monit. 2005, 7, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Asadi, M.; Taghizadeh, S.; Kaviani, E.; Vakili, O.; Taheri-Anganeh, M.; Tahamtan, M.; Savardashtaki, A. Caspase-3: Structure, Function, and Biotechnological Aspects. Biotechnol. Appl. Biochem. 2021, 1–13. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Number of Reads | Number of Reads after Trim |
|---|---|---|
| Coelomocyte | 20,682,190 | 19,948,624 |
| Coelomocyte (replicate) | 16,865,448 | 15,699,186 |
| Intestine | 12,145,212 | 11,748,696 |
| Intestine (replicate) | 11,348,164 | 10,620,928 |
| Gonad | 18,495,858 | 17,901,954 |
| Gonad (replicate) | 16,208,768 | 15,199,912 |
| Total | 95,745,640 | 91,119,300 |
| Transcriptome | De novo Assembly | After Filter |
| (Trinity) | (Corset) | |
| Total contigs | 278,803 | 185,239 |
| Average large contig (bp) | 326 | 929 |
| Coverage contig | 708.32 | - |
| %GC | 38.2 | 38.81 |
| N10 (bp) | 5015 | 5328 |
| N30 (bp) | 2645 | 2945 |
| N50 (bp) | 1418 | 1769 |
| Total bases | 197,480,887 | 172,122,576 |
| Coelomocytes vs. Intestine | ||||
| KEGG Term (ID) | Number of Genes | Percentage of Genes | Fold Enrichment | p Value |
| cAMP signaling pathway (4024) | 13 | 2.9% | 2.4 | 8.1 × 10−3 |
| Platelet activation (4611) | 10 | 2.2% | 2.8 | 9.3 × 10−3 |
| Neurotrophin signaling pathway (4722) | 9 | 2.0% | 2.7 | 1.7 × 10−2 |
| Regulation of lipolysis in adipocytes (4923) | 6 | 1.4% | 3.8 | 1.8 × 10−2 |
| Proteoglycans in cancer (5205) | 12 | 2.7% | 2.2 | 2.1 × 10−2 |
| Focal adhesion (4510) | 12 | 2.7% | 2.1 | 2.6 × 10−2 |
| Insulin secretion (4911) | 7 | 1.6% | 3.0 | 2.9 × 10−2 |
| cGMP-PKG signaling pathway (4022) | 10 | 2.2% | 2.3 | 3.0 × 10−2 |
| Coelomocytes vs. Gonad | ||||
| KEGG Term (ID) | Number of Genes | Percentage of Genes | Fold Enrichment | p Value |
| Fc gamma R-mediated phagocytosis (4666) | 15 | 2.5% | 4.9 | 1.7 × 10−6 |
| Platelet activation (4611) | 16 | 2.6% | 3.6 | 7.0 × 10−5 |
| Pathogenic Escherichia coli infection (5130) | 10 | 1.6% | 5.4 | 7.9 × 10−5 |
| cGMP-PKG signaling pathway (4022) | 17 | 2.8% | 2.9 | 1.9 × 10−4 |
| Gap junction (4540) | 12 | 1.9% | 3.7 | 3.3 × 10−4 |
| Proteoglycans in cancer (5205) | 19 | 2.0% | 2.6 | 3.5 × 10−4 |
| Focal adhesion (4510) | 19 | 3.1% | 2.5 | 4.9 × 10−4 |
| Bacterial invasion of epithelial cells (5100) | 11 | 1.8% | 3.8 | 4.9 × 10−4 |
| Gonad vs. Intestine | ||||
| KEGG Term (ID) | Number of Genes | Percentage of Genes | Fold Enrichment | p Value |
| Purine metabolism (230) | 10 | 2.9% | 2.4 | 6.9 × 10−3 |
| Spliceosome (3040) | 8 | 2.3% | 2.8 | 1.4 × 10−2 |
| Huntington’s disease (5016) | 9 | 2.6% | 2.7 | 3.2 × 10−2 |
| p53 signaling pathway (4115) | 5 | 1.5% | 3.8 | 4.0 × 10−2 |
| Focal adhesion (4510) | 9 | 2.6% | 2.1 | 4.6 × 10−2 |
| Metabolic pathways (1100) | 32 | 9.2% | 3.0 | 5.9 × 10−2 |
| Pathogenic Escherichia coli infection (5130) | 4 | 1.5% | 2.3 | 7.5 × 10−2 |
| Gap junction (4540) | 5 | 1.7% | 2.4 | 9.0 × 10−2 |
| Gonad vs. Coelomocytes | ||||
| KEGG Term (ID) | Number of Genes | Percentage of Genes | Fold Enrichment | p Value |
| Biosynthesis of antibiotics (1130) | 20 | 4.5% | 3.4 | 4.5 × 10−6 |
| Gap junction (4540) | 10 | 2.3% | 4.1 | 6.2 × 10−4 |
| Fatty acid degradation (71) | 7 | 1.6% | 6.1 | 8.9 × 10−4 |
| Valine, leucine, and isoleucine degradation (280) | 7 | 1.6% | 5.4 | 1.6 × 10−3 |
| Fatty acid metabolism (1212) | 7 | 1.6% | 5.3 | 1.8 × 10−3 |
| Phagosome (4145) | 12 | 2.7% | 2.9 | 2.6 × 10−3 |
| Metabolic pathways (1100) | 47 | 10.6% | 1.4 | 1.1 × 10−2 |
| DNA replication (3030) | 5 | 1.1% | 5.1 | 1.6 × 10−2 |
| Intestine vs. Coelomocytes | ||||
| KEGG Term (ID) | Number of Genes | Percentage of Genes | Fold Enrichment | p Value |
| Adherens junction (4520) | 13 | 2.1% | 5.0 | 7.7 × 10−6 |
| Pathogenic Escherichia coli infection (5130) | 11 | 1.7% | 5.9 | 1.2 × 10−5 |
| ABC transporters (2010) | 10 | 1.6% | 6.2 | 2.2 × 10−5 |
| Lysosome (4142) | 16 | 2.5% | 3.6 | 2.9 × 10−5 |
| Metabolic pathways (1100) | 68 | 10.7% | 1.5 | 1.8 × 10−4 |
| Gap junction (4540) | 11 | 1.7% | 3.4 | 1.2 × 10−3 |
| Chemical carcinogenesis (5204) | 9 | 1.4% | 3.1 | 8.2 × 10−3 |
| cAMP signaling pathway (4024) | 15 | 2.3% | 2.1 | 1.3 × 10−2 |
| Intestine vs. Gonad | ||||
| KEGG Term (ID) | Number of Genes | Percentage of Genes | Fold Enrichment | p Value |
| ABC transporters (2010) | 11 | 1.9% | 7.9 | 7.1 × 10−7 |
| Chemical carcinogenesis (5204) | 12 | 2.2% | 4.8 | 3.4 × 10−5 |
| Pathogenic Escherichia coli infection (5130) | 9 | 1.6% | 5.6 | 1.6 × 10−4 |
| Lysosome (4142) | 12 | 2.2% | 3.2 | 1.3 × 10−3 |
| Gap junction (4540) | 10 | 1.8% | 3.6 | 1.6 × 10−3 |
| Galactose metabolism (52) | 6 | 1.1% | 6.4 | 2.1 × 10−3 |
| Starch and sucrose metabolism (500) | 6 | 1.1% | 5.8 | 3.3 × 10−3 |
| Amino sugar and nucleotide sugar metabolism (520) | 7 | 1.3% | 4.6 | 3.6 × 10−3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antiqueo, P.; Zuloaga, R.; Bastias-Molina, M.; Meneses, C.; Estrada, J.M.; Molina, A.; Valdés, J.A. De novo Assembly and Analysis of Tissue-Specific Transcriptomes of the Edible Red Sea Urchin Loxechinus albus Using RNA-Seq. Biology 2021, 10, 995. https://doi.org/10.3390/biology10100995
Antiqueo P, Zuloaga R, Bastias-Molina M, Meneses C, Estrada JM, Molina A, Valdés JA. De novo Assembly and Analysis of Tissue-Specific Transcriptomes of the Edible Red Sea Urchin Loxechinus albus Using RNA-Seq. Biology. 2021; 10(10):995. https://doi.org/10.3390/biology10100995
Chicago/Turabian StyleAntiqueo, Paulette, Rodrigo Zuloaga, Macarena Bastias-Molina, Claudio Meneses, Juan Manuel Estrada, Alfredo Molina, and Juan Antonio Valdés. 2021. "De novo Assembly and Analysis of Tissue-Specific Transcriptomes of the Edible Red Sea Urchin Loxechinus albus Using RNA-Seq" Biology 10, no. 10: 995. https://doi.org/10.3390/biology10100995