
The Impact of Apolipoprotein E (APOE) Epigenetics on Aging and Sporadic Alzheimer’s Disease
 ,
,  ,
,  , ,
, ,  ,
,
Abstract
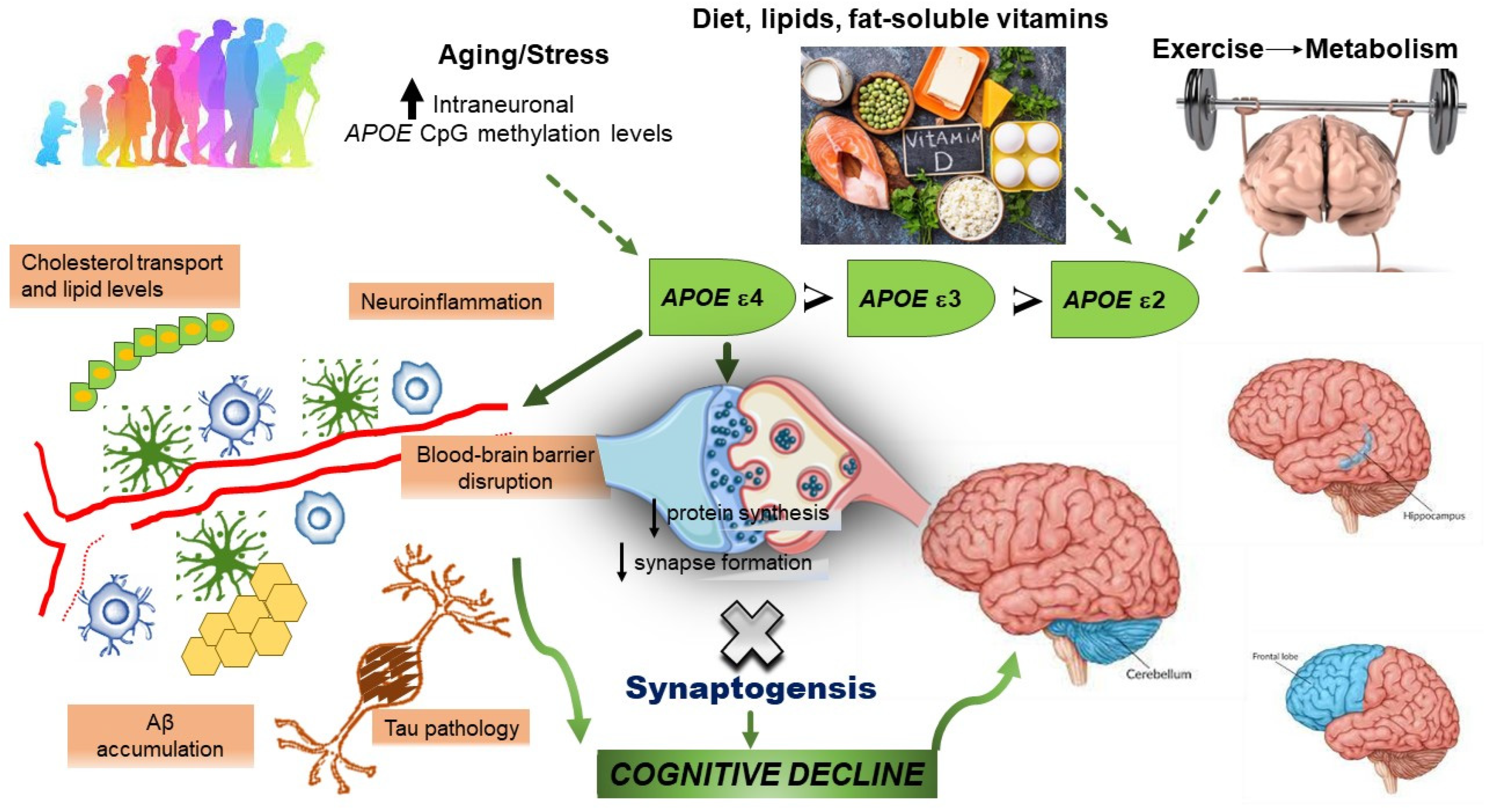
:Simple Summary
Abstract
1. Introduction
2. The Role of Apolipoprotein E in Alzheimer’s Disease Pathogenesis
3. Apolipoprotein E, Human Longevity, and Alzheimer’s Disease
4. Specific Epigenetic Modifications of Apolipoprotein E in Alzheimer’s Disease

{kind=link}
{kind=link}
| APOE Exons, Promoter, and CGI | ||||
|---|---|---|---|---|
| Study | Study Design | Sample Size | Age or Mean Age at Death (Years) | Principal Findings |
| Foraker et al., 2015 [40] | Cross-sectional | AD: 15 Controls: 10 | AD: 82.7 ± 9.3 Controls: 84.9 ± 8.9 | In the cerebellum there was the highest levels of methylation (marginal mean = 93%), with lower levels in the hippocampus (marginal mean = 85%), and the lowest levels in the frontal lobe (marginal mean = 77%) of AD brain compared to controls. There was a complex interaction among the presence of the APOE ε4 allele, AD status, and DNA methylation levels in the APOE CpG islands. AD-specific methylation differences were mainly attributed to the ε3/ε4 heterozygous subjects |
| Lambert et al., 1997 [60] | Cross-sectional | Frontal lobe ε3ε4, ε2ε4, ε2ε3 AD cases: 14 Controls: 12 | AD: 74.1 ± 11.8 (five male and nine female) controls: 83.0 ± 10.6 | In heterozygotes AD, APOE ε4 mRNA expression is increased in patients with AD compared with healthy controls: genetic variability in the neural expression at the APOE locus contributes to AD risk. APOE ε3ε4 heterozygote subjects (high ε4 expressors and/or low ε3 expressors) were more likely to develop AD than subjects with high ε3 expressors and/or low ε4 expressors |
| Lee et al., 2020 [69] | Cross-sectional | Frontal lobe AD: 44 Controls: 21 Cerebellum AD: 51 Controls: 25 | Frontal lobe AD: 86.8 ± 6.9 Controls: 87.9 ± 8.6 Cerebellum AD: 74.6 ± 9.3 Controls: 73.5 ± 10.9 | APOE has a single CpG island overlapping with its 3′-exon. APOE circular RNA and full-length mRNA each constitute one third of the total APOE RNA, with truncated mRNAs likely constituting some of the missing fraction. All APOE RNA species had significantly higher expression in AD frontal lobe than in controls, suggesting a possible modified mechanism of gene action for APOE in AD involving also an epigenetically regulated transcriptional program driven by DNA methylation in the APOE CpG island |
| Yu et al., 2013 [73] | Cross-sectional | Frontal lobe AD: 9 Controls: 6 | AD: 86.8 ± 6.9 Controls: 87.9 ± 8.6 | APOE 3′-exon CpG island exhibited transcriptional enhancer/silencer activity, modulating expression of genes at the APOE locus in a cell type-, DNA methylation- and ε2/ε3/ε4 allele-specific manner. These results suggested a novel functional role for a 3′-exon CpG island involving the protein isoforms and also an epigenetically regulated transcriptional program |
| Ma et al., 2015 [76] | Cross-sectional | 475 men and 518 women | 18–87 | The 13 APOE CpG sites were categorized into three groups: Group 1 exhibited hypermethylation (>50%, in the promoter region), Group 2 showed hypomethylation (<50%, in the first two exons and introns), and Group 3 exhibited hypermethylation (>50%, in the exon 4). APOE methylation was significantly associated to age and plasma total cholesterol. APOE methylation patterns differed across APOE ε variants and the promoter variant rs405509, which further had a significant interaction with age |
| Wang et al., 2008 [77] | Cross-sectional | Prefrontal cortex AD: 24 Matched controls: 10 Blood samples AD: 6 Matched controls: 6 | AD: 80.9 ± 9.3 matched controls: 80.0 ± 9.8 Blood samples AD: 81 ± 4.5 matched controls: 80.0 ± 5.2 | In the AD brain samples, a notably age-specific epigenetic drift was identified, suggesting a role of epigenetic effects in the AD development. APOE gene is of bimodal structure, with a hypomethylated CpG-poor promoter and a fully methylated 39-CpG-island, containing the sequences for the ε4-haplotype |
5. Epigenetics of Apolipoprotein E and Cognitive Function: Contrasting Evidence in Alzheimer’s Disease
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Heyn, H. A symbiotic liaison between the genetic and epigenetic code. Front. Genet. 2014, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Mill, J.; Heijmans, B.T. From promises to practical strategies in epigenetic epidemiology. Nat. Rev. Genet. 2013, 14, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Frisoni, G.B.; Altomare, D.; Thal, D.R.; Ribaldi, F.; van der Kant, R.; Ossenkoppele, R.; Blennow, K.; Cummings, J.; van Duijn, C.; Nilsson, P.M.; et al. The probabilistic model of Alzheimer disease: The amyloid hypothesis revised. Nat. Rev. Neurosci. 2022, 23, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, N.; Dragunow, M. Epigenetics in Alzheimer’s disease: A focus on DNA modifications. Curr. Pharm. Des. 2011, 17, 3398–3412. [Google Scholar] [CrossRef] [PubMed]
- Seripa, D.; Panza, F.; Paroni, G.; D’Onofrio, G.; Bisceglia, P.; Gravina, C.; Urbano, M.; Lozupone, M.; Solfrizzi, V.; Bizzarro, A.; et al. Role of CLU, PICALM, and TNK1 Genotypes in Aging with and without Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 4333–4344. [Google Scholar] [CrossRef]
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef]
- Zhang, D.; Cheng, L.; Badner, J.A.; Chen, C.; Chen, Q.; Luo, W.; Craig, D.W.; Redman, M.; Gershon, E.S.; Liu, C. Genetic control of individual differences in gene-specific methylation in human brain. Am. J. Hum. Genet. 2010, 86, 411–419. [Google Scholar] [CrossRef]
- Gibbs, J.R.; van der Brug, M.P.; Hernandez, D.G.; Traynor, B.J.; Nalls, M.A.; Lai, S.L.; Arepalli, S.; Dillman, A.; Rafferty, I.P.; Troncoso, J.; et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010, 6, e1000952. [Google Scholar] [CrossRef]
- Shoemaker, R.; Deng, J.; Wang, W.; Zhang, K. Allele-specific methylation is prevalent and is contributed by CpG-SNPs in the human genome. Genome Res. 2010, 20, 883–889. [Google Scholar] [CrossRef]
- Seripa, D.; D’Onofrio, G.; Panza, F.; Cascavilla, L.; Masullo, C.; Pilotto, A. The genetics of the human APOE polymorphism. Rejuvenation Res. 2011, 14, 491–500. [Google Scholar] [CrossRef]
- Nickerson, D.A.; Taylor, S.L.; Fullerton, S.M.; Weiss, K.M.; Clark, A.G.; Stengård, J.H.; Salomaa, V.; Boerwinkle, E.; Sing, C.F. Sequence diversity and large-scale typing of SNPs in the human apolipoprotein E gene. Genome Res. 2000, 10, 1532–1545. [Google Scholar] [CrossRef]
- Yu, C.E.; Foraker, J. Epigenetic considerations of the APOE gene. Biomol. Concepts 2015, 6, 77–84. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein e type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Chartier-Harlin, M.C.; Parfitt, M.; Legrain, S.; Pérez-Tur, J.; Brousseau, T.; Evans, A.; Berr, C.; Odile, V.; Roques, P.; Gourlet, V.; et al. Apolipoprotein E, epsilon 4 allele as a major risk factor for sporadic early and late-onset forms of alzheimer’s disease: Analysis of the 19q13,2 chromosomal region. Hum. Mol. Genet. 1994, 3, 569–574. [Google Scholar] [CrossRef]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein e genotype and alzheimer disease: A meta-analysis. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef]
- Genin, E.; Hannequin, D.; Wallon, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Bullido, M.J.; Engelborghs, S.; De Deyn, P.; Berr, C.; et al. APOE and Alzheimer disease: A major gene with semi-dominant inheritance. Mol. Psychiatry 2011, 16, 903–907. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Risch, N.J.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C., Jr.; Rimmler, J.B.; Locke, P.A.; Conneally, P.M.; Schmader, K.E.; et al. Protective effect of apolipoprotein e type 2 allele for late onset Alzheimer disease. Nat. Genet. 1994, 7, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Solfrizzi, V.; Torres, F.; Mastroianni, F.; Colacicco, A.M.; Basile, A.M.; Capurso, C.; D’Introno, A.; Del Parigi, A.; Capurso, A. Apolipoprotein E in Southern Italy: Protective effect of epsilon 2 allele in early- and late-onset sporadic Alzheimer’s disease. Neurosci. Lett. 2000, 292, 79–82. [Google Scholar] [CrossRef]
- Mayeux, R.; Saunders, A.M.; Shea, S.; Mirra, S.; Evans, D.; Roses, A.D.; Hyman, B.T.; Crain, B.; Tang, M.X.; Phelps, C.H. Utility of the apolipoprotein E genotype in the diagnosis of Alzheimer’s disease. Alzheimer’s disease centers consortium on apolipoprotein E and Alzheimer’s disease. N. Engl. J. Med. 1998, 338, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, M.; Imbimbo, B.P.; Balducci, C.; Lo Vecchio, F.; Bisceglia, P.; Latino, R.R.; Leone, M.; Dibello, V.; Solfrizzi, V.; Greco, A.; et al. Does the imbalance in the apolipoprotein E isoforms underlie the pathophysiological process of sporadic Alzheimer’s disease? Alzheimer’s Dement. 2023, 19, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Das, S.; Hyman, B.T. APOE and Alzheimer’s disease: Advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 2021, 20, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Arboleda-Velasquez, J.F.; Quiroz, Y.T.; Huentelman, M.J.; Beach, T.G.; Caselli, R.J.; Chen, Y.; Su, Y.; Myers, A.J.; Hardy, J.; et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5000-person neuropathological study. Nat. Commun. 2020, 11, 667. [Google Scholar] [CrossRef]
- Salvadó, G.; Grothe, M.J.; Groot, C.; Moscoso, A.; Schöll, M.; Gispert, J.D.; Ossenkoppele, R.; Alzheimer’s Disease Neuroimaging Initiative. Differential associations of APOE-ε2 and APOE-ε4 alleles with PET-measured amyloid-β and tau deposition in older individuals without dementia. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2212–2224. [Google Scholar] [CrossRef] [PubMed]
- Therriault, J.; Benedet, A.L.; Pascoal, T.A.; Mathotaarachchi, S.; Chamoun, M.; Savard, M.; Thomas, E.; Kang, M.S.; Lussier, F.; Tissot, C.; et al. Association of Apolipoprotein E 4 With Medial Temporal Tau Independent of Amyloid-β. JAMA Neurol. 2020, 77, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hong, T.; Chen, F.; Sun, Y.; Wang, Y.; Cui, L. Interplay between microglia and Alzheimer’s disease-focus on the most relevant risks: APOE genotype, sex and age. Front. Aging Neurosci. 2021, 13, 631827. [Google Scholar] [CrossRef]
- Chung, W.S.; Verghese, P.B.; Chakraborty, C.; Joung, J.; Hyman, B.T.; Ulrich, J.D.; Holtzman, D.M.; Barres, B.A. Novel allele-dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 10186–10191. [Google Scholar] [CrossRef]
- Blanchard, J.W.; Bula, M.; Davila-Velderrain, J.; Akay, L.A.; Zhu, L.; Frank, A.; Victor, M.B.; Bonner, J.M.; Mathys, H.; Lin, Y.T.; et al. Reconstruction of the human blood-brain barrier in vitro reveals a pathogenic mechanism of APOE4 in pericytes. Nat. Med. 2020, 26, 952–963. [Google Scholar] [CrossRef]
- Riphagen, J.M.; Ramakers, I.H.G.M.; Freeze, W.M.; Pagen, L.H.G.; Hanseeuw, B.J.; Verbeek, M.M.; Verhey, F.R.J.; Jacobs, H.I.L. Linking APOE-ε4, blood-brain barrier dysfunction, and inflammation to Alzheimer’s pathology. Neurobiol. Aging 2020, 85, 96–103. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein e: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef]
- Fagan, A.M.; Holtzman, D.M. Astrocyte lipoproteins, effects of ApoE on neuronal function, and role of ApoE in amyloid-beta deposition in vivo. Microsc. Res. Tech. 2000, 50, 297–304. [Google Scholar] [CrossRef]
- Tokuda, T.; Calero, M.; Matsubara, E.; Vidal, R.; Kumar, A.; Permanne, B.; Zlokovic, B.; Smith, J.D.; Ladu, M.J.; Rostagno, A.; et al. Lipidation of apolipoprotein e influences its isoform-specific interaction with Alzheimer’s amyloid beta peptides. Biochem. J. 2002, 348, 359–365. [Google Scholar] [CrossRef]
- Deane, R.; Sagare, A.; Hamm, K.; Parisi, M.; Lane, S.; Finn, M.B.; Holtzman, D.M.; Zlokovic, B.V. ApoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J. Clin. Investig. 2008, 118, 4002–40013. [Google Scholar] [CrossRef]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human ApoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci. Transl. Med. 2011, 3, 89ra57. [Google Scholar] [CrossRef]
- Huang, Y.A.; Zhou, B.; Wernig, M.; Sudhof, T.C. ApoE2, ApoE3, and ApoE4 differentially stimulate APP transcription and A-β secretion. Cell 2017, 168, 427.e21–441.e21. [Google Scholar] [CrossRef]
- Sala Frigerio, C.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.T.; Woodbury, M.E.; Srivastava, G.; et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Rep. 2019, 27, 1293–1306.e6. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Holtzman, D.M. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 2018, 18, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Liu, C.C.; Van Ingelgom, A.J.; Linares, C.; Kurti, A.; Knight, J.A.; Heckman, M.G.; Diehl, N.N.; Shinohara, M.; Martens, Y.A.; et al. APOE ε2 is associated with increased tau pathology in primary tauopathy. Nat. Commun. 2018, 9, 4388. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, M.; Panza, F. Impact of apolipoprotein E isoforms on sporadic Alzheimer’s disease: Beyond the role of amyloid beta. Neural Regen. Res. 2024, 19, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Foraker, J.; Millard, S.P.; Leong, L.; Thomson, Z.; Chen, S.; Keene, C.D.; Bekris, L.M.; Yu, C.E. The APOE Gene is Differentially Methylated in Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 48, 745–755. [Google Scholar] [CrossRef]
- Rueter, J.; Rimbach, G.; Huebbe, P. Allelic variation within the major APOE CpG island affects its methylation in the brain of targeted replacement mice expressing human APOE. Biochim. Biophys. Acta Gene Regul. Mech. 2023, 1866, 194942. [Google Scholar] [CrossRef]
- Lefterov, I.; Fitz, N.F.; Lu, Y.; Koldamova, R. APOEε4 and risk of Alzheimer’s disease—Time to move forward. Front. Neurosci. 2023, 17, 1195724. [Google Scholar] [CrossRef]
- Sullivan, P.M.; Mezdour, H.; Aratani, Y.; Knouff, C.; Najib, J.; Reddick, R.L.; Quarfordt, S.H.; Maeda, N. Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J. Biol. Chem. 1997, 272, 17972–17980. [Google Scholar] [CrossRef]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]
- Reas, E.T.; Laughlin, G.A.; Bergstrom, J.; Kritz-Silverstein, D.; Barrett-Connor, E.; McEvoy, L.K. Effects of APOE on cognitive aging in community-dwelling older adults. Neuropsychology 2019, 33, 406–416. [Google Scholar] [CrossRef]
- Mahley, R.W. Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science 1988, 240, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Siest, G.; Pillot, T.; Regis-Bailly, A.; Leininger-Muller, B.; Steinmetz, J.; Galteau, M.M.; Visvikis, S. Apolipoprotein E: An important gene and protein to follow in laboratory medicine. Clin. Chem. 1995, 41, 1068–1086. [Google Scholar] [CrossRef] [PubMed]
- Seripa, D.; Franceschi, M.; Matera, M.G.; Panza, F.; Kehoe, P.G.; Gravina, C.; Orsitto, G.; Solfrizzi, V.; Di Minno, G.; Dallapiccola, B.; et al. Sex differences in the association of apolipoprotein e and angiotensin-converting enzyme gene polymorphisms with healthy aging and longevity: A population-based study from Southern Italy. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 918–923. [Google Scholar] [CrossRef]
- Seripa, D.; Panza, F.; Franceschi, M.; D’Onofrio, G.; Solfrizzi, V.; Dallapiccola, B.; Pilotto, A. Non-apolipoprotein E and apolipoprotein E genetics of sporadic Alzheimer’s disease. Ageing Res. Rev. 2009, 8, 214–236. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W.; Rall, S.C., Jr. Apolipoprotein E: Far more than a lipid transport protein. Annu. Rev. Genom. Hum. Genet. 2000, 1, 507–537. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, K. Cholesterol and pathological processes in Alzheimer’s disease. J. Neurosci. Res. 2002, 70, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Ordovas, J.M.; Lopez-Miranda, J.; Mata, P.; Perez-Jimenez, F.; Lichtenstein, A.H.; Schaefer, E.J. Gene-diet interaction in determining plasma lipid response to dietary intervention. Atherosclerosis 1995, 118, S11–S27. [Google Scholar] [CrossRef]
- Grimm, M.O.W.; Michaelson, D.M.; Hartmann, T. Omega-3 fatty acids, lipids, and ApoE lipidation in Alzheimer’s disease: A rationale for multi-nutrient dementia prevention. J. Lipid Res. 2017, 58, 2083–2101. [Google Scholar] [CrossRef]
- Bos, M.M.; Noordam, R.; Blauw, G.J.; Slagboom, P.E.; Rensen, P.C.N.; van Heemst, D. The ApoE ε4 Isoform: Can the Risk of Diseases be Reduced by Environmental Factors? J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 99–107. [Google Scholar] [CrossRef]
- Brooks-Wilson, A.R. Genetics of healthy aging and longevity. Hum. Genet. 2013, 132, 1323–1338. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, P.; Gurinovich, A.; Nygaard, M.; Sasaki, T.; Sweigart, B.; Bae, H.; Andersen, S.L.; Villa, F.; Atzmon, G.; Christensen, K.; et al. APOE alleles and extreme human longevity. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 74, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.D. Apolipoproteins and aging: Emerging mechanisms. Ageing Res. Rev. 2002, 1, 345–365. [Google Scholar] [CrossRef]
- Feng, J.; Xiang, L.; Wan, G.; Qi, K.; Sun, L.; Huang, Z.; Zheng, C.; Lv, Z.; Hu, C.; Yang, Z. Is APOE ε3 a favorable factor for the longevity: An association study in Chinese population. J. Genet. 2011, 90, 343–347. [Google Scholar] [CrossRef]
- Paik, Y.K.; Chang, D.J.; Reardon, C.A.; Walker, M.D.; Taxman, E.; Taylor, J.M. Identification and characterization of transcriptional regulatory regions associated with expression of the human apolipoprotein E gene. J. Biol. Chem. 1988, 263, 13340–13349. [Google Scholar] [CrossRef]
- Lambert, J.C.; Perez-Tur, J.; Dupire, M.J.; Galasko, D.; Mann, D.; Amouyel, P.; Hardy, J.; Delacourte, A.; Chartier-Harlin, M.C. Distortion of allelic expression of apolipoprotein E in Alzheimer’s disease. Hum. Mol. Genet. 1997, 6, 2151–2154. [Google Scholar] [CrossRef]
- Mui, S.; Briggs, M.; Chung, H.; Wallace, R.B.; Gomez-Isla, T.; Rebeck, G.W.; Hyman, B.T. A newly identified polymorphism in the apolipoprotein E enhancer gene region is associated with Alzheimer’s disease and strongly with the epsilon 4 allele. Neurology 1996, 47, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Berr, C.; Pasquier, F.; Delacourte, A.; Frigard, B.; Cottel, D.; Pérez-Tur, J.; Mouroux, V.; Mohr, M.; Cécyre, D.; et al. Pronounced impact of Th1/E47cs mutation compared with-491 AT mutation on neural APOE gene expression and risk of developing Alzheimer’s disease. Hum. Mol. Genet. 1998, 7, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Brousseau, T.; Defosse, V.; Evans, A.; Arveiler, D.; Ruidavets, J.B.; Haas, B.; Cambou, J.P.; Luc, G.; Ducimetière, P.; et al. Independent association of an APOE gene promoter polymorphism with increased risk of myocardial infarction and decreased APOE plasma concentrations-the ECTIM study. Hum. Mol. Genet. 2000, 9, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Pasquier, F.; Cottel, D.; Frigard, B.; Amouyel, P.; Chartier-Harlin, M.C. A new polymorphism in the APOE promoter associated with risk of developing Alzheimer’s disease. Hum. Mol. Genet. 1998, 7, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Bullido, M.A.J.; and Valdivieso, F. Apolipoprotein E gene promoter polymorphisms in Alzheimer’s disease. Microsc. Res. Tech. 2000, 50, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Lumsden, A.L.; Mulugeta, A.; Zhou, A.; Hypponen, E. Apolipoprotein E (APOE) genotype-associated disease risks: A phenome-wide, registry-based, case-control study utilising the UK biobank. eBioMedicine 2020, 59, 102954. [Google Scholar] [CrossRef] [PubMed]
- Artiga, M.J.; Bullido, M.J.; Frank, A.; Sastre, I.; Recuero, M.; García, M.A.; Lendon, C.L.; Han, S.W.; Morris, J.C.; Vázquez, J.; et al. Risk for Alzheimer’s disease correlates with transcriptional activity of the APOE gene. Hum. Mol. Genet. 1998, 7, 1887–1892. [Google Scholar] [CrossRef]
- Sims, R.; van der Lee, S.J.; Naj, A.C.; Bellenguez, C.; Badarinarayan, N.; Jakobsdottir, J.; Kunkle, B.W.; Boland, A.; Raybould, R.; Bis, J.C.; et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet. 2017, 49, 1373–1384. [Google Scholar] [CrossRef]
- Lee, E.G.; Tulloch, J.; Chen, S.; Leong, L.; Saxton, A.D.; Kraemer, B.; Darvas, M.; Keene, C.D.; Shutes-David, A.; Todd, K.; et al. Redefining transcriptional regulation of the APOE gene and its association with Alzheimer’s disease. PLoS ONE 2020, 15, e0227667. [Google Scholar] [CrossRef]
- Medvedeva, Y.A.; Fridman, M.V.; Oparina, N.J.; Malko, D.B.; Ermakova, E.O.; Kulakovskiy, I.V.; Heinzel, A.; Makeev, V.J. Intergenic, gene terminal, and intragenic CpG islands in the human genome. BMC Genom. 2010, 11, 48. [Google Scholar] [CrossRef]
- Maunakea, A.K.; Nagarajan, R.P.; Bilenky, M.; Ballinger, T.J.; D’Souza, C.; Fouse, S.D.; Johnson, B.E.; Hong, C.; Nielsen, C.; Zhao, Y.; et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010, 466, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Gellersen, H.M.; Guell, X.; Sami, S. Differential vulnerability of the cerebellum in healthy ageing and Alzheimer’s disease. Neuroimage Clin. 2021, 30, 102605. [Google Scholar] [CrossRef]
- Yu, C.E.; Cudaback, E.; Foraker, J.; Thomson, Z.; Leong, L.; Lutz, F.; Gill, J.A.; Saxton, A.; Kraemer, B.; Navas, P.; et al. Epigenetic signature and enhancer activity of the human APOE gene. Hum. Mol. Genet. 2013, 22, 5036–5047. [Google Scholar] [CrossRef] [PubMed]
- Hendrich, B.; Bird, A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol. Cell. Biol. 1998, 18, 6538–6547. [Google Scholar] [CrossRef] [PubMed]
- Cedar, H.; Bergman, Y. Programming of DNA methylation patterns. Annu. Rev. Biochem. 2012, 81, 97–117. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Smith, C.E.; Lai, C.Q.; Irvin, M.R.; Parnell, L.D.; Lee, Y.C.; Pham, L.; Aslibekyan, S.; Claas, S.A.; Tsai, M.Y.; et al. Genetic variants modify the effect of age on APOE methylation in the Genetics of Lipid Lowering Drugs and Diet Network study. Aging Cell 2015, 14, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Oelze, B.; Schumacher, A. Age-specific epigenetic drift in late-onset Alzheimer’s disease. PLoS ONE 2008, 3, e2698. [Google Scholar] [CrossRef]
- Lin, L.; Liu, X.; Cheng, X.; Li, Y.; Gearing, M.; Levey, A.; Huang, X.; Li, Y.; Jin, P.; Li, X. MicroRNA-650 Regulates the Pathogenesis of Alzheimer’s Disease Through Targeting Cyclin-Dependent Kinase 5. Mol. Neurobiol. 2023, 60, 2426–2441. [Google Scholar] [CrossRef]
- De Marco, M.; Clough, P.J.; Dyer, C.E.; Vince, R.V.; Waby, J.S.; Midgley, A.W.; Venneri, A. Apolipoprotein E epsilon allele modulates the immediate impact of acute exercise on prefrontal function. Behav. Genet. 2014, 45, 106–116. [Google Scholar] [CrossRef]
- Cook, C.J.; Fletcher, J.M. Can education rescue genetic liability for cognitive decline? Social Sci. Med. 2015, 127, 159–170. [Google Scholar] [CrossRef]
- Maddock, J.; Cavadino, A.; Power, C.; Hyppönen, E. 25-hydroxyvitamin D, APOE ε4 genotype and cognitive function: Findings from the 1958 British birth cohort. Eur. J. Clin. Nutr. 2015, 69, 505–508. [Google Scholar] [CrossRef]
- Egert, S.; Rimbach, G.; Huebbe, P. Symposium on ‘Metabolic flexibility in animal and human nutrition’ Session IV: Nutritional compounds for optimized healthspan and life performance: ApoE genotype: From geographic distribution to function and responsiveness to dietary factors. Proc. Nutr. Soc. 2012, 71, 410–424. [Google Scholar] [CrossRef]
- Panza, F.; La Montagna, M.; Lampignano, L.; Zupo, R.; Bortone, I.; Castellana, F.; Sardone, R.; Borraccino, L.; Dibello, V.; Resta, E.; et al. Vitamin D in the development and progression of alzheimer’s disease: Implications for clinical management. Expert Rev. Neurother. 2021, 2, 287–301. [Google Scholar] [CrossRef]
- Suzuki, K.; Hirakawa, A.; Ihara, R.; Iwata, A.; Ishii, K.; Ikeuchi, T.; Sun, C.K.; Donohue, M.; Iwatsubo, T.; Alzheimer’s Disease Neuroimaging Initiative, Japanese Alzheimer’s Disease Neuroimaging Initiative. Effect of apolipoprotein E ε4 allele on the progression of cognitive decline in the early stage of Alzheimer’s disease. Alzheimer’s Dement. 2020, 6, e12007. [Google Scholar] [CrossRef]
- Karlsson, I.K.; Ploner, A.; Wang, Y.; Gatz, M.; Pedersen, N.L.; Hagg, S. Apolipoprotein E DNA methylation and late-life disease. Int. J. Epidemiol. 2018, 47, 899–907. [Google Scholar] [CrossRef]
- Shao, Y.; Shaw, M.; Todd, K.; Khrestian, M.; D’Aleo, G.; Barnard, P.J.; Zahratka, J.; Pillai, J.; Yu, C.E.; Keene, C.D.; et al. DNA methyla-tion of TOMM40-APOE-APOC2 in Alzheimer’s disease. J. Hum. Genet. 2018, 63, 459–471. [Google Scholar] [CrossRef]
- Mancera-Paez, O.; Estrada-Orozco, K.; Mahecha, M.F.; Cruz, F.; Bonilla-Vargas, K.; Sandoval, N.; Guerrero, E.; Salcedo-Tacuma, D.; Melgarejo, J.D.; Vega, E.; et al. Differential methylation in APOE (Chr19; exon four; from 44,909,188 to 44,909,373/hg38) and increased apolipoprotein E plasma levels in subjects with mild cognitive impairment. Int. J. Mol. Sci. 2019, 20, 1394. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhao, W.; Ware, E.B.; Turner, S.T.; Mosley, T.H.; Smith, J.A. DNA methylation in the APOE genomic region is associated with cognitive function in African Americans. BMC Med. Genom. 2018, 11, 43. [Google Scholar] [CrossRef] [PubMed]
- Mur, J.; McCartney, D.L.; Walker, R.M.; Campbell, A.; Bermingham, M.L.; Morris, S.W.; Porteous, D.J.; McIntosh, A.M.; Deary, I.J.; Evans, K.L.; et al. DNA methylation in APOE: The relationship with Alzheimer’s and with cardiovascular health. Alzheimer’s Dement. 2020, 6, e12026. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Griseta, C.; Battista, P.; Castellana, F.; Colonna, I.; Sciarra, S.; Zupo, R.; Bortone, I.; Lampignano, L.; Tirelli, S.; Bernardino, G.; et al. Serum levels of IL-6 are associated with cognitive impairment in the salus in apulia population-based study. Heliyon 2023, 9, e13972. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Hernandez, D.G.; Chitrala, K.N.; Fanelli-Kuczmarski, M.T.; Noren Hooten, N.; Pacheco, N.L.; Mode, N.A.; Zonderman, A.B.; Ezike, N.; Evans, M.K. APOE gene region methylation is associated with cognitive performance in middle-aged urban adults. Neurobiol. Aging 2022, 116, 41–48. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Tulloch, J.; Leong, L.; Thomson, Z.; Chen, S.; Lee, E.G.; Keene, C.D.; Millard, S.P.; Yu, C.E. Glia-specific APOE epigenetic changes in the Alzheimer’s disease brain. Brain Res. 2018, 1698, 179–186. [Google Scholar] [CrossRef]
- Di Lena, P.; Sala, C.; Prodi, A.; Nardini, C. Missing value estimation methods for DNA methylation data. Bioinformatics 2019, 35, 3786–3793. [Google Scholar] [CrossRef]
- Slooter, A.J.C.; Cruts, M.; Kalmijn, S.; Hofman, A.; Breteler, M.M.; Van Broeckhoven, C.; van Duijn, C.M. Risk estimates of dementia by apolipoprotein E genotypes from a population-based incidence study: The Rotterdam Study. Arch. Neurol. 1998, 55, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Pogribny, I.P.; Vanyushin, B.F. Age-related Genomic Hypomethylation. In Epigenetics of Aging; Tollefsbol, T.O., Ed.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 11–27. [Google Scholar]
- Cummings, J.L.; Osse, A.M.L.; Kinney, J.W. Alzheimer’s Disease: Novel Targets and Investigational Drugs for Disease Modification. Drugs 2023, 83, 1387–1408. [Google Scholar] [CrossRef]
- Lozupone, M.; Dibello, V.; Sardone, R.; Castellana, F.; Zupo, R.; Lampignano, L.; Bortone, I.; Stallone, R.; Altamura, M.; Bellomo, A.; et al. The development of peptide- and oligonucleotide-based drugs to prevent the formation of abnormal tau in tauopathies. Expert Opin. Drug Discov. 2023, 18, 515–526. [Google Scholar] [CrossRef]
- Costa, D.; Scognamiglio, M.; Fiorito, C.; Benincasa, G.; Napoli, C. Genetic background, epigenetic factors and dietary interventions which influence human longevity. Biogerontology 2019, 20, 605–626. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lozupone, M.; Dibello, V.; Sardone, R.; Castellana, F.; Zupo, R.; Lampignano, L.; Bortone, I.; Daniele, A.; Bellomo, A.; Solfrizzi, V.; et al. The Impact of Apolipoprotein E (APOE) Epigenetics on Aging and Sporadic Alzheimer’s Disease. Biology 2023, 12, 1529. https://doi.org/10.3390/biology12121529
Lozupone M, Dibello V, Sardone R, Castellana F, Zupo R, Lampignano L, Bortone I, Daniele A, Bellomo A, Solfrizzi V, et al. The Impact of Apolipoprotein E (APOE) Epigenetics on Aging and Sporadic Alzheimer’s Disease. Biology. 2023; 12(12):1529. https://doi.org/10.3390/biology12121529
Chicago/Turabian StyleLozupone, Madia, Vittorio Dibello, Rodolfo Sardone, Fabio Castellana, Roberta Zupo, Luisa Lampignano, Ilaria Bortone, Antonio Daniele, Antonello Bellomo, Vincenzo Solfrizzi, and et al. 2023. "The Impact of Apolipoprotein E (APOE) Epigenetics on Aging and Sporadic Alzheimer’s Disease" Biology 12, no. 12: 1529. https://doi.org/10.3390/biology12121529