
Gender Differences in the Pathogenesis and Risk Factors of Hepatocellular Carcinoma
 , , , , , , , ,
, , , , , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Epidemiological and Clinical Gender Difference in HCC
3. Gender Differences in Risk Factors of Hepatocellular Carcinoma
3.1. Alcohol
3.2. Obesity and Metabolic Syndrome
3.3. Viral Hepatitis
3.4. Other Causes of Cirrhosis
3.5. Cigarette Smoking
4. Genetic Gender Differences in the Risk of HCC Development
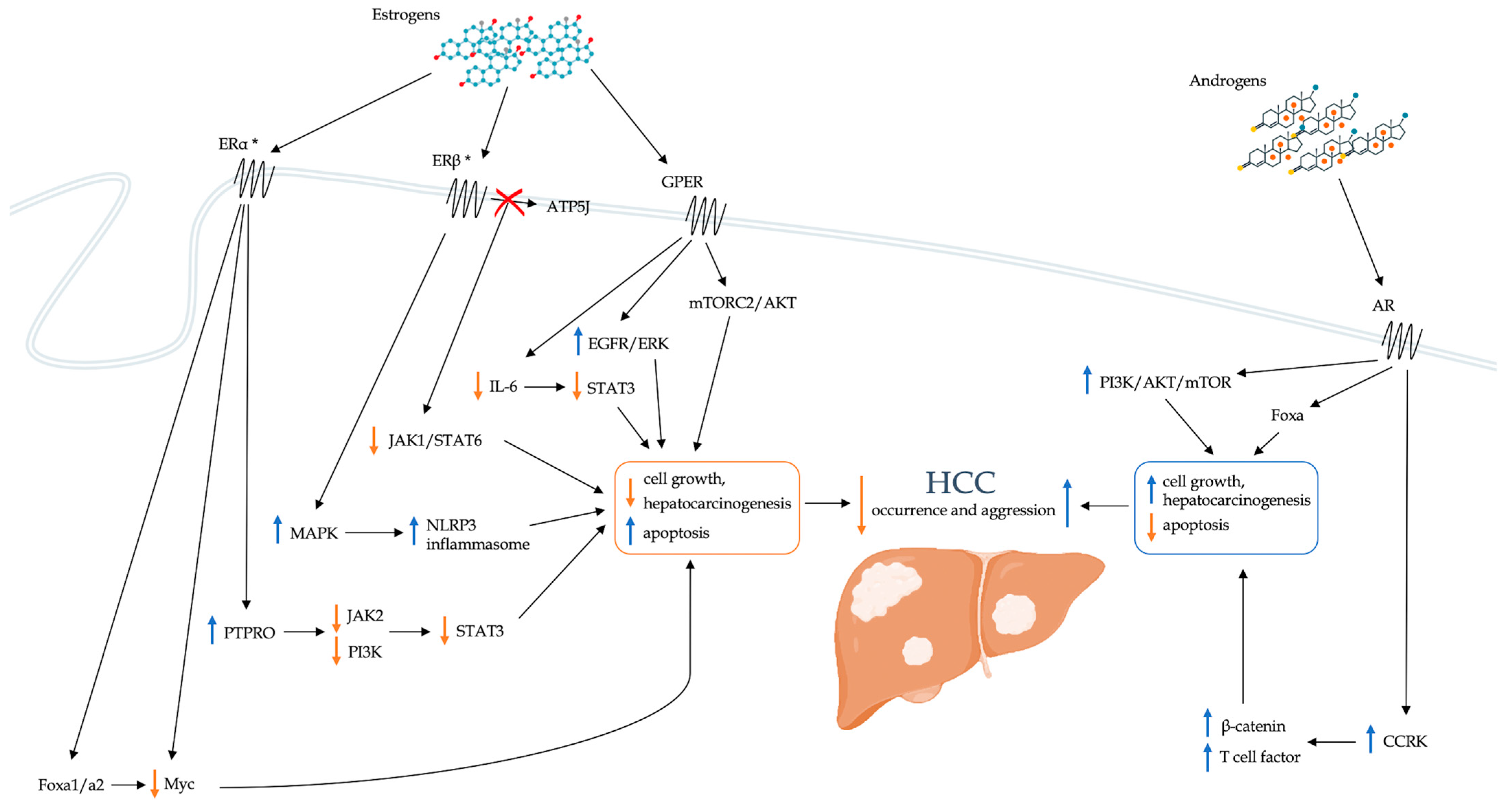
5. Impact of Sex Hormones on the Development of Hepatocellular Carcinoma
5.1. Estrogens
5.1.1. PTPRO
5.1.2. Foxa1 and Foxa2
5.1.3. GPER
5.1.4. Inflammation
IL-6
Tumor-Associated Macrophages
NLRP3
5.2. Androgens
5.3. Influence of Sex Hormones on Other Risk Factors for HCC
5.3.1. Chronic Viral Hepatitis
5.3.2. Obesity
5.4. Interaction between Sex Hormones and miRNAs
6. Therapeutic Implications
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, R.; Bandoh, S.; Roberts, L.R. Molecular pathogenesis of hepatocellular carcinoma and impact of therapeutic advances. F1000Research 2016, 5, F1000 Faculty Rev-879. [Google Scholar] [CrossRef] [PubMed]
- Baecker, A.; Liu, X.; La Vecchia, C.; Zhang, Z.F. Worldwide incidence of hepatocellular carcinoma cases attributable to major risk factors. Eur. J. Cancer Prev. 2018, 27, 205–212. [Google Scholar] [CrossRef]
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma. Hepatology 2021, 73 (Suppl. S1), 4–13. [Google Scholar] [CrossRef]
- Petrick, J.L.; Florio, A.A.; Znaor, A.; Ruggieri, D.; Laversanne, M.; Alvarez, C.S.; Ferlay, J.; Valery, P.C.; Bray, F.; McGlynn, K.A. International trends in hepatocellular carcinoma incidence, 1978–2012. Int. J. Cancer 2020, 147, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.K.; Chatterjee, B. Sexual dimorphism in the liver. Annu. Rev. Physiol. 1983, 45, 37–50. [Google Scholar] [CrossRef]
- Liu, Y.; Zheng, J.; Hao, J.; Wang, R.R.; Liu, X.; Gu, P.; Yu, H.; Yu, Y.; Wu, C.; Ou, B.; et al. Global burden of primary liver cancer by five etiologies and global prediction by 2035 based on global burden of disease study 2019. Cancer Med. 2022, 11, 1310–1323. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Shao, X.; Wang, X.; Liu, L.; Liang, H. Sex disparities in cancer. Cancer Lett. 2019, 466, 35–38. [Google Scholar] [CrossRef]
- Choi, Y.; Kim, N.; Kim, K.W.; Jo, H.H.; Park, J.; Yoon, H.; Shin, C.M.; Park, Y.S.; Lee, D.H.; Oh, H.J.; et al. Sex-based differences in histology, staging, and prognosis among 2983 gastric cancer surgery patients. World J. Gastroenterol. 2022, 28, 933–947. [Google Scholar] [CrossRef]
- Behbahani, S.; Maddukuri, S.; Cadwell, J.B.; Lambert, W.C.; Schwartz, R.A. Gender differences in cutaneous melanoma: Demographics, prognostic factors, and survival outcomes. Dermatol. Ther. 2020, 33, e14131. [Google Scholar] [CrossRef]
- Farinati, F.; Sergio, A.; Giacomin, A.; Di Nolfo, M.A.; Del Poggio, P.; Benvegnù, L.; Rapaccini, G.; Zoli, M.; Borzio, F.; Giannini, E.G.; et al. Is female sex a significant favorable prognostic factor in hepatocellular carcinoma? Eur. J. Gastroenterol. Hepatol. 2009, 21, 1212–1218. [Google Scholar] [CrossRef]
- Rich, N.E.; Murphy, C.C.; Yopp, A.C.; Tiro, J.; Marrero, J.A.; Singal, A.G. Sex disparities in presentation and prognosis of 1110 patients with hepatocellular carcinoma. Aliment. Pharmacol. Ther. 2020, 52, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Tangkijvanich, P.; Mahachai, V.; Suwangool, P.; Poovorawan, Y. Gender difference in clinicopathologic features and survival of patients with hepatocellular carcinoma. World J. Gastroenterol. 2004, 10, 1547–1550. [Google Scholar] [CrossRef] [PubMed]
- Ng, I.O.; Ng, M.M.; Lai, E.C.; Fan, S.T. Better survival in female patients with hepatocellular carcinoma. Possible causes from a pathologic approach. Cancer 1995, 75, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Nevola, R.; Ruocco, R.; Criscuolo, L.; Villani, A.; Alfano, M.; Beccia, D.; Imbriani, S.; Claar, E.; Cozzolino, D.; Sasso, F.C.; et al. Predictors of early and late hepatocellular carcinoma recurrence. World J. Gastroenterol. 2023, 29, 1243–1260. [Google Scholar] [CrossRef]
- Wu, E.M.; Wong, L.L.; Hernandez, B.Y.; Ji, J.F.; Jia, W.; Kwee, S.A.; Kalathil, S. Gender differences in hepatocellular cancer: Disparities in nonalcoholic fatty liver disease/steatohepatitis and liver transplantation. Hepatoma. Res. 2018, 4, 66. [Google Scholar] [CrossRef]
- Sobotka, L.; Hinton, A.; Conteh, L. Women receive more inpatient resections and ablations for hepatocellular carcinoma than men. World J. Hepatol. 2017, 9, 1346–1351. [Google Scholar] [CrossRef]
- Dohmen, K.; Shigematsu, H.; Irie, K.; Ishibashi, H. Longer survival in female than male with hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2003, 18, 267–272. [Google Scholar] [CrossRef]
- Liang, T.; He, Y.; Mo, S.; Chen, Z.; Liao, X.; Zhou, X.; Yang, C.; Zhao, S.; Han, C.; Zhu, G.; et al. Gender disparity in hepatocellular carcinoma recurrence after curative hepatectomy. Ann. Hepatol. 2022, 27, 100695. [Google Scholar] [CrossRef]
- Davis, J.L.; Buchanan, K.L.; Katz, R.V.; Green, B.L. Gender differences in cancer screening beliefs, behaviors, and willingness to participate: Implications for health promotion. Am. J. Mens. Health 2012, 6, 211–217. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Qin, L.X.; Gong, X.; Zhou, J.; Sun, H.C.; Wang, L.; Qiu, S.J.; Ye, Q.H.; Fan, J. Clinical characteristics, outcome, and risk factors for early and late intrahepatic recurrence of female patients after curative resection of hepatocellular carcinoma. Surgery 2014, 156, 651–660. [Google Scholar] [CrossRef]
- Yang, D.; Hanna, D.L.; Usher, J.; LoCoco, J.; Chaudhari, P.; Lenz, H.J.; Setiawan, V.W.; El-Khoueiry, A. Impact of sex on the survival of patients with hepatocellular carcinoma: A Surveillance, Epidemiology, and End Results analysis. Cancer 2014, 120, 3707–3716. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.M.; Yong, J.L.; Chan, A.O.; Ng, K.K.; Poon, R.T.; Liu, C.L.; Lo, C.M.; Fan, S.T. Better survival in female patients with hepatocellular carcinoma: Oral contraceptive pills related? J. Clin. Gastroenterol. 2005, 39, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.M.; Botrus, G.; Abdel-Wahab, R.; Wolff, R.A.; Li, D.; Tweardy, D.; Phan, A.T.; Hawk, E.; Javle, M.; Lee, J.S.; et al. Estrogen Replacement Reduces Risk and Increases Survival Times of Women with Hepatocellular Carcinoma. Clin. Gastroenterol. Hepatol. 2017, 15, 1791–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, M.W.; Chu, Y.D.; Lin, C.L.; Chien, R.N.; Yeh, T.S.; Pan, T.L.; Ke, P.Y.; Lin, K.H.; Yeh, C.T. Is there a sex difference in postoperative prognosis of hepatocellular carcinoma? BMC Cancer 2019, 19, 250. [Google Scholar] [CrossRef]
- Ladenheim, M.R.; Kim, N.G.; Nguyen, P.; Le, A.; Stefanick, M.L.; Garcia, G.; Nguyen, M.H. Sex differences in disease presentation, treatment and clinical outcomes of patients with hepatocellular carcinoma: A single-centre cohort study. BMJ Open Gastroenterol. 2016, 3, e000107. [Google Scholar] [CrossRef]
- Rich, N.E.; Hester, C.; Odewole, M.; Murphy, C.C.; Parikh, N.D.; Marrero, J.A.; Yopp, A.C.; Singal, A.G. Racial and Ethnic Differences in Presentation and Outcomes of Hepatocellular Carcinoma. Clin. Gastroenterol. Hepatol. 2019, 17, 551–559.e1. [Google Scholar] [CrossRef] [Green Version]
- Nevola, R.; Rinaldi, L.; Giordano, M.; Marrone, A.; Adinolfi, L.E. Mechanisms and clinical behavior of hepatocellular carcinoma in HBV and HCV infection and alcoholic and non-alcoholic fatty liver disease. Hepatoma. Res. 2018, 4, 55. [Google Scholar] [CrossRef]
- Liu, S.Y.; Tsai, I.T.; Hsu, Y.C. Alcohol-Related Liver Disease: Basic Mechanisms and Clinical Perspectives. Int. J. Mol. Sci. 2021, 22, 5170. [Google Scholar] [CrossRef]
- Huang, D.Q.; Mathurin, P.; Cortez-Pinto, H.; Loomba, R. Global epidemiology of alcohol-associated cirrhosis and HCC: Trends, projections and risk factors. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 37–49. [Google Scholar] [CrossRef]
- White, A.M. Gender Differences in the Epidemiology of Alcohol Use and Related Harms in the United States. Alcohol. Res. 2020, 40, 1. [Google Scholar] [CrossRef] [PubMed]
- Baraona, E.; Abittan, C.S.; Dohmen, K.; Moretti, M.; Pozzato, G.; Chayes, Z.W.; Schaefer, C.; Lieber, C.S. Gender differences in pharmacokinetics of alcohol. Alcohol. Clin. Exp. Res. 2001, 25, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Jepsen, P.; Ott, P.; Andersen, P.K.; Sørensen, H.T.; Vilstrup, H. Risk for hepatocellular carcinoma in patients with alcoholic cirrhosis: A Danish nationwide cohort study. Ann. Intern. Med. 2012, 156, 841–847. [Google Scholar] [CrossRef]
- Ganne-Carrié, N.; Chaffaut, C.; Bourcier, V.; Archambeaud, I.; Perarnau, J.M.; Oberti, F.; Roulot, D.; Moreno, C.; Louvet, A.; Dao, T.; et al. Estimate of hepatocellular carcinoma incidence in patients with alcoholic cirrhosis. J. Hepatol. 2018, 69, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riazi, K.; Azhari, H.; Charette, J.H.; Underwood, F.E.; King, J.A.; Afshar, E.E.; Swain, M.G.; Congly, S.E.; Kaplan, G.G.; Shaheen, A.A. The prevalence and incidence of NAFLD worldwide: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2022, 7, 851–861. [Google Scholar] [CrossRef]
- Acierno, C.; Caturano, A.; Pafundi, P.C.; Nevola, R.; Adinolfi, L.E.; Sasso, F.C. Nonalcoholic fatty liver disease and type 2 diabetes: Pathophysiological mechanisms shared between the two faces of the same coin. Explor. Med. 2020, 1, 287–306. [Google Scholar] [CrossRef]
- Vetrano, E.; Rinaldi, L.; Mormone, A.; Giorgione, C.; Galiero, R.; Caturano, A.; Nevola, R.; Marfella, R.; Sasso, F.C. Non-alcoholic Fatty Liver Disease (NAFLD), Type 2 Diabetes, and Non-viral Hepatocarcinoma: Pathophysiological Mechanisms and New Therapeutic Strategies. Biomedicines 2023, 11, 468. [Google Scholar] [CrossRef]
- Gupta, A.; Das, A.; Majumder, K.; Arora, N.; Mayo, H.G.; Singh, P.P.; Beg, M.S.; Singh, S. Obesity is Independently Associated With Increased Risk of Hepatocellular Cancer-related Mortality: A Systematic Review and Meta-Analysis. Am. J. Clin. Oncol. 2018, 41, 874–881. [Google Scholar] [CrossRef]
- Raff, E.J.; Kakati, D.; Bloomer, J.R.; Shoreibah, M.; Rasheed, K.; Singal, A.K. Diabetes Mellitus Predicts Occurrence of Cirrhosis and Hepatocellular Cancer in Alcoholic Liver and Non-alcoholic Fatty Liver Diseases. J. Clin. Transl. Hepatol. 2015, 3, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Sung, H.; Siegel, R.L.; Torre, L.A.; Pearson-Stuttard, J.; Islami, F.; Fedewa, S.A.; Goding Sauer, A.; Shuval, K.; Gapstur, S.M.; Jacobs, E.J.; et al. Global patterns in excess body weight and the associated cancer burden. CA Cancer J. Clin. 2019, 69, 88–112. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, B.D.; Goncalves, M.D.; Cantley, L.C. Obesity and Cancer Mechanisms: Cancer Metabolism. J. Clin. Oncol. 2016, 34, 4277–4283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutari, C.; Mantzoros, C.S. A 2022 update on the epidemiology of obesity and a call to action: As its twin COVID-19 pandemic appears to be receding, the obesity and dysmetabolism pandemic continues to rage on. Metabolism 2022, 133, 155217. [Google Scholar] [CrossRef] [PubMed]
- Wild, S.; Roglic, G.; Green, A.; Sicree, R.; King, H. Global prevalence of diabetes: Estimates for the year 2000 and projections for 2030. Diabetes Care 2004, 27, 1047–1053. [Google Scholar] [CrossRef] [Green Version]
- Menke, A.; Casagrande, S.; Geiss, L.; Cowie, C.C. Prevalence of and Trends in Diabetes among Adults in the United States, 1988-2012. JAMA 2015, 314, 1021–1029. [Google Scholar] [CrossRef] [Green Version]
- Shepard, B.D. Sex differences in diabetes and kidney disease: Mechanisms and consequences. Am. J. Physiol. Renal. Physiol. 2019, 317, F456–F462. [Google Scholar] [CrossRef]
- Gambineri, A.; Pelusi, C. Sex hormones, obesity and type 2 diabetes: Is there a link? Endocr. Connect. 2019, 8, R1–R9. [Google Scholar] [CrossRef]
- Nevola, R.; Messina, V.; Marrone, A.; Coppola, N.; Rescigno, C.; Esposito, V.; Sangiovanni, V.; Claar, E.; Pisaturo, M.; Fusco, F.M.; et al. Epidemiology of HCV and HBV in a High Endemic Area of Southern Italy: Opportunities from the COVID-19 Pandemic-Standardized National Screening or One Tailored to Local Epidemiology? Biology 2022, 11, 609. [Google Scholar] [CrossRef]
- Nevola, R.; Beccia, D.; Rosato, V.; Ruocco, R.; Mastrocinque, D.; Villani, A.; Perillo, P.; Imbriani, S.; Delle Femine, A.; Criscuolo, L.; et al. HBV Infection and Host Interactions: The Role in Viral Persistence and Oncogenesis. Int. J. Mol. Sci. 2023, 24, 7651. [Google Scholar] [CrossRef]
- Leung, J.; Peacock, A.; Colledge, S.; Grebely, J.; Cunningham, E.B.; Hickman, M.; Vickerman, P.; Stone, J.; Trickey, A.; Dumchev, K.; et al. A Global Meta-analysis of the Prevalence of HIV, Hepatitis C Virus, and Hepatitis B Virus Among People Who Inject Drugs-Do Gender-Based Differences Vary by Country-Level Indicators? J. Infect. Dis. 2019, 220, 78–90. [Google Scholar] [CrossRef] [Green Version]
- Chidambaranathan-Reghupaty, S.; Fisher, P.B.; Sarkar, D. Hepatocellular carcinoma (HCC): Epidemiology, etiology and molecular classification. Adv. Cancer Res. 2021, 149, 1–61. [Google Scholar] [CrossRef] [PubMed]
- Nevola, R.; Rinaldi, L.; Zeni, L.; Romano, C.; Marrone, A.; Galiero, R.; Pafundi, P.C.; Acierno, C.; Vetrano, E.; Adinolfi, L.E. Changes in clinical scenarios, management, and perspectives of patients with chronic hepatitis C after viral clearance by direct-acting antivirals. Expert Rev. Gastroenterol. Hepatol. 2021, 15, 643–656. [Google Scholar] [CrossRef] [PubMed]
- Galoosian, A.; Hanlon, C.; Zhang, J.; Holt, E.W.; Yimam, K.K. Clinical Updates in Primary Biliary Cholangitis: Trends, Epidemiology, Diagnostics, and New Therapeutic Approaches. J. Clin. Transl. Hepatol. 2020, 8, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Premkumar, M.; Anand, A.C. Tobacco, Cigarettes, and the Liver: The Smoking Gun. J. Clin. Exp. Hepatol. 2021, 11, 700–712. [Google Scholar] [CrossRef]
- Lee, Y.C.; Cohet, C.; Yang, Y.C.; Stayner, L.; Hashibe, M.; Straif, K. Meta-analysis of epidemiologic studies on cigarette smoking and liver cancer. Int. J. Epidemiol. 2009, 38, 1497–1511. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Chuang, Y.H.; Wu, C.F.; Jan, M.C.; Wu, W.J.; Lin, C.L.; Liu, C.J.; Yang, Y.C.; Chen, P.J.; Lin, S.M.; et al. Smoking and Hepatitis B Virus-Related Hepatocellular Carcinoma Risk: The Mediating Roles of Viral Load and Alanine Aminotransferase. Hepatology 2019, 69, 1412–1425. [Google Scholar] [CrossRef]
- Yu, M.C.; Yuan, J.M. Environmental factors and risk for hepatocellular carcinoma. Gastroenterology 2004, 127, S72–S78. [Google Scholar] [CrossRef]
- GBD 2019 Tobacco Collaborators. Spatial, temporal, and demographic patterns in prevalence of smoking tobacco use and attributable disease burden in 204 countries and territories, 1990–2019: A systematic analysis from the Global Burden of Disease Study 2019. Lancet 2021, 397, 2337–2360, Erratum in Lancet 2021, 397, 2336. [Google Scholar] [CrossRef]
- Nagahama, Y.; Chakraborty, T.; Paul-Prasanth, B.; Ohta, K.; Nakamura, M. Sex determination, gonadal sex differentiation, and plasticity in vertebrate species. Physiol. Rev. 2021, 101, 1237–1308. [Google Scholar] [CrossRef]
- Turner, M.E.; Ely, D.; Prokop, J.; Milsted, A. Sry, more than testis determination? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R561–R571. [Google Scholar] [CrossRef] [Green Version]
- Xue, T.C.; Zhang, L.; Ren, Z.G.; Chen, R.X.; Cui, J.F.; Ge, N.L.; Ye, S.L. Sex-determination gene SRY potentially associates with poor prognosis but not sex bias in hepatocellular carcinoma. Dig. Dis. Sci. 2015, 60, 427–435. [Google Scholar] [CrossRef]
- Liu, C.; Ren, Y.F.; Dong, J.; Ke, M.Y.; Ma, F.; Monga, S.P.S.; Wu, R.; Lv, Y.; Zhang, X.F. Activation of SRY accounts for male-specific hepatocarcinogenesis: Implication in gender disparity of hepatocellular carcinoma. Cancer Lett. 2017, 410, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Tanimizu, N.; Nishikawa, Y.; Ichinohe, N.; Akiyama, H.; Mitaka, T. Sry HMG box protein 9-positive (Sox9+) epithelial cell adhesion molecule-negative (EpCAM-) biphenotypic cells derived from hepatocytes are involved in mouse liver regeneration. J. Biol. Chem. 2014, 289, 7589–7598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, T.R.; Munro, T.; Mattick, J.S. The potential of long noncoding RNA therapies. Trends Pharmacol. Sci. 2022, 43, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Furlan, G.; Gutierrez Hernandez, N.; Huret, C.; Galupa, R.; van Bemmel, J.G.; Romito, A.; Heard, E.; Morey, C.; Rougeulle, C. The Ftx Noncoding Locus Controls X Chromosome Inactivation Independently of Its RNA Products. Mol. Cell. 2018, 70, 462–472.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Qu, T.; Li, Y.; Ma, J.; Yu, H. Biological role of long non-coding RNA FTX in cancer progression. Biomed. Pharmacother. 2022, 153, 113446. [Google Scholar] [CrossRef]
- Liu, F.; Yuan, J.H.; Huang, J.F.; Yang, F.; Wang, T.T.; Ma, J.Z.; Zhang, L.; Zhou, C.C.; Wang, F.; Yu, J.; et al. Long noncoding RNA FTX inhibits hepatocellular carcinoma proliferation and metastasis by binding MCM2 and miR-374a. Oncogene 2016, 35, 5422–5434. [Google Scholar] [CrossRef]
- Wu, H.; Zhong, Z.; Wang, A.; Yuan, C.; Ning, K.; Hu, H.; Wang, C.; Yin, X. LncRNA FTX represses the progression of non-alcoholic fatty liver disease to hepatocellular carcinoma via regulating the M1/M2 polarization of Kupffer cells. Cancer Cell. Int. 2020, 20, 266. [Google Scholar] [CrossRef]
- Porter, L.E.; Elm, M.S.; Van Thiel, D.H.; Eagon, P.K. Hepatic estrogen receptor in human liver disease. Gastroenterology 1987, 92, 735–745. [Google Scholar] [CrossRef]
- Kur, P.; Kolasa-Wołosiuk, A.; Misiakiewicz-Has, K.; Wiszniewska, B. Sex Hormone-Dependent Physiology and Diseases of Liver. Int. J. Environ. Res. Public Health 2020, 17, 2620. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Shen, Y.; Li, R. Estrogen synthesis and signaling pathways during aging: From periphery to brain. Trends Mol. Med. 2013, 19, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holinka, C.F.; Diczfalusy, E.; Coelingh Bennink, H.J. Estetrol: A unique steroid in human pregnancy. J. Steroid. Biochem. Mol. Biol. 2008, 110, 138–143. [Google Scholar] [CrossRef]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar] [CrossRef]
- Xu, S.; Yu, S.; Dong, D.; Lee, L.T.O. G Protein-Coupled Estrogen Receptor: A Potential Therapeutic Target in Cancer. Front. Endocrinol. 2019, 10, 725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinge, C.M. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001, 29, 2905–2919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrtačnik, P.; Ostanek, B.; Mencej-Bedrač, S.; Marc, J. The many faces of estrogen signaling. Biochem. Med. 2014, 24, 329–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björnström, L.; Sjöberg, M. Mechanisms of estrogen receptor signaling: Convergence of genomic and nongenomic actions on target genes. Mol. Endocrinol. 2005, 19, 833–842. [Google Scholar] [CrossRef] [Green Version]
- Iorga, A.; Cunningham, C.M.; Moazeni, S.; Ruffenach, G.; Umar, S.; Eghbali, M. The protective role of estrogen and estrogen receptors in cardiovascular disease and the controversial use of estrogen therapy. Biol. Sex Differ. 2017, 8, 33. [Google Scholar] [CrossRef] [Green Version]
- Bustamante-Barrientos, F.A.; Méndez-Ruette, M.; Ortloff, A.; Luz-Crawford, P.; Rivera, F.J.; Figueroa, C.D.; Molina, L.; Bátiz, L.F. The Impact of Estrogen and Estrogen-Like Molecules in Neurogenesis and Neurodegeneration: Beneficial or Harmful? Front. Cell. Neurosci. 2021, 15, 636176. [Google Scholar] [CrossRef]
- Knowlton, A.A.; Lee, A.R. Estrogen and the cardiovascular system. Pharmacol. Ther. 2012, 135, 54–70. [Google Scholar] [CrossRef] [Green Version]
- Wehbe, Z.; Nasser, S.A.; El-Yazbi, A.; Nasreddine, S.; Eid, A.H. Estrogen and Bisphenol A in Hypertension. Curr. Hypertens. Rep. 2020, 22, 23. [Google Scholar] [CrossRef] [PubMed]
- Mahboobifard, F.; Pourgholami, M.H.; Jorjani, M.; Dargahi, L.; Amiri, M.; Sadeghi, S.; Tehrani, F.R. Estrogen as a key regulator of energy homeostasis and metabolic health. Biomed. Pharmacother. 2022, 156, 113808. [Google Scholar] [CrossRef]
- Fischer, V.; Haffner-Luntzer, M. Interaction between bone and immune cells: Implications for postmenopausal osteoporosis. Semin. Cell. Dev. Biol. 2022, 123, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Bagit, A.; Hayward, G.C.; MacPherson, R.E.K. Exercise and estrogen: Common pathways in Alzheimer’s disease pathology. Am. J. Physiol. Endocrinol. Metab. 2021, 321, E164–E168. [Google Scholar] [CrossRef]
- Hwang, W.J.; Lee, T.Y.; Kim, N.S.; Kwon, J.S. The Role of Estrogen Receptors and Their Signaling across Psychiatric Disorders. Int. J. Mol. Sci. 2020, 22, 373. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Shang, Y. Estrogen and cancer. Annu. Rev. Physiol. 2013, 75, 225–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggieri, A.; Barbati, C.; Malorni, W. Cellular and molecular mechanisms involved in hepatocellular carcinoma gender disparity. Int. J. Cancer 2010, 127, 499–504. [Google Scholar] [CrossRef]
- Tautz, L.; Critton, D.A.; Grotegut, S. Protein tyrosine phosphatases: Structure, function, and implication in human disease. Methods Mol. Biol. 2013, 1053, 179–221. [Google Scholar] [CrossRef]
- Hou, J.; Xu, J.; Jiang, R.; Wang, Y.; Chen, C.; Deng, L.; Huang, X.; Wang, X.; Sun, B. Estrogen-sensitive PTPRO expression represses hepatocellular carcinoma progression by control of STAT3. Hepatology 2013, 57, 678–688. [Google Scholar] [CrossRef]
- Ming, F.; Sun, Q. Epigenetically silenced PTPRO functions as a prognostic marker and tumor suppressor in human lung squamous cell carcinoma. Mol. Med. Rep. 2017, 16, 746–754. [Google Scholar] [CrossRef] [Green Version]
- Gan, J.; Zhang, H. PTPRO predicts patient prognosis and correlates with immune infiltrates in human clear cell renal cell carcinoma. Transl. Cancer Res. 2020, 9, 4800–4810. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Xiang, W.; Han, L.; Yuan, Z.; Wang, R.; Ma, Y.; Yang, Y.; Cai, S.; Xu, Y.; Mo, S.; et al. PTPRO represses colorectal cancer tumorigenesis and progression by reprogramming fatty acid metabolism. Cancer Commun. 2022, 42, 848–867. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, Y.; Yan, Z.; Yang, H.; Sun, W.; Yao, Y.; Chen, Y.; Jiang, R. IL-6 promotes PD-L1 expression in monocytes and macrophages by decreasing protein tyrosine phosphatase receptor type O expression in human hepatocellular carcinoma. J. Immunother. Cancer 2020, 8, e000285. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Cheung, S.T. STAT3: An Emerging Therapeutic Target for Hepatocellular Carcinoma. Cancers 2019, 11, 1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castaneda, M.; Hollander, P.D.; Mani, S.A. Forkhead Box Transcription Factors: Double-Edged Swords in Cancer. Cancer Res. 2022, 82, 2057–2065. [Google Scholar] [CrossRef]
- Gong, Z.; Yu, J.; Yang, S.; Lai, P.B.S.; Chen, G.G. FOX transcription factor family in hepatocellular carcinoma. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188376. [Google Scholar] [CrossRef]
- Golson, M.L.; Kaestner, K.H. Fox transcription factors: From development to disease. Development 2016, 143, 4558–4570. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Huang, W.; He, Q.; Li, D.; Wang, Z.; Feng, Y.; Liu, D.; Zhang, T.; Wang, Y.; Xie, M.; et al. FOXC1 promotes HCC proliferation and metastasis by Upregulating DNMT3B to induce DNA Hypermethylation of CTH promoter. J. Exp. Clin. Cancer Res. 2021, 40, 50. [Google Scholar] [CrossRef]
- Lee, C.S.; Friedman, J.R.; Fulmer, J.T.; Kaestner, K.H. The initiation of liver development is dependent on Foxa transcription factors. Nature 2005, 435, 944–947. [Google Scholar] [CrossRef]
- Li, Z.; Tuteja, G.; Schug, J.; Kaestner, K.H. Foxa1 and Foxa2 are essential for sexual dimorphism in liver cancer. Cell 2012, 148, 72–83. [Google Scholar] [CrossRef] [Green Version]
- Wei, T.; Chen, W.; Wen, L.; Zhang, J.; Zhang, Q.; Yang, J.; Liu, H.; Chen, B.W.; Zhou, Y.; Feng, X.; et al. G protein-coupled estrogen receptor deficiency accelerates liver tumorigenesis by enhancing inflammation and fibrosis. Cancer Lett. 2016, 382, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.A.; Xiong, J.; Fu, Q.; Dong, Y.; Liu, M.; Peng, M.; Jin, W.; Zhou, L.; Xu, X.; Huang, X.; et al. GPER-Induced ERK Signaling Decreases Cell Viability of Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 638171. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Cai, J.; Huang, Y.; Zhu, X.; Gong, B.; Yang, Z.; Yan, C.; Hu, Z.; Yang, L.; Wang, Z. G-Protein-Coupled Estrogen Receptor 1 Promotes Gender Disparities in Hepatocellular Carcinoma via Modulation of SIN1 and mTOR Complex 2 Activity. Mol. Cancer Res. 2020, 18, 1863–1875. [Google Scholar] [CrossRef]
- Yang, Y.M.; Kim, S.Y.; Seki, E. Inflammation and Liver Cancer: Molecular Mechanisms and Therapeutic Targets. Semin. Liver Dis. 2019, 39, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Parlar, Y.E.; Ayar, S.N.; Cagdas, D.; Balaban, Y.H. Liver immunity, autoimmunity, and inborn errors of immunity. World J. Hepatol. 2023, 15, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Refolo, M.G.; Messa, C.; Guerra, V.; Carr, B.I.; D’Alessandro, R. Inflammatory Mechanisms of HCC Development. Cancers 2020, 12, 641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, T. IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 2021, 33, 127–148. [Google Scholar] [CrossRef]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Lin, H.; Wu, G.; Zhu, M.; Li, M. IL-6/STAT3 Is a Promising Therapeutic Target for Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 760971. [Google Scholar] [CrossRef]
- Soresi, M.; Giannitrapani, L.; D’Antona, F.; Florena, A.M.; La Spada, E.; Terranova, A.; Cervello, M.; D’Alessandro, N.; Montalto, G. Interleukin-6 and its soluble receptor in patients with liver cirrhosis and hepatocellular carcinoma. World J. Gastroenterol. 2006, 12, 2563–2568. [Google Scholar] [CrossRef]
- Nakagawa, H.; Maeda, S.; Yoshida, H.; Tateishi, R.; Masuzaki, R.; Ohki, T.; Hayakawa, Y.; Kinoshita, H.; Yamakado, M.; Kato, N.; et al. Serum IL-6 levels and the risk for hepatocarcinogenesis in chronic hepatitis C patients: An analysis based on gender differences. Int. J. Cancer 2009, 125, 2264–2269. [Google Scholar] [CrossRef] [PubMed]
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007, 317, 121–124, Erratum in Science 2009, 326, 1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falleti, E.; Fabris, C.; Toniutto, P.; Fontanini, E.; Cussigh, A.; Bitetto, D.; Fumolo, E.; Fornasiere, E.; Bragagnini, W.; Pinato, D.J.; et al. Interleukin-6 polymorphisms and gender: Relationship with the occurrence of hepatocellular carcinoma in patients with end-stage liver disease. Oncology 2009, 77, 304–313. [Google Scholar] [CrossRef]
- Stein, B.; Yang, M.X. Repression of the interleukin-6 promoter by estrogen receptor is mediated by NF-kappa B and C/EBP beta. Mol. Cell. Biol. 1995, 15, 4971–4979. [Google Scholar] [CrossRef] [Green Version]
- Galdiero, M.R.; Garlanda, C.; Jaillon, S.; Marone, G.; Mantovani, A. Tumor associated macrophages and neutrophils in tumor progression. J. Cell. Physiol. 2013, 228, 1404–1412. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Lu, Y.; Xu, Y.; Xu, L.; Zheng, W.; Wu, Y.; Li, L.; Shen, P. Estrogen represses hepatocellular carcinoma (HCC) growth via inhibiting alternative activation of tumor-associated macrophages (TAMs). J. Biol. Chem. 2012, 287, 40140–40149. [Google Scholar] [CrossRef] [Green Version]
- Sharma, B.R.; Kanneganti, T.D. NLRP3 inflammasome in cancer and metabolic diseases. Nat. Immunol. 2021, 22, 550–559. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Wei, Q.; Mu, K.; Li, T.; Zhang, Y.; Yang, Z.; Jia, X.; Zhao, W.; Huai, W.; Guo, P.; Han, L. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab. Investig. 2014, 94, 52–62. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.; Guo, P.; Mu, K.; Zhang, Y.; Zhao, W.; Huai, W.; Qiu, Y.; Li, T.; Ma, X.; Liu, Y.; et al. Estrogen suppresses hepatocellular carcinoma cells through ERβ-mediated upregulation of the NLRP3 inflammasome. Lab. Investig. 2015, 95, 804–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naamneh Elzenaty, R.; du Toit, T.; Flück, C.E. Basics of androgen synthesis and action. Best Pract. Res. Clin. Endocrinol. Metab. 2022, 36, 101665. [Google Scholar] [CrossRef] [PubMed]
- Fanelli, F.; Baronio, F.; Ortolano, R.; Mezzullo, M.; Cassio, A.; Pagotto, U.; Balsamo, A. Normative Basal Values of Hormones and Proteins of Gonadal and Adrenal Functions from Birth to Adulthood. Sex. Dev. 2018, 12, 50–94. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, E.J.; Xing, E.; Campbell, M.J.; Li, P.K.; Blachly, J.S.; Tsung, A.; Coss, C.C. Constitutively Active Androgen Receptor in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2022, 23, 13768. [Google Scholar] [CrossRef]
- Ma, W.L.; Hsu, C.L.; Wu, M.H.; Wu, C.T.; Wu, C.C.; Lai, J.J.; Jou, Y.S.; Chen, C.W.; Yeh, S.; Chang, C. Androgen receptor is a new potential therapeutic target for the treatment of hepatocellular carcinoma. Gastroenterology 2008, 135, 947–955.e5, Erratum in Gastroenterology 2008, 135, 1805. Ma, Cheng-Lung [corrected to Ma, Wen-Lung]. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Li, X.X.; Yang, Y.; Zhang, Y.; Wang, H.Y.; Zheng, X.F.S. Significance and mechanism of androgen receptor overexpression and androgen receptor/mechanistic target of rapamycin cross-talk in hepatocellular carcinoma. Hepatology 2018, 67, 2271–2286. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Cheng, A.S.; Tsang, D.P.; Li, M.S.; Go, M.Y.; Cheung, Y.S.; Zhao, G.J.; Ng, S.S.; Lin, M.C.; Yu, J.; et al. Cell cycle-related kinase is a direct androgen receptor-regulated gene that drives β-catenin/T cell factor-dependent hepatocarcinogenesis. J. Clin. Investig. 2011, 121, 3159–3175. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.H.; Chen, P.J.; Yeh, S.H. Gender disparity in chronic hepatitis B: Mechanisms of sex hormones. J. Gastroenterol. Hepatol. 2015, 30, 1237–1245. [Google Scholar] [CrossRef] [Green Version]
- Di Maio, M.; Daniele, B.; Pignata, S.; Gallo, C.; De Maio, E.; Morabito, A.; Piccirillo, M.C.; Perrone, F. Is human hepatocellular carcinoma a hormone-responsive tumor? World J. Gastroenterol. 2008, 14, 1682–1689. [Google Scholar] [CrossRef] [Green Version]
- Nevola, R.; Delle Femine, A.; Rosato, V.; Kondili, L.A.; Alfano, M.; Mastrocinque, D.; Imbriani, S.; Perillo, P.; Beccia, D.; Villani, A.; et al. Neoadjuvant and Adjuvant Systemic Therapies in Loco-Regional Treatments for Hepatocellular Carcinoma: Are We at the Dawn of a New Era? Cancers 2023, 15, 2950. [Google Scholar] [CrossRef]
- Lee, C.M.; Lu, S.N.; Changchien, C.S.; Yeh, C.T.; Hsu, T.T.; Tang, J.H.; Wang, J.H.; Lin, D.Y.; Chen, C.L.; Chen, W.J. Age, gender, and local geographic variations of viral etiology of hepatocellular carcinoma in a hyperendemic area for hepatitis B virus infection. Cancer 1999, 86, 1143–1150. [Google Scholar] [CrossRef]
- Wang, S.H.; Yeh, S.H.; Lin, W.H.; Wang, H.Y.; Chen, D.S.; Chen, P.J. Identification of androgen response elements in the enhancer I of hepatitis B virus: A mechanism for sex disparity in chronic hepatitis B. Hepatology 2009, 50, 1392–1402. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.J.; Chang, C.J.; Yeh, S.H.; Lin, W.H.; Wang, S.H.; Tsai, T.F.; Chen, D.S.; Chen, P.J. Hepatitis B virus X protein enhances the transcriptional activity of the androgen receptor through c-Src and glycogen synthase kinase-3beta kinase pathways. Hepatology 2009, 49, 1515–1524. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Zhu, Y.J.; Wang, H.Y.; Chen, L. Gender disparity in hepatocellular carcinoma (HCC): Multiple underlying mechanisms. Sci. China Life Sci. 2017, 60, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Yeh, S.H.; Lin, W.H.; Yeh, K.H.; Yuan, Q.; Xia, N.S.; Chen, D.S.; Chen, P.J. Estrogen receptor α represses transcription of HBV genes via interaction with hepatocyte nuclear factor 4α. Gastroenterology 2012, 142, 989–998.e4. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.J.; Lai, K.P.; Zeng, W.; Chuang, K.H.; Altuwaijri, S.; Chang, C. Androgen receptor influences on body defense system via modulation of innate and adaptive immune systems: Lessons from conditional AR knockout mice. Am. J. Pathol. 2012, 181, 1504–1512. [Google Scholar] [CrossRef] [Green Version]
- Poynard, T.; Ratziu, V.; Charlotte, F.; Goodman, Z.; McHutchison, J.; Albrecht, J. Rates and risk factors of liver fibrosis progression in patients with chronic hepatitis c. J. Hepatol. 2001, 34, 730–739. [Google Scholar] [CrossRef]
- Kanda, T.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus core protein augments androgen receptor-mediated signaling. J. Virol. 2008, 82, 11066–11072. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Du, Q.; Cao, Z.; Guo, Z.; Evankovich, J.; Yan, W.; Chang, Y.; Shao, L.; Stolz, D.B.; Tsung, A.; et al. Interferon-γ induces autophagy with growth inhibition and cell death in human hepatocellular carcinoma (HCC) cells through interferon-regulatory factor-1 (IRF-1). Cancer Lett. 2012, 314, 213–222. [Google Scholar] [CrossRef] [Green Version]
- Ulitzky, L.; Lafer, M.M.; KuKuruga, M.A.; Silberstein, E.; Cehan, N.; Taylor, D.R. A New Signaling Pathway for HCV Inhibition by Estrogen: GPR30 Activation Leads to Cleavage of Occludin by MMP-9. PLoS ONE 2016, 11, e0145212. [Google Scholar] [CrossRef] [Green Version]
- Marengo, A.; Rosso, C.; Bugianesi, E. Liver Cancer: Connections with Obesity, Fatty Liver, and Cirrhosis. Annu. Rev. Med. 2016, 67, 103–117. [Google Scholar] [CrossRef]
- Al-Hussaniy, H.A.; Alburghaif, A.H.; Naji, M.A. Leptin hormone and its effectiveness in reproduction, metabolism, immunity, diabetes, hopes and ambitions. J. Med. Life 2021, 14, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Zavos, C.; Deretzi, G. The potential adverse role of leptin resistance in nonalcoholic fatty liver disease: A hypothesis based on critical review of the literature. J. Clin. Gastroenterol. 2011, 45, 50–54. [Google Scholar] [CrossRef]
- Rotundo, L.; Persaud, A.; Feurdean, M.; Ahlawat, S.; Kim, H.S. The Association of leptin with severity of non-alcoholic fatty liver disease: A population-based study. Clin. Mol. Hepatol. 2018, 24, 392–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, N.K.; Sharma, D.; Ding, X.; Lin, S.; Marra, F.; Merlin, D.; Anania, F.A. Concomitant activation of the JAK/STAT, PI3K/AKT, and ERK signaling is involved in leptin-mediated promotion of invasion and migration of hepatocellular carcinoma cells. Cancer Res. 2007, 67, 2497–2507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.; Shi, H. Estradiol and Estrogen Receptor Agonists Oppose Oncogenic Actions of Leptin in HepG2 Cells. PLoS ONE 2016, 11, e0151455. [Google Scholar] [CrossRef]
- Choi, H.M.; Doss, H.M.; Kim, K.S. Multifaceted Physiological Roles of Adiponectin in Inflammation and Diseases. Int. J. Mol. Sci. 2020, 21, 1219. [Google Scholar] [CrossRef] [Green Version]
- Xu, A.; Wang, Y.; Keshaw, H.; Xu, L.Y.; Lam, K.S.; Cooper, G.J. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J. Clin. Investig. 2003, 112, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Manieri, E.; Herrera-Melle, L.; Mora, A.; Tomás-Loba, A.; Leiva-Vega, L.; Fernández, D.I.; Rodríguez, E.; Morán, L.; Hernández-Cosido, L.; Torres, J.L.; et al. Adiponectin accounts for gender differences in hepatocellular carcinoma incidence. J. Exp. Med. 2019, 216, 1108–1119. [Google Scholar] [CrossRef]
- Nishizawa, H.; Shimomura, I.; Kishida, K.; Maeda, N.; Kuriyama, H.; Nagaretani, H.; Matsuda, M.; Kondo, H.; Furuyama, N.; Kihara, S.; et al. Androgens decrease plasma adiponectin, an insulin-sensitizing adipocyte-derived protein. Diabetes 2002, 51, 2734–2741. [Google Scholar] [CrossRef] [Green Version]
- Diener, C.; Keller, A.; Meese, E. Emerging concepts of miRNA therapeutics: From cells to clinic. Trends Genet. 2022, 38, 613–626. [Google Scholar] [CrossRef]
- Yamada, H.; Suzuki, K.; Ichino, N.; Ando, Y.; Sawada, A.; Osakabe, K.; Sugimoto, K.; Ohashi, K.; Teradaira, R.; Inoue, T.; et al. Associations between circulating microRNAs (miR-21, miR-34a, miR-122 and miR-451) and non-alcoholic fatty liver. Clin. Chim. Acta 2013, 424, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Ayub, H.; Khan, T.; Wahid, F. MicroRNA biogenesis, gene silencing mechanisms and role in breast, ovarian and prostate cancer. Biochimie 2019, 167, 12–24. [Google Scholar] [CrossRef]
- Morishita, A.; Oura, K.; Tadokoro, T.; Fujita, K.; Tani, J.; Masaki, T. MicroRNAs in the Pathogenesis of Hepatocellular Carcinoma: A Review. Cancers 2021, 13, 514. [Google Scholar] [CrossRef] [PubMed]
- Morishita, A.; Iwama, H.; Fujihara, S.; Sakamoto, T.; Fujita, K.; Tani, J.; Miyoshi, H.; Yoneyama, H.; Himoto, T.; Masaki, T. MicroRNA profiles in various hepatocellular carcinoma cell lines. Oncol. Lett. 2016, 12, 1687–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.J.; Yeh, S.H.; Liu, W.H.; Lin, C.C.; Huang, H.C.; Chen, C.L.; Chen, D.S.; Chen, P.J. Androgen pathway stimulates microRNA-216a transcription to suppress the tumor suppressor in lung cancer-1 gene in early hepatocarcinogenesis. Hepatology 2012, 56, 632–643. [Google Scholar] [CrossRef]
- Liu, W.H.; Yeh, S.H.; Lu, C.C.; Yu, S.L.; Chen, H.Y.; Lin, C.Y.; Chen, D.S.; Chen, P.J. MicroRNA-18a prevents estrogen receptor-alpha expression, promoting proliferation of hepatocellular carcinoma cells. Gastroenterology 2009, 136, 683–693. [Google Scholar] [CrossRef]
- Pandey, D.P.; Picard, D. miR-22 inhibits estrogen signaling by directly targeting the estrogen receptor alpha mRNA. Mol. Cell. Biol. 2009, 29, 3783–3790, Erratum in: Mol Cell Biol 2009, 29, 4873. [Google Scholar] [CrossRef] [Green Version]
- Jiang, R.; Deng, L.; Zhao, L.; Li, X.; Zhang, F.; Xia, Y.; Gao, Y.; Wang, X.; Sun, B. miR-22 promotes HBV-related hepatocellular carcinoma development in males. Clin. Cancer Res. 2011, 17, 5593–5603. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, T.; He, G.; Matsuzawa, A.; Yu, G.Y.; Maeda, S.; Hardiman, G.; Karin, M. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell 2008, 14, 156–165. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Li, T.; Qi, J.; Liu, J.; Qin, C. The miR-545/374a cluster encoded in the Ftx lncRNA is overexpressed in HBV-related hepatocellular carcinoma and promotes tumorigenesis and tumor progression. PLoS ONE 2014, 9, e109782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez Cerezo, F.J.; Tomás, A.; Donoso, L.; Enríquez, J.; Guarner, C.; Balanzó, J.; Martínez Nogueras, A.; Vilardell, F. Controlled trial of tamoxifen in patients with advanced hepatocellular carcinoma. J. Hepatol. 1994, 20, 702–706. [Google Scholar] [CrossRef]
- Farinati, F.; Salvagnini, M.; de Maria, N.; Fornasiero, A.; Chiaramonte, M.; Rossaro, L.; Naccarato, R. Unresectable hepatocellular carcinoma: A prospective controlled trial with tamoxifen. J. Hepatol. 1990, 11, 297–301. [Google Scholar] [CrossRef]
- Chow, P.K.; Tai, B.C.; Tan, C.K.; Machin, D.; Win, K.M.; Johnson, P.J.; Soo, K.C.; Asian-Pacific Hepatocellular Carcinoma Trials Group. High-dose tamoxifen in the treatment of inoperable hepatocellular carcinoma: A multicenter randomized controlled trial. Hepatology 2002, 36, 1221–1226. [Google Scholar] [CrossRef] [PubMed]
- Gallo, C.; De Maio, E.; Di Maio, M.; Signoriello, G.; Daniele, B.; Pignata, S.; Annunziata, A.; Perrone, F.; CLIP (Cancer of the Liver Italian Programme) Investigators. Tamoxifen is not effective in good prognosis patients with hepatocellular carcinoma. BMC Cancer 2006, 6, 196. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Yao, S.; Zhang, S.; Wang, J.R.; Guo, P.D.; Li, X.M.; Gan, W.J.; Mei, L.; Gao, T.M.; Li, J.M. Elevated expression of Erbin destabilizes ERα protein and promotes tumorigenesis in hepatocellular carcinoma. J. Hepatol. 2017, 66, 1193–1204. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Thandra, K.C.; Vellipuram, A.; Ali, C.D.M. Efficacy and safety of megestrol in the management of hepatocellular carcinoma: A systematic review of the literature. Contemp. Oncol. 2018, 22, 209–214. [Google Scholar] [CrossRef] [PubMed]
- McGlynn, K.A.; Hagberg, K.; Chen, J.; Braunlin, M.; Graubard, B.I.; Suneja, N.; Jick, S.; Sahasrabuddhe, V.V. Menopausal hormone therapy use and risk of primary liver cancer in the clinical practice research datalink. Int. J. Cancer 2016, 138, 2146–2153. [Google Scholar] [CrossRef] [Green Version]
- Tempfer, C.B.; Hilal, Z.; Kern, P.; Juhasz-Boess, I.; Rezniczek, G.A. Menopausal Hormone Therapy and Risk of Endometrial Cancer: A Systematic Review. Cancers 2020, 12, 2195. [Google Scholar] [CrossRef]
- Chao, Y.; Chan, W.K.; Huang, Y.S.; Teng, H.C.; Wang, S.S.; Lui, W.Y.; Whang-Peng, J.; Lee, S.D. Phase II study of flutamide in the treatment of hepatocellular carcinoma. Cancer 1996, 77, 635–639. [Google Scholar] [CrossRef]
- Forbes, A.; Wilkinson, M.L.; Iqbal, M.J.; Johnson, P.J.; Williams, R. Response to cyproterone acetate treatment in primary hepatocellular carcinoma is related to fall in free 5 alpha-dihydrotestosterone. Eur. J. Cancer Clin. Oncol. 1987, 23, 1659–1664. [Google Scholar] [CrossRef]
- Gupta, S.; Korula, J. Failure of ketoconazole as anti-androgen therapy in nonresectable primary hepatocellular carcinoma. J. Clin. Gastroenterol. 1988, 10, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Guéchot, J.; Peigney, N.; Ballet, F.; Vaubourdolle, M.; Giboudeau, J.; Poupon, R. Effect of D-tryptophan-6-luteinizing hormone-releasing hormone on the tumoral growth and plasma sex steroid levels in cirrhotic patients with hepatocellular carcinoma. Hepatology 1989, 10, 346–348. [Google Scholar] [CrossRef] [PubMed]
- Dauki, A.M.; Blachly, J.S.; Kautto, E.A.; Ezzat, S.; Abdel-Rahman, M.H.; Coss, C.C. Transcriptionally Active Androgen Receptor Splice Variants Promote Hepatocellular Carcinoma Progression. Cancer Res. 2020, 80, 561–575. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Spencer, K.; Burley, S.K.; Zheng, X.F.S. Toward improving androgen receptor-targeted therapies in male-dominant hepatocellular carcinoma. Drug Discov. Today 2021, 26, 1539–1546. [Google Scholar] [CrossRef]
- Hwang, D.J.; He, Y.; Ponnusamy, S.; Mohler, M.L.; Thiyagarajan, T.; McEwan, I.J.; Narayanan, R.; Miller, D.D. New Generation of Selective Androgen Receptor Degraders: Our Initial Design, Synthesis, and Biological Evaluation of New Compounds with Enzalutamide-Resistant Prostate Cancer Activity. J. Med. Chem. 2019, 62, 491–511. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Men | Women | Comments | ||
|---|---|---|---|---|
| Prevalence of risk factors | HCV | Lower | Higher | Wide regional variations |
| HBV | Higher | Lower | Wide regional variations | |
| Alcohol | Higher | Lower | Women show more susceptibility to alcohol-related liver injury than men | |
| Obesity | Lower | Higher | The gender gap is greatest in low-income countries. Visceral obesity is significantly more frequent in men than in women. | |
| T2DM | Higher | Lower | ||
| PBC/AIH | Lower | Higher | The overall incidence rate of PBC/AIH is low. | |
| Smoke | Higher | Lower | The gender gap is greatest in Asia and Africa. | |
| Clinical features | Occurrence | Higher | Lower | Occurrence up to 5 times higher in men, depending on region and clinical |
| Age of development | Earlier | Later | Peak incidence in men aged 50 to 69 years. In women similar incidence between 50–69 years and > 69 years. | |
| Size (diagnosis) | Larger | Smaller | Male gender is independently associated with a more advanced stage of HCC at diagnosis (size, vascular invasion, multifocality, metastasis). | |
| Encapsulation rate (diagnosis) | Lower | Higher | ||
| Multifocality rate (diagnosis) | Higher | Lower | ||
| Vascular invasion (diagnosis) | Higher | Lower | ||
| Metastasis rate (diagnosis) | Higher | Lower | ||
| Overall stage (diagnosis) | Most advanced | Earliest | ||
| Course | More aggressive | Less aggressive | ||
| Prognosis | Worst | Better | The mortality rate is 2–3 times higher in men than in women. Female gender is independently associated with lower mortality rate. | |
| Treatment response | Response to treatment | Worst | Better | Female gender is independently associated with a higher first therapy response rate. |
| Recurrence rate | Higher | Lower | Male gender is an independent risk factor for early recurrence (OR: 1.864). DFS is, on average, higher in women than in men (4.5 vs. 19.5 months). | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nevola, R.; Tortorella, G.; Rosato, V.; Rinaldi, L.; Imbriani, S.; Perillo, P.; Mastrocinque, D.; La Montagna, M.; Russo, A.; Di Lorenzo, G.; et al. Gender Differences in the Pathogenesis and Risk Factors of Hepatocellular Carcinoma. Biology 2023, 12, 984. https://doi.org/10.3390/biology12070984
Nevola R, Tortorella G, Rosato V, Rinaldi L, Imbriani S, Perillo P, Mastrocinque D, La Montagna M, Russo A, Di Lorenzo G, et al. Gender Differences in the Pathogenesis and Risk Factors of Hepatocellular Carcinoma. Biology. 2023; 12(7):984. https://doi.org/10.3390/biology12070984
Chicago/Turabian StyleNevola, Riccardo, Giovanni Tortorella, Valerio Rosato, Luca Rinaldi, Simona Imbriani, Pasquale Perillo, Davide Mastrocinque, Marco La Montagna, Antonio Russo, Giovanni Di Lorenzo, and et al. 2023. "Gender Differences in the Pathogenesis and Risk Factors of Hepatocellular Carcinoma" Biology 12, no. 7: 984. https://doi.org/10.3390/biology12070984