
Vascular Dysfunctions Contribute to the Long-Term Cognitive Deficits Following COVID-19

1
Center for Cerebrovascular Research, University of California (San Francisco), San Francisco, CA 94131, USA
2
Department of Anesthesia and Perioperative Care, University of California (San Francisco), San Francisco, CA 94131, USA
3
Department of Neurosurgery, University of California (San Francisco), San Francisco, CA 94131, USA
*
Author to whom correspondence should be addressed.
Biology 2023, 12(8), 1106; https://doi.org/10.3390/biology12081106
Submission received: 30 May 2023
/
Revised: 31 July 2023
/
Accepted: 3 August 2023
/
Published: 9 August 2023
(This article belongs to the Special Issue Post-COVID-19 Era: Infectious Diseases from One Health and Global Health Perspectives)
{kind=link}
{kind=link}
Abstract
:Simple Summary
Although SARS-CoV-2 primarily affects the upper respiratory system and the lungs, it can also affect vasculature, leading to the impairment of endothelial and multi-organ function. SARS-CoV-2 can trigger the release of pro-inflammatory cytokines, leading to disruption of tight junction proteins between endothelial cells and impairment of the blood-brain barrier (BBB). This process further allows the infiltration of immune cells and other particles into the brain, worsening brain injury. Prolonged neuro-inflammation and disruption of the BBB have been postulated as the potential primary causes of both acute and chronic cognitive dysfunction in COVID-19 patients. This review explores the effects of COVID-19 on vascular dysfunction and consequent cognitive impairment in patients.
Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a single-stranded RNA virus and a member of the corona virus family, primarily affecting the upper respiratory system and the lungs. Like many other respiratory viruses, SARS-CoV-2 can spread to other organ systems. Apart from causing diarrhea, another very common but debilitating complication caused by SARS-CoV-2 is neurological symptoms and cognitive difficulties, which occur in up to two thirds of hospitalized COVID-19 patients and range from shortness of concentration and overall declined cognitive speed to executive or memory function impairment. Neuro-cognitive dysfunction and “brain fog” are frequently present in COVID-19 cases, which can last several months after the infection, leading to disruption of daily life. Cumulative evidence suggests that SARS-CoV-2 affects vasculature in the extra-pulmonary systems directly or indirectly, leading to impairment of endothelial function and even multi-organ damage. The post COVID-19 long-lasting neurocognitive impairments have not been studied fully and their underlying mechanism remains elusive. In this review, we summarize the current understanding of the effects of COVID-19 on vascular dysfunction and how vascular dysfunction leads to cognitive impairment in patients.
1. Introduction
In late 2019, a new variant of coronaviruses, SARS-CoV-2, emerged as a cause of novel severe acute respiratory syndrome, which quickly spread around the entire world and became the newest global health concern [1,2]. SARS-CoV-2 is primarily characterized as a respiratory pathogen transmitted via respiratory droplets, although abnormalities of other organs including the brain, heart, kidneys, liver, skeletal muscle, and skin have also been noticed [3]. A meta-analysis study of 24,410 patients with COVID-19 reported fever, cough, fatigue, and hyposmia as the most common symptoms [4]. Compared to common symptoms of impaired olfaction and gustation that are present in 85% of COVID-19 patients, 36.4% of patients exhibited neurological abnormalities, such as dizziness, headache, and weakened consciousness, during hospitalization [3]. A study using murine models indicated that coronavirus could enter the CNS after infection [5], corroborating the neurological symptoms observed clinically. In addition, long-term cognitive dysfunction has been detected in patients who have recovered from COVID-19 infection and patients with persistent infection [6].
Cognition is defined as the conceptual processing of information that is mandatory for acquiring knowledge and expression of a response. This process includes obtaining information (perception), selecting necessary data (attention), and representing (understanding) and retaining information (memory), leading to behavior control (reasoning and judgment) [7]. Fatigue and lasting impairment of memory, decision making, and concentration are the most common neurological injuries reported in non-hospitalized COVID-19 patients. Additionally, numerous patients have described subtle cognitive and behavioral deficits that are difficult to characterize. These symptoms are commonly described as “brain fog” or “mental clouding” [8]. A recent guideline on long-term effects of COVID-19 published by the National Institute of Health and Care Excellence (NICE) suggests using the term “ongoing symptomatic COVID-19” for indications lasting between 4 and 12 weeks after the acute onset and “post-COVID-19-syndrome” for symptoms lasting more than 12 weeks [9]. Evidence from cohort studies implies the relationship between continuing systemic inflammation during SARS-CoV-2 infectivity and subsequent cognitive failure and hippocampal atrophy. Hence, in cognitive decline caused by COVID-19, attention should be paid to the role of inflammation. Through pro-inflammatory cytokines’ release, SARS-CoV-2 can trigger signaling pathways, finally resulting in disrupted tight junction (TJ) proteins and an impaired blood-brain barrier (BBB). This process further allows the infiltration of immune cells and other particles into the brain, worsening brain injury [10,11]. In this review, we discuss the probable role of vascular dysfunction in causing the cognitive deficiency of COVID-19 patients.
2. Long-Term Neurological and Cognitive Dysfunction Following COVID-19
During the COVID-19 pandemic, cognitive abnormalities were commonly detected. Long-term cognitive deficiency is a common manifestation of COVID-19, resulting in problems with memory, concentration, and receptive language, executive function difficulties, and fatigue. Psychiatric indications and cognitive impairment may progress and persist months after the infection. Therefore, this condition is called “Long COVID” [12]. Long-term cognitive outcomes can affect patients’ daily living functions, employment, and the ability to return to work, leading to a large disease burden [13]. Several mechanisms are suggested for long-term cognitive impairment, including disruption of the BBB, neuro-inflammation, synaptic dysfunction, disturbed neurotransmitter release, and neuronal loss. Targeting these processes may have treatment potential for long-term cognitive difficulties [14].
Interests of clinical and public health are therefore no longer restricted to the mortality rate and clinical consequences of hospitalized patients but extend to long-term adverse outcomes of COVID-19 disease, even after recovery and discharge from the hospital. There is a growing body of evidence for the persistent neurological and cognitive symptoms after COVID-19 illness. Given the persistent neurological features after SARS-CoV-2 infection, several studies have reported on the onset of neurodegenerative disease in patients without any previous history. Accordingly, 15% of COVID-19 cases showed one or more neurological signs for the first time, including 13% polyneuro/myopathy, 1% Guillain-Barré syndrome, 2% mild encephalopathy, 1% parkinsonism, and 1% ischemic stroke in the 3 months after infection [15]. In addition, a cohort study of 143 patients revealed headache, hyposmia, and myalgia at least in 5% of the population 2 months after SARS-CoV-2 infection [16].
In the context of cognitive deficits, it has been reported that 20–70% of COVID-19 cases display a quantitative or qualitative disturbance of consciousness during the acute phase [17,18]. In one study, the psychopathological and cognitive status of 226 COVID-19 pneumonia survivors were prospectively assessed one- and three-months following hospital discharge. Regardless of clinical and physical severity, 78% of the participants presented poor function in at least one cognitive domain, while 50 to 57% of the sample displayed impaired executive functions and psychomotor coordination [19]. In another case report, cognitive screens from recovered non-hospitalized COVID-19 cases showed persistent neuro-cognitive symptoms, particularly difficulties in working memory and executive function [20]. Furthermore, a follow-up study of 13,001 participants with relatively mild COVID-19 disease reported a significantly higher prevalence of memory problems eight months after the positive SARS-CoV-2 test compared to the control group in favor of the worsening of health even after recovery from infection [21]. In a separate cohort study, only long-term anosmia, ageusia, memory loss, and headache remained in COVID-19 cases after 60 days, yet memory deficit continued in these patients into the third month of disease [22]. Similarly, according to a follow-up study of 18 mild to moderate COVID-19 patients 85 days after recovery, over 75% of patients reported problems in episodic memory, attention, and concentration [23]. In addition, a comparison of cognitive function in SARS-CoV-2 patients to matched controls suggested significant differences in sustained attention [24], executive function and visuospatial processing [25], attention, memory, and language [26].
Interestingly, a case report describes progression of disease from mild common symptoms like myalgia, fatigue, smelling loss, and memory deficit at the beginning to severe outcomes including right-sided weakness, sensory loss, and worsening cognitive impairment. Neuroimaging confirmed an ischemic infarct in the middle cerebral artery of this case [1,27]. In addition, a recent cross-sectional report studied the connection between COVID-19 disease and cognitive signs in 57 hospitalized participants and found a high prevalence of cognitive impairment (>80% of the sample), primarily in the fields of attention and executive function [28].
Among cognitive performances, fatigue, memory, and concentration difficulties were reported as the most common long-lasting symptoms by 36% of symptomatic COVID-19 patients after 4 weeks and mostly observed in women [9,29,30,31].
According to another study, cases with a history of mild symptomatic SARS-CoV-2 infections have more than 18 times higher risk for cognitive deficits than individuals without clinical manifestations of the infection [32]. In addition, a recent longitudinal study showed the important role of COVID-19 severity in modulating the long-term cognitive outcomes of the disease [33]. Therefore, cognitive changes can occur even after mild COVID-19 illness. However, hospitalized and severely affected patients are at higher risk for persistent cognitive and neurological dysfunction. Moreover, amongst diverse cognitive domains, impairment of episodic and working memory, attention and concentration deficits, consciousness, and executive function difficulty are the major cognitive complications in patients suffering COVID-19 infection.
3. Molecules Contribute to COVID-19 Penetration into the CNS
Since none of the COVID-19 cases with severe neurological complaints reveal evidence of SARS-CoV-2 virus in the cerebrospinal fluid, both direct and indirect mechanisms may participate in CNS dysfunction [20]. The COVID-19 virus infects via the spike glycoprotein (S protein) of the virus binding to the angiotensin converting enzyme 2 (ACE2) receptor on the host cell surface, followed by the incorporation of the viral genome into the host cell genome. ACE2, a cardio-cerebro-vascular protective factor that converts angiotensin (Ang) I to Ang II to stimulate Ang II receptor type1 (AT1R), is expressed by neurons, astrocytes, oligodendrocytes, and endothelial cells of the CNS and is highly concentrated in the olfactory bulb, substantia nigra, middle temporal gyrus, and posterior cingulate gyrus [34]. Activated AT1R can promote inflammation and neurodegeneration and increase blood pressure [35]. Therefore, the widespread presentation of ACE2 as a SARS-CoV-2 receptor on the neural and endothelial cells makes the CNS susceptible to SARS-CoV-2 invasion [36,37]. Since direct access of the SARS-CoV-2 virus is possible through BBB-damaged sites in the brain, its interaction with ACE-2 of endothelial cells in the disrupted BBB provides another penetration route for the virus [38], while transmembrane protease serine 2 (TMPRSS2) facilitates virus entrance through aiding S protein priming. TMPRSS2 inhibitors are beneficial for mental recovery [34]. TMPRSS2 is detectable all over the olfactory epithelium and choroid plexus of rodents and humans [39,40]. In addition to ACE2, basigin or CD147 and neuropilin-1 (NRP1) also operate as docking receptors for the SARS-CoV-2 virus. Furthermore, several proteases like cathepsin B and L (CatB/L) and furin contribute to the viral entrance into cells and its replication [41]. NRP1, a transmembrane receptor without cytosolic protein kinase domain, is highly expressed in the olfactory epithelium and mediates the SARS-CoV-2 invasion into the CNS [42]. As mentioned above, an alternative receptor for the penetration of SARS-CoV-2 into the brain is CD147, which is expressed in neural cells [43]. Additionally, CatB/L, an endosomal cysteine protease, mediates the priming of the SARS-CoV-2 spike protein [44]. It has been suggested that TMPRSS2 and lysosomal cathepsins are able to trigger SARS-CoV-2 invasion; likewise, furin facilitates this process [45].
4. Endothelial Cell Infection and Vascular Dysfunction in COVID-19
The vascular endothelia cells are the innermost lining of blood vessels and fundamental for maintaining vessel structural and functional integrity by regulating vascular tone and permeabilization and responses to oxidative stress or inflammation to achieve tissue homeostasis [46].
A recent study displayed the relationship between the levels of endothelia biomarkers and the levels of pro-inflammatory cytokines and chemokines [47]. Along the same vein, recent research indicated that pro-inflammatory mediators released by spike-activated macrophages augment endothelia cells’ activation, likely contributing to the impairment of vascular integrity and development of a pro-coagulative endothelium [48].
Accumulating evidence indicates the presence of extensive microthrombi and endothelia cell damage all over the pulmonary vasculature, suggesting the involvement of vasculopathy in the pathogenesis of COVID-19. Although one study failed to find viral RNA in endothelial cells from COVID-19 patients’ autopsy tissue [49], another study detected SARS-CoV-2 RNA in endothelial cells through in situ hybridization (ISH) in 2 out of 32 cases [47,50]. It has been shown that endothelial cells and pericytes throughout the body express ACE-2 as well as BSG, CD147, and NRP1 [41,51,52,53], supporting the trophism for SARS-CoV-2. Autopsy tissues from COVID-19 patients show high expression of ACE2, TMPRSS2, and endothelial cell inflammation agents in capillaries but less presence in arteries and veins. The existent data suggest that the development of COVID-19 causes endotheliitis in small vessels like capillaries; nevertheless, the involvement of main coronary vessels principally arises from indirect mechanisms of SARS-CoV-2 infection [54].
A recent study utilizing the COVID-19 rhesus macaque model showed that SARS-CoV-2 infection can mediate indirect endothelial cell dysfunction through immune and inflammatory pathways interaction [55]. In another COVID-19 study, the combined activation of the NF-κB signaling cascade and changes in mitochondrial quality control led to endothelia cells’ activation and enhanced neuroinflammation [56]. As a result, SARS-CoV-2-infected human brain microvascular endothelial cells showed augmented caspase 3 cleavage and apoptotic cell death of endothelial cells.
RNA sequencing data of COVID-19 subjects showed higher expression of the endothelia cell activation markers RELB and TNF-α [57]. Accordingly, it is assumed that hyper-inflammation is responsible for endothelia cell injury and death following SARS-CoV-2 infection. In addition, significantly high levels of Cxcl9 and Cxcl10 in the endothelia cell cluster in SARS-CoV-2-infected K18 mice (SARS-CoV-2-infected human cytokeratin 18 (K18)-hACE2) were reported compared to infected WT mice, suggesting interferon pathway upregulation. The Kras pathway was also upregulated. Additionally, upregulation of the Vcam1 and Icam1 genes in the lysosome/apoptosis and complement pathways in viral-RNA-positive cells was evident [58]. A more recent study reported signs of damage and activation of endothelia cells in the form of enhanced gene expression cascades regarding EMT/EndoMT in the hearts of COVID-19-infected patients followed by an upregulation of tissue hypoxia-related pathways and the formation of ultra-structurally detectable thrombi (uTh). Endothelia cells induce the recruitment of monocytes/macrophages to the site of injury through the upregulation of adhesion-molecules and activation of SDF-1/CXCR4 signaling [59]. Overall, these results shed light on the importance of SARS-CoV-2-mediated immune activation in endothelial dysfunction and injury.
5. Degradation of Endothelial Glycocalyx Makes Them Vulnerable to SARS-CoV-2 Entry
The glycocalyx is a layer covering the luminal surface of vascular endothelia cells, which contributes to the maintenance of vascular homeostasis via regulating vascular tone, permeability, thrombosis, and leukocyte adhesion to the endothelium [60]. The glycocalyx is comprised of numerous proteoglycans, glycosaminoglycans, glycoproteins, and associated plasma proteins. The intact highly sulfated glycocalyx structure of the endothelium can prevent SARS-CoV-2 but its disruption increases susceptibility to SARS-CoV-2 infection, resulting in hyperinflammation and oxidative stress [61]. In this regard, a recent study has reported the interaction of intact glycocalyx with the Spike protein and its ability to prohibit the binding of the S protein and ACE2. Conversely, damaged glycocalyx encouraged S protein and ACE2 binding and enabled viral entry [62]. In vivo evidence suggests that COVID-19 patients with severe symptoms showed endothelial glycocalyx disruption and syndecan-1 (SDC-1) secretion (Yamaoka-Tojo 2020). Even among patients who had recovered from COVID-19, the levels of SDC-1 were significantly elevated compared to healthy controls, exhibiting the existence of persistent endothelial damage after COVID-19 progression [63].
6. Vascular Inflammation and Blood-Brain Barrier Disruption in COVID-19
Cranial nerves and the BBB are the main avenues between the brain and SARS-CoV-2. However, viruses such as SARS-CoV-2 cannot easily enter the brain parenchyma through the endothelial cells that line the lumen side of the capillaries of the systemic circulatory system due to the unique physiology of the BBB. The BBB is a functional unit that mostly consists of brain endothelia cells linked by TJ proteins, vascular basement membrane, pericytes, astrocytes end processes, neural cells, and microglia [64]. This dynamic structure operates as a bridge between the plasma and brain parenchyma, which are jointly considered as the neurovascular units (NVUs). In the BBB, endothelial cells are fastened by TJ proteins including ZO scaffolding proteins, claudin-5, and occludin as well as junctional adhesion molecules to prevent the extracellular and transcellular diffusion of molecules in the CNS [65]. Hence, in addition to neuroinvasion, disturbance of BBB integrity including endothelial TJ disruption may expose the brain to the danger of SARS-CoV-2 penetration from infected blood, influencing neuronal function in the CNS [66]. Hyper-inflammatory conditions after COVID-19 infection may contribute to BBB damage [67]. In support of this notion, human brain microvascular endothelia cells were reported to overexpress pro-inflammatory cytokines, chemokines, and adhesion molecules and to have lower expression of the TJ protein, which coincided with increased BBB permeability following SARS-CoV-2 infection [68].
Increased pro-inflammatory cytokine levels cause TJ function alteration and disruption of the BBB. For example, IL-1 enhancement damages BBB integrity [69], whereas IL-1β upregulates matrix metalloproteinase (MMP)-9 and promotes TJ protein disruption via activating extracellular signal regulated kinases [70]. Additionally, increased TNF-α, IL-6, and IL-12 cytokine levels result in the deprivation of TJ proteins and consequent BBB permeability impairment [71]. Cytokines likely disturb the integrity of different sorts of junctional proteins such as VE-cadherin, ZO-1, β-catenin, and gap junction, leading to the penetration of inflammatory and immune cells [72]. BBB injury enhances the passing of immune cells through the BBB and increases viral particles and proinflammatory cytokines in the CNS, causing cytokine activation and vascular endothelial growth factor (VEGF) production in astrocytes [73]. By triggering the phosphoinositide 3 (PI3)-kinase and AKT signaling cascades and MMP-9 upregulation, VEGF is capable of disrupting TJ proteins in brain capillary endothelia cells and causing BBB deficiency [74] (Figure 1).
7. Disseminating Intravascular Coagulation and BBB Disruption in COVID-19
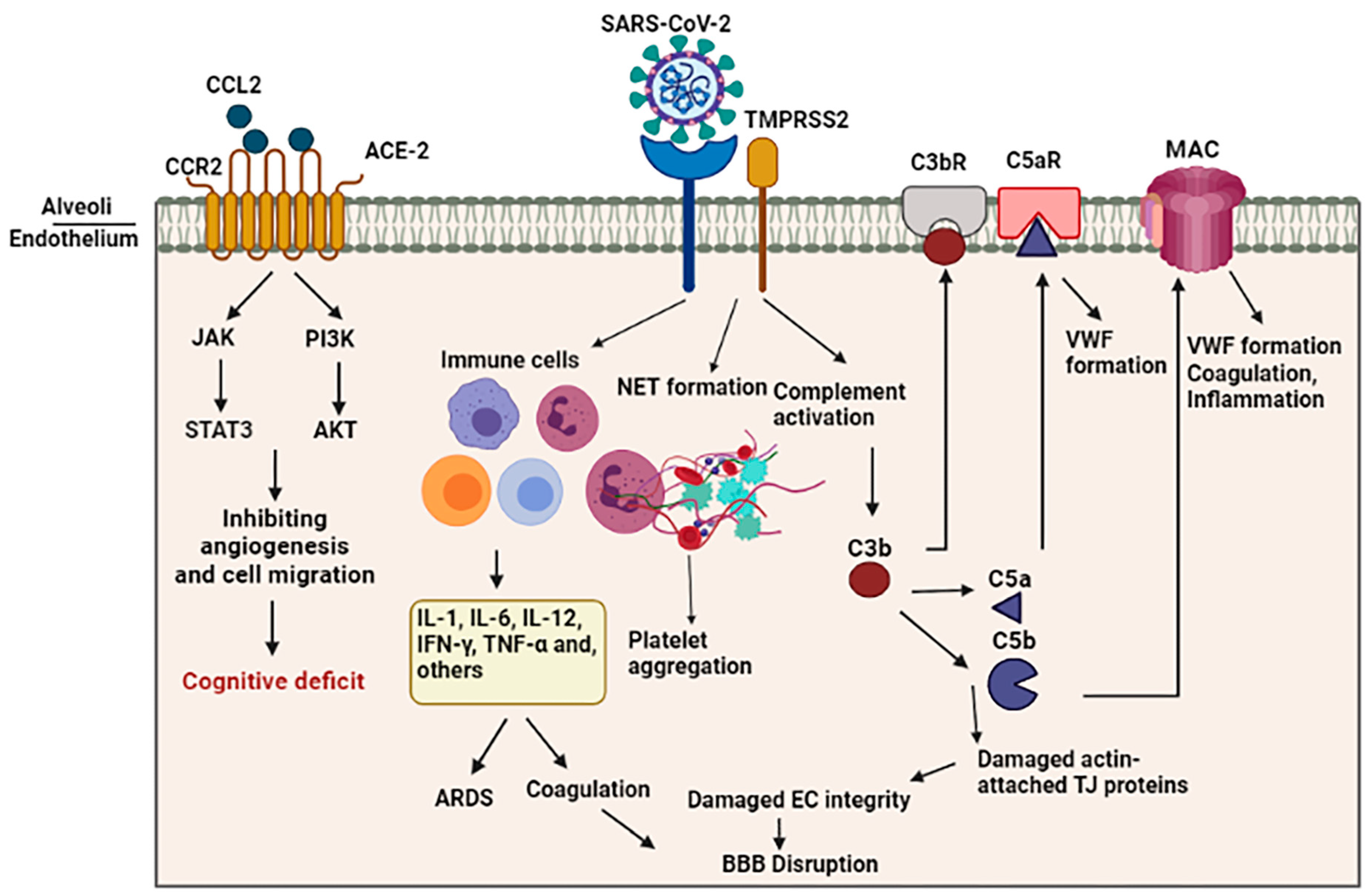
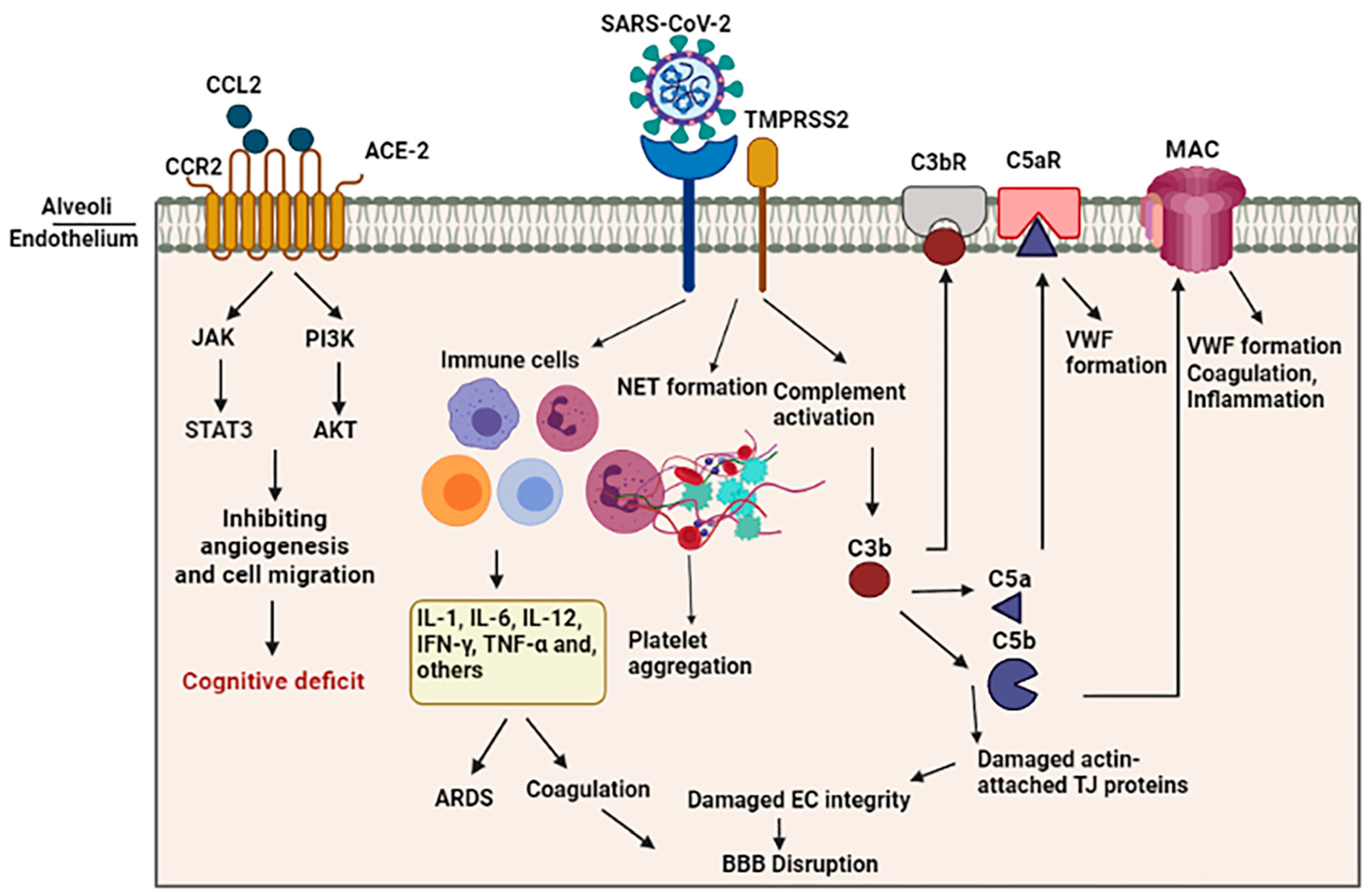
Both overactive coagulation and hyper-inflammation have been proposed as the main mechanisms underlying endothelia cell damage and thrombus development in COVID-19 [75]. Coagulation is frequently impaired in COVID-19 patients, resulting in a common hypercoagulable state in patients, which may be related to the incidence of stroke. Scientific records imply increased intravascular coagulation, blood clot formation, and bleeding in severe COVID-19 patients. Since thromboembolic single or multiple infarcts are reported in nearly 20% of dementia cases [76], thromboembolic occlusion of cerebral blood vessels is a potential causative of neurological manifestations, including cognitive deficit or dementia, especially in younger healthy adult COVID-19 subjects [77]. As shown in Figure 2, several pathways are involved in intravascular coagulation in COVID-19 patients. (1) The release of pro-inflammatory cytokines can cause platelets’ release, activation, and accumulation. Amongst cytokines, some agents like IL-6 and Cathepsin G, a serine protease generated by neutrophils, have more thrombogenic capacity and can stimulate platelets’ aggregation. In addition, TNF-α induces the surge of plasminogen-activator inhibitor-1 (PAI-1), leading to subsequent decreased activity of plasmin and reduced fibrinolysis [78]. (2) Activation of the complement system is a principal inducer of coagulation. SARS-CoV2 interacts with ACE-2 and activates the complement system including the lectin and classical pathways, resulting in the production of C3a and C3b. C3a mediates inflammation and activates platelets, while C3b contributes to the production of C5a and C5b. The binding of C5a to the C5 receptor mediates the platelet activation, aggregations, discharge of procoagulant microparticles (PMPs), and the development of blood clots. In addition, it may also contribute to the recruitment of neutrophils. C5b generates membrane attack complexes (MACs), serving as a transmembrane channel to initiate the lysis of the embedded cells. The MACs activate the microvascular complement deposition, coagulation, and inflammation. Furthermore, cell lysis and death of target cells are involved in coagulopathy through the enhancement of prothrombin activity as well as von Willebrand factor (VWF) formation [79]. Another mechanism by which complement activation is involved in coagulation is the binding of C3b to the CR1 receptor on the platelet’s membrane. This process triggers the release of short-chain polyphosphate (polyP) from platelets, inducing the expression of tissue factor (TF). (3) Whereas the formation of neutrophil extracellular traps (NETs), composed of chromatin and microbicidal proteins as well as neutrophils, is a key mechanism of conglutination since neutrophils are a crucial player in the production of thromboses, NETs participate in the pathobiology of thrombosis, through which histones, as a main element of NETs, attract and bind to platelets, leading to their aggregation [80]. As indicated in a recent study, neutrophils contribute to the immune response to SARS-CoV-2 invasion. Since these cells are much bigger than erythrocytes and the average capillary diameter, neutrophils can plug capillaries and cause significant blood flow disruption [81]. The adhesion of hyper-activated neutrophils in brain capillaries diminishes the cerebral blood flow in animal models of AD and consequently causes memory dysfunction [82]. Neutrophil-induced disruption of capillary blood flow within the lungs, brain, heart, and other organs is implicated in the poor prognosis of COVID-19 illness [83].
A study utilizing a rat model of intraventricular hemorrhage indicated BBB disruption followed by thrombin-caused activation of Src kinase phosphorylation. Src triggering increases BBB permeability through MMP phosphorylation, TJ protein disruption, and VEGF upregulation [84,85]. In addition, fibrinogen can harm endothelia cells’ integrity by damaging actin filament-attached TJ proteins [86]. On the other hand, enhanced generation of actin likely results in cellular stiffness, actin filament retraction, and spreading of endothelia cells junctions, thus interrupting endothelia cell integrity [87]. In this regard, Yepes et al. discovered vascular leakage in a dose-dependent manner following the intraventricular infusion of endogenous tissue plasminogen activator (tPA) [88] (Figure 2).
8. Pneumonia and BBB Disruption in COVID-19
It has been suggested that cerebral hypoxia triggers hypometabolic, cognitive, and degenerative changes in the brain and is contributory to the pathology of Alzheimer’s disease (AD) [89].
A large body of evidence suggests the bidirectional relationship between cognitive outcomes and ARDS, so pneumonia may influence cognitive performance depending on the patient populations and clinical contexts [90]. In contrast, pre-existing cognitive dysfunction is linked to an enhanced rate of mortality by pneumonia [91]. Hypoxemia and hypoxia are considered the causes of neuronal atrophy, consequent enlargement of ventricles, and related cognitive dysfunction, mostly memory loss [92]. In this line, CT scans of patients with ARDS exhibited widespread cerebral atrophy and widening of the bilateral temporal horn compared to the control group [93]. Consistently, pneumonia has been reported to increase the risk of developing dementia among elderly patients [94]. Even non-elderly patients with a single episode of pneumonia without main medical comorbidities are vulnerable to enhanced danger of cognitive decline [94]. According to the report by Wilcox et al., 70 to 100% of ARDS-hospitalized patients indicated cognitive impairment, ranging from difficulties in attention, concentration, and memory to executive function. These changes persisted in 46–78% one year, 25–47% two years, and almost 20% of cases five years after infection [95]. The weakening of memory may result from the susceptibility of hippocampal neurons to oxygen shortage [96]. Therefore, ARDS and hypoxia are vital contributors to the cognitive deficits, especially memory loss, in COVID-19 subjects.
Blood vessels supply oxygen and nutrients to neurons. Continuous hypoxic condition of brain tissue will ultimately cause irreversible neural damage [97]. Postmortem analyses of COVID-19 patients with hypoxic brain damage indicated neuronal injure in the neocortex, hippocampus, and cerebellum areas. Oligodendrocyte demise and widespread gliosis were also reported [98]. A body of evidence demonstrates an important role of hypoxia in BBB disruption [99]. Based on evidence, through changing the actin distribution and attenuating TJ proteins, hypoxia raises the paracellular permeability of brain capillary endothelial cells (Figure 2).
Mitochondria are organelles operating as an energy delivery system. Mitochondrial energy metabolism is directly associated with the hypoxic condition. In addition, hypoxia in the brain may strengthen the proliferative ability of the virus [100]. After hypoxia, a high virus presence in COVID-19 subjects with CNS involvement leads to the compromise of neurons with high-level energy metabolism. Hence, it has been indicated that targeting selective neuronal mitochondrial in SARS-CoV-2 infection induces ‘brain fog’ and causes cognitive and behavioral deficits [8].
In another way, the SARS-CoV-2 virus can result in mitochondrial energy metabolism impairment through targeting oxygen availability and consumption. Notably, the integration of the viral genome into the host cell mitochondrial matrix and creation of a viral-mitochondrial interaction leads to these effects. The interaction between the virus and mitochondria increases energy and reduces host immune reaction, resulting in enhanced replication and survival of the virus [101,102]. Thus, this pathological influence of SARS-CoV-2 infection may elucidate the long-term psychiatric, cognitive, and neurodegenerative outcomes. Furthermore, the reduction in available mitochondrial energy leads to undesired host immune response [103]. Therefore, impaired mitochondrial energy metabolism may be considered as a chief factor in cognitive manifestations in COVID-19 patients.
9. Vascular Dysfunction, Brain Inflammation, and Cognitive Impairment
Normally, viruses such as SARS-CoV-2 cannot easily enter the brain parenchyma through the endothelial cells that line inside of the capillaries in the systemic circulatory system due to the unique physiology of the BBB. VE-cadherin, ZO-1, β-catenin, and gap junction caused by the hyper-inflammatory condition of the SARS-CoV-2 virus results in dysfunction and enhanced leakage of the BBB, which exacerbates the penetration of the virus through the disrupted BBB [66]. Once SARS-CoV-2 reaches cerebral tissue, it causes neuro-inflammation through the activation of microglia and macrophages, leading to the release of local pro-inflammatory cytokines in addition to circulatory cytokines. Due to the very high amount of pro-inflammatory cytokines in the circulation, the integrity of the BBB is disrupted, and the brain becomes more vulnerable to ischemic, hypoxic, and thrombolytic threats along with the invasion of various pathogens [104,105] (Figure 2).
Based on research, neurological dysfunction levels and BBB injury seem to be associated with the grade of cognitive loss and of COVID-19 infection severity. A recent study found BBB permeability in 58% of COVID-19 subjects in 31 cases with neurological indications [106], suggesting that SARS-CoV-2 can cause BBB dysfunction. The data of another recent study suggested that the loss of BBB integrity might contribute to the progressive impairment of cognition in diabetic rats. The increase in TNF-α and IL-6 expression might trigger the disruption of the BBB in the brain, which eventually caused cognitive impairment in 8-week-old STZ rats [107]. In support of BBB disruption’s influence on cognitive impairment, a recent work suggested that BBB breakdown takes part in APOE4-connected cognitive weakening independently of AD pathology and might be a therapeutic target in APOE4 carriers [108]. Nevertheless, the accumulation of extracellular β-amyloid (Aβ) plaques and intracellular tau neurofibrillary tangles are considered a main characteristic of the preclinical phase of AD and are important predictors of mild cognitive deficit in cognitively normal persons [109]. SARS-CoV-2 infection is involved in cognitive impairment partially via increased Aβ accumulation. The virus leads to increased neuro-inflammation, changing brain structure, and finally abnormal gathering of neurodegenerative Aβ and phosphorylated tau, resulting in an enhanced risk of cognitive deficiency in patients with COVID-19 [110]. In this regard, a post-mortem investigation of COVID-19 cases indicated significant activation of microglia and neuro-inflammation associated with brain pathology [111]. Microglia, as innate immune cells in the CNS, take part in neurodegeneration pathology. A study showed that microglial distribution and activation in the hippocampus is associated with SARS-CoV-2-virus-induced cognitive impairment [112]. Data revealed that the activation of microglia is a main source of neurotoxic agents, like pro-inflammatory cytokines and reactive oxygen and nitrogen species, causing neural damage progression [113]. It has been illustrated that activating cultured human microglia with neuropeptides causes IL-1β and CXCL8 release [114], which induces pro-inflammatory responses. Microglial-derived pro-inflammatory cytokines and chemokines are able to encourage astrogliosis and Aβ deposition and consequently worsen neuro-inflammation [115].
Likewise, regarding the regulatory role of ACE2 in controlling blood pressure, the occupying of ACE2 by the viral spike protein may result in an imbalance of angiotensin system and affect normal blood pressure. Primary pneumonia and pulmonary infection also cause oxygen deprivation in the brain parenchyma and produce hypoxic conditions and subsequent metabolic disruption [116].
The cytokine storm phenomenon is an increase in pro-inflammatory cytokine levels in the serum, such as IL-2, IL-6 and IL-1β, IL-17, IL-8, G-CSF, GM-CSF, IP10, MCP1, MIP1α (also known as CCL3), and TNF [36], and causes acute respiratory distress syndrome (ARDS) in severe COVID-19 cases [117]. It has been indicated that increased cytokine concentration, especially IL-6, TNFα, and IL-1β, has a strong effect on working memory and attention. Damage of these cognitive abilities is a typical feature of delirium and supports the key role of these cytokines in the etiology of COVID-19-associated cognitive impairments [118].
Prolonged neuro-inflammation and continued hypoxia have been postulated as the potential primary causes of both acute and chronic cognitive features of COVID-19 [105].
Chemokines are small molecules primarily known for regulating the chemoattraction of leukocytes and modulating immune reaction [119]. Additionally, chemokines are involved in different phases of CNS development via supporting cells’ migration, proliferation, and survival [120,121]. Four families of chemokines include C, CC, CXC, and CX3C, characterized based on the conserved cysteine residue position [122]. CCL11 acts through activating the PI3K/AKT, MAPK/p38, and JAK/STAT3 signaling pathways to prevent apoptosis and promote angiogenesis and cell migration [123]. CCL11 also can trigger oxidative stress via microglial NOX1 stimulation and potentiate glutamate-mediated neurotoxicity [124]. During adulthood, the CCL2/CCR2 axis is able to modulate neurotransmission and neuromodulation [123]. It has been indicated that treatment with CCL11 is capable of inhibiting neurogenesis in the adult brain, resulting in cognitive weakening [125].
Enhanced CCL11 concentration was found in plasma samples of long COVID cases suffering from cognitive symptoms, and white matter microglial reactivity was also detected in patients with SARS-CoV-2 infection [126]. In addition, elevated levels of CCL11 are noted in AD and schizophrenia. Accordingly, targeted therapy to normalize CCL11 levels might develop mental and physical health among patients with schizophrenia and AD [127].
A recent study reported white matter microglial reactivity in a mouse model of mild SARS-CoV-2 infection. Furthermore, the same pattern of prominent white-matter-selective microglial responses was found in SARS-CoV-2-infected human brain tissue. In the mouse model, pro-inflammatory CSF CCL11 was elevated at least 7 weeks after infection. In addition, humans suffering from long COVID with cognitive deficits showed higher CCL11 levels than the cases with long COVID without cognitive indications. Likewise, following mild SARS-CoV-2 infection in mice, hippocampal neurogenesis impairment, decreased oligodendrocytes, and myelin loss in subcortical white matter were detected after 1 week of infection, which persevered till at least 7 weeks [128]. The results of this study revealed a crucial role for long-term enhanced CCL11 levels in astrocyte-mediated microglial activation during SARS-CoV-2-induced CNS inflammation, leading to the extended mental and cognitive dysfunction of COVID-19 patients [129]. It has also been suggested that cerebral hypoxia triggers hypometabolic, cognitive, and degenerative changes in the brain and is contributory to the pathology of AD and related cognitive impairment [130].
10. Conclusions and Future Directions
In summary, the SARS-CoV-2 virus can invade the brain and exert its neurological manifestation through binding to ACE2 on nerve cells and endothelial cells. A sound body of evidence shows that SARS-CoV-2 impairs vascular integrity through direct or indirect viral infection, leading to endothelium damage and augmenting vascular penetrability in peripheral vessels [131], disrupting the BBB integrity and the CNS function [132].
Given the evidence, the SARS-CoV-2 pathogen can induce cognitive impairment via vascular dysfunction, disruption of the BBB, interruption of oxygen supply, dissemination of intravascular coagulation, and neuro-inflammation. Taken together, the long-term cognitive consequences of SARS-CoV-2 infection, to some extent, may be due to disruption of micro-structural and functional brain vasculatures during COVID-19 illness and in the recovery stages. In addition to the present evidence, future studies are needed to discover the exact long-term cognitive deficits in patients with COVID-19 and their probable mediator mechanisms.
Author Contributions
Z.S.: conceptualization, writing, original draft preparation, review, and editing; J.L.: conceptualization, writing, review, and editing; H.S.: supervision, conceptualization, original draft, and writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the National Institutes of Health R01 HL122774 (H.S.) NS027713 (H.S.), NS112819 (H.S.), NIH NINDS R01NS102886 (J.L.), Department of Veterans Affairs Administration BLR&D 2I01BX003335 (J.L.), and IK6BX004600 (J.L.) and by the Michael Ryan Zodda Foundation (H.S.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Garg, A.; Marji, A.; Goyal, S.; Ismail, R. A case of COVID-19 with memory impairment and delayed presentation as stroke. Cureus 2020, 12, e10025. [Google Scholar]
- Wu, Z.; McGoogan, J.M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: Summary of a report of 72 314 cases from the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar]
- Pilotto, A.; Cristillo, V.; Cotti Piccinelli, S.; Zoppi, N.; Bonzi, G.; Sattin, D.; Schiavolin, S.; Raggi, A.; Canale, A.; Gipponi, S. Long-term neurological manifestations of COVID-19: Prevalence and predictive factors. Neurol. Sci. 2021, 42, 4903–4907. [Google Scholar] [PubMed]
- Grant, M.C.; Geoghegan, L.; Arbyn, M.; Mohammed, Z.; McGuinness, L.; Clarke, E.L.; Wade, R.G. The prevalence of symptoms in 24,410 adults infected by the novel coronavirus (SARS-CoV-2; COVID-19): A systematic review and meta-analysis of 148 studies from 9 countries. PLoS ONE 2020, 15, e0234765. [Google Scholar]
- Dubé, M.; Le Coupanec, A.; Wong, A.H.; Rini, J.M.; Desforges, M.; Talbot, P.J. Axonal transport enables neuron-to-neuron propagation of human coronavirus OC43. J. Virol. 2018, 92, e00404–e00418. [Google Scholar] [PubMed] [Green Version]
- Goërtz, Y.M.; Van Herck, M.; Delbressine, J.M.; Vaes, A.W.; Meys, R.; Machado, F.V.; Houben-Wilke, S.; Burtin, C.; Posthuma, R.; Franssen, F.M. Persistent symptoms 3 months after a SARS-CoV-2 infection: The post-COVID-19 syndrome? ERJ Open Res. 2020, 6, 542–2020. [Google Scholar]
- Raja, C.P.; Bharathi, B.; Chandrasekaran, C.V.; Muruganantham, N.; Deepak, M.; Amit, A. Science behind Usefulness of Bacopa monnieri for Memory and Cognition. In Phytopharmaceuticals for Brain Health; CRC Press: Boca Raton, FL, USA, 2017; pp. 225–250. [Google Scholar]
- Stefano, G.B.; Ptacek, R.; Ptackova, H.; Martin, A.; Kream, R.M. Selective neuronal mitochondrial targeting in SARS-CoV-2 infection affects cognitive processes to induce ‘brain fog’and results in behavioral changes that favor viral survival. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2021, 27, e930886-1. [Google Scholar]
- Bliddal, S.; Banasik, K.; Pedersen, O.B.; Nissen, J.; Cantwell, L.; Schwinn, M.; Tulstrup, M.; Westergaard, D.; Ullum, H.; Brunak, S. Acute and persistent symptoms in non-hospitalized PCR-confirmed COVID-19 patients. Sci. Rep. 2021, 11, 13153. [Google Scholar] [CrossRef]
- Iwashyna, T.J.; Ely, E.W.; Smith, D.M.; Langa, K.M. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA 2010, 304, 1787–1794. [Google Scholar] [CrossRef] [Green Version]
- Semmler, A.; Widmann, C.N.; Okulla, T.; Urbach, H.; Kaiser, M.; Widman, G.; Mormann, F.; Weide, J.; Fliessbach, K.; Hoeft, A. Persistent cognitive impairment, hippocampal atrophy and EEG changes in sepsis survivors. J. Neurol. Neurosurg. Psychiatry 2013, 84, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Penninx, B.W.J.H. Psychiatric symptoms and cognitive impairment in “Long COVID”: The relevance of immunopsychiatry. World Psychiatry 2021, 20, 357. [Google Scholar] [PubMed]
- Kozik, V.; Reuken, P.; Utech, I.; Gramlich, J.; Stallmach, Z.; Demeyere, N.; Rakers, F.; Schwab, M.; Stallmach, A.; Finke, K. Subtle cognitive impairments in memory, attention, and executive functioning in patients with post-COVID syndrome and their relationships with clinical variables and subjective complaints. medRxiv 2022. [Google Scholar] [CrossRef]
- Li, Y.; Ji, M.; Yang, J. Current understanding of long-term cognitive impairment after sepsis. Front. Immunol. 2022, 13, 855006. [Google Scholar] [CrossRef] [PubMed]
- Rass, V.; Beer, R.; Schiefecker, A.J.; Kofler, M.; Lindner, A.; Mahlknecht, P.; Heim, B.; Limmert, V.; Sahanic, S.; Pizzini, A. Neurological outcome and quality of life 3 months after COVID-19: A prospective observational cohort study. Eur. J. Neurol. 2021, 28, 3348–3359. [Google Scholar]
- Carfì, A.; Bernabei, R.; Landi, F. Persistent symptoms in patients after acute COVID-19. JAMA 2020, 324, 603–605. [Google Scholar] [CrossRef]
- Helms, J.; Kremer, S.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Kummerlen, C.; Collange, O.; Boulay, C.; Fafi-Kremer, S.; Ohana, M. Neurologic features in severe SARS-CoV-2 infection. N. Engl. J. Med. 2020, 382, 2268–2270. [Google Scholar] [CrossRef]
- Mao, L.; Jin, H.; Wang, M.; Hu, Y.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. 2020, 77, 683–690. [Google Scholar]
- Mazza, M.G.; Palladini, M.; De Lorenzo, R.; Magnaghi, C.; Poletti, S.; Furlan, R.; Ciceri, F.; Rovere-Querini, P.; Benedetti, F.; The COVID-19 BioB Outpatient Clinic Study Group. Persistent psychopathology and neurocognitive impairment in COVID-19 survivors: Effect of inflammatory biomarkers at three-month follow-up. Brain Behav. Immun. 2021, 94, 138–147. [Google Scholar]
- Hellmuth, J.; Barnett, T.A.; Asken, B.M.; Kelly, J.D.; Torres, L.; Stephens, M.L.; Greenhouse, B.; Martin, J.N.; Chow, F.C.; Deeks, S.G. Persistent COVID-19-associated neurocognitive symptoms in non-hospitalized patients. J. Neurovirol. 2021, 27, 191–195. [Google Scholar] [CrossRef]
- Garrigues, E.; Janvier, P.; Kherabi, Y.; Le Bot, A.; Hamon, A.; Gouze, H.; Doucet, L.; Berkani, S.; Oliosi, E.; Mallart, E. Post-discharge persistent symptoms and health-related quality of life after hospitalization for COVID-19. J. Infect. 2020, 81, e4–e6. [Google Scholar] [CrossRef]
- Han, Q.; Zheng, B.; Daines, L.; Sheikh, A. Long-term sequelae of COVID-19: A systematic review and meta-analysis of one-year follow-up studies on post-COVID symptoms. Pathogens 2022, 11, 269. [Google Scholar]
- Ritchie, K.; Chan, D. The emergence of cognitive COVID. World Psychiatry 2021, 20, 52. [Google Scholar] [CrossRef]
- Zhou, H.; Lu, S.; Chen, J.; Wei, N.; Wang, D.; Lyu, H.; Shi, C.; Hu, S. The landscape of cognitive function in recovered COVID-19 patients. J. Psychiatr. Res. 2020, 129, 98–102. [Google Scholar]
- Raman, B.; Cassar, M.P.; Tunnicliffe, E.M.; Filippini, N.; Griffanti, L.; Alfaro-Almagro, F.; Okell, T.; Sheerin, F.; Xie, C.; Mahmod, M. Medium-term effects of SARS-CoV-2 infection on multiple vital organs, exercise capacity, cognition, quality of life and mental health, post-hospital discharge. EClinicalMedicine 2021, 31, 100683. [Google Scholar] [PubMed]
- Woo, M.S.; Malsy, J.; Pöttgen, J.; Seddiq Zai, S.; Ufer, F.; Hadjilaou, A.; Schmiedel, S.; Addo, M.M.; Gerloff, C.; Heesen, C. Frequent neurocognitive deficits after recovery from mild COVID-19. Brain Commun. 2020, 2, fcaa205. [Google Scholar]
- Kumar, V.; Singla, S.; Gupta, N.; Bharati, S.J.; Garg, R.; Pandit, A.; Vig, S.; Mishra, S.; Bhatnagar, S. The incidence of anosmia in patients with laboratory-confirmed COVID 19 infection in India: An observational study. J. Anaesthesiol. Clin. Pharmacol. 2021, 37, 51. [Google Scholar] [PubMed]
- Jaywant, A.; Vanderlind, W.M.; Alexopoulos, G.S.; Fridman, C.B.; Perlis, R.H.; Gunning, F.M. Frequency and profile of objective cognitive deficits in hospitalized patients recovering from COVID-19. Neuropsychopharmacology 2021, 46, 2235–2240. [Google Scholar] [PubMed]
- Villani, E.R.; Vetrano, D.L.; Damiano, C.; Paola, A.D.; Ulgiati, A.M.; Martin, L.; Hirdes, J.P.; Fratiglioni, L.; Bernabei, R.; Onder, G. Impact of COVID-19-related lockdown on psychosocial, cognitive, and functional well-being in adults with down syndrome. Front. Psychiatry 2020, 11, 578686. [Google Scholar] [PubMed]
- Zhou, J.; Liu, C.; Sun, Y.; Huang, W.; Ye, K. Cognitive disorders associated with hospitalization of COVID-19: Results from an observational cohort study. Brain Behav. Immun. 2021, 91, 383–392. [Google Scholar]
- Heesakkers, H.; van der Hoeven, J.G.; Corsten, S.; Janssen, I.; Ewalds, E.; Simons, K.S.; Westerhof, B.; Rettig, T.C.; Jacobs, C.; van Santen, S. Clinical outcomes among patients with 1-year survival following intensive care unit treatment for COVID-19. JAMA 2022, 327, 559–565. [Google Scholar]
- Del Brutto, O.H.; Wu, S.; Mera, R.M.; Costa, A.F.; Recalde, B.Y.; Issa, N.P. Cognitive decline among individuals with history of mild symptomatic SARS-CoV-2 infection: A longitudinal prospective study nested to a population cohort. Eur. J. Neurol. 2021, 28, 3245–3253. [Google Scholar] [CrossRef]
- Cristillo, V.; Pilotto, A.; Cotti Piccinelli, S.; Bonzi, G.; Canale, A.; Gipponi, S.; Bezzi, M.; Leonardi, M.; Padovani, A.; Neuro Covid Next Study Group. Premorbid vulnerability and disease severity impact on Long-COVID cognitive impairment. Aging Clin. Exp. Res. 2022, 34, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar]
- Sparks, M.A.; Crowley, S.D.; Gurley, S.B.; Mirotsou, M.; Coffman, T.M. Classical renin-angiotensin system in kidney physiology. Compr. Physiol. 2014, 4, 1201. [Google Scholar]
- Shabani, Z. Demyelination as a result of an immune response in patients with COVID-19. Acta Neurol. Belg. 2021, 121, 859–866. [Google Scholar] [CrossRef]
- Doobay, M.F.; Talman, L.S.; Obr, T.D.; Tian, X.; Davisson, R.L.; Lazartigues, E. Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin-angiotensin system. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2007, 292, R373–R381. [Google Scholar] [PubMed] [Green Version]
- Baig, A.M.; Khaleeq, A.; Ali, U.; Syeda, H. Evidence of the COVID-19 virus targeting the CNS: Tissue distribution, host–virus interaction, and proposed neurotropic mechanisms. ACS Chem. Neurosci. 2020, 11, 995–998. [Google Scholar] [PubMed] [Green Version]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.-Y. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef] [PubMed]
- Seyran, M.; Takayama, K.; Uversky, V.N.; Lundstrom, K.; Palù, G.; Sherchan, S.P.; Attrish, D.; Rezaei, N.; Aljabali, A.A.; Ghosh, S. The structural basis of accelerated host cell entry by SARS-CoV-2. FEBS J. 2021, 288, 5010–5020. [Google Scholar] [CrossRef]
- Iadecola, C.; Anrather, J.; Kamel, H. Effects of COVID-19 on the nervous system. Cell 2020, 183, 16–27.e1. [Google Scholar] [CrossRef]
- Davies, J.; Randeva, H.S.; Chatha, K.; Hall, M.; Spandidos, D.A.; Karteris, E.; Kyrou, I. Neuropilin-1 as a new potential SARS-CoV-2 infection mediator implicated in the neurologic features and central nervous system involvement of COVID-19. Mol. Med. Rep. 2020, 22, 4221–4226. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, H.; Pillat, M.M. CD147 as a target for COVID-19 treatment: Suggested effects of azithromycin and stem cell engagement. Stem Cell Rev. Rep. 2020, 16, 434–440. [Google Scholar] [PubMed]
- Istifli, E.S.; Tepe, A.Ş.; SarikÜrkcÜ, C.; Tepe, B. Interaction of certain monoterpenoid hydrocarbons with the receptor binding domain of 2019 novel coronavirus (2019-nCoV), transmembrane serine protease 2 (TMPRSS2), cathepsin B, and cathepsin L (CatB/L) and their pharmacokinetic properties. Turk. J. Biol. 2020, 44, 242–264. [Google Scholar]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Shabani, Z.; Schuerger, J.; Su, H. Cellular loci involved in the development of brain arteriovenous malformations. Front. Hum. Neurosci. 2022, 16, 968369. [Google Scholar] [CrossRef] [PubMed]
- Osburn, W.O.; Smith, K.; Yanek, L.; Amat-Alcaron, N.; Thiemann, D.R.; Cox, A.L.; Leucker, T.M.; Lowenstein, C.J. Markers of endothelial cell activation are associated with the severity of pulmonary disease in COVID-19. PLoS ONE 2022, 17, e0268296. [Google Scholar]
- Rotoli, B.M.; Barilli, A.; Visigalli, R.; Ferrari, F.; Dall’Asta, V. Endothelial cell activation by sars-cov-2 spike s1 protein: A crosstalk between endothelium and innate immune cells. Biomedicines 2021, 9, 1220. [Google Scholar]
- Szekely, L.; Bozoky, B.; Bendek, M.; Ostad, M.; Lavignasse, P.; Haag, L.; Wu, J.; Jing, X.; Gupta, S.; Saccon, E. Pulmonary stromal expansion and intra-alveolar coagulation are primary causes of COVID-19 death. Heliyon 2021, 7, e07134. [Google Scholar] [PubMed]
- Bhatnagar, J.; Gary, J.; Reagan-Steiner, S.; Estetter, L.B.; Tong, S.; Tao, Y.; Denison, A.M.; Lee, E.; DeLeon-Carnes, M.; Li, Y. Evidence of severe acute respiratory syndrome coronavirus 2 replication and tropism in the lungs, airways, and vascular endothelium of patients with fatal coronavirus disease 2019: An autopsy case series. J. Infect. Dis. 2021, 223, 752–764. [Google Scholar]
- Hamming, I.; Timens, W.; Bulthuis, M.; Lely, A.; Navis, G.v.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. A J. Pathol. Soc. Great Br. Irel. 2004, 203, 631–637. [Google Scholar]
- Chen, L.; Hao, G. The role of angiotensin-converting enzyme 2 in coronaviruses/influenza viruses and cardiovascular disease. Cardiovasc. Res. 2020, 116, 1932–1936. [Google Scholar] [CrossRef]
- Wang, K.; Chen, W.; Zhou, Y.-S.; Lian, J.-Q.; Zhang, Z.; Du, P.; Gong, L.; Zhang, Y.; Cui, H.-Y.; Geng, J.-J. SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Maccio, U.; Zinkernagel, A.S.; Shambat, S.M.; Zeng, X.; Cathomas, G.; Ruschitzka, F.; Schuepbach, R.A.; Moch, H.; Varga, Z. SARS-CoV-2 leads to a small vessel endotheliitis in the heart. EBioMedicine 2021, 63, 103182. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Kanevsky, I.; Yildiz, S.; Sellers, R.S.; Swanson, K.A.; Franks, T.; Rathnasinghe, R.; Munoz-Moreno, R.; Jangra, S.; Gonzalez, O. Modeling SARS-CoV-2: Comparative pathology in rhesus macaque and Golden Syrian hamster models. Toxicol. Pathol. 2022, 50, 280–293. [Google Scholar] [PubMed]
- Adesse, D.; Gladulich, L.; Alvarez-Rosa, L.; Siqueira, M.; Marcos, A.C.; Heider, M.; Motta, C.S.; Torices, S.; Toborek, M.; Stipursky, J. Role of aging in Blood–Brain Barrier dysfunction and susceptibility to SARS-CoV-2 infection: Impacts on neurological symptoms of COVID-19. Fluids Barriers CNS 2022, 19, 63. [Google Scholar]
- Motta, C.S.; Torices, S.; da Rosa, B.G.; Marcos, A.C.; Alvarez-Rosa, L.; Siqueira, M.; Moreno-Rodriguez, T.; Matos, A.d.R.; Caetano, B.C.; Martins, J.S.C.d.C. Human Brain Microvascular Endothelial Cells Exposure to SARS-CoV-2 Leads to Inflammatory Activation through NF-κB Non-Canonical Pathway and Mitochondrial Remodeling. Viruses 2023, 15, 745. [Google Scholar] [CrossRef]
- Qin, Z.; Liu, F.; Blair, R.; Wang, C.; Yang, H.; Mudd, J.; Currey, J.M.; Iwanaga, N.; He, J.; Mi, R. Endothelial cell infection and dysfunction, immune activation in severe COVID-19. Theranostics 2021, 11, 8076. [Google Scholar] [CrossRef]
- Werlein, C.; Ackermann, M.; Stark, H.; Shah, H.R.; Tzankov, A.; Haslbauer, J.D.; von Stillfried, S.; Bülow, R.D.; El-Armouche, A.; Kuenzel, S. Inflammation and vascular remodeling in COVID-19 hearts. Angiogenesis 2022, 26, 233–248. [Google Scholar]
- Potje, S.R.; Costa, T.J.; Fraga-Silva, T.F.; Martins, R.B.; Benatti, M.N.; Almado, C.E.; de Sa, K.S.; Bonato, V.L.; Arruda, E.; Louzada-Junior, P. Heparin prevents in vitro glycocalyx shedding induced by plasma from COVID-19 patients. Life Sci. 2021, 276, 119376. [Google Scholar]
- du Preez, H.N.; Aldous, C.; Hayden, M.R.; Kruger, H.G.; Lin, J. Pathogenesis of COVID-19 described through the lens of an undersulfated and degraded epithelial and endothelial glycocalyx. FASEB J. 2022, 36, e22052. [Google Scholar] [CrossRef]
- Targosz-Korecka, M.; Kubisiak, A.; Kloska, D.; Kopacz, A.; Grochot-Przeczek, A.; Szymonski, M. Endothelial glycocalyx shields the interaction of SARS-CoV-2 spike protein with ACE2 receptors. Sci. Rep. 2021, 11, 12157. [Google Scholar] [CrossRef]
- Vollenberg, R.; Tepasse, P.-R.; Ochs, K.; Floer, M.; Strauss, M.; Rennebaum, F.; Kabar, I.; Rovas, A.; Nowacki, T. Indications of persistent glycocalyx damage in convalescent COVID-19 patients: A prospective multicenter study and hypothesis. Viruses 2021, 13, 2324. [Google Scholar] [CrossRef] [PubMed]
- Langen, U.H.; Ayloo, S.; Gu, C. Development and cell biology of the blood-brain barrier. Annu. Rev. Cell Dev. Biol. 2019, 35, 591–613. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.; Campbell, M. Tight junction modulation of the blood brain barrier: CNS delivery of small molecules. Tissue Barriers 2016, 4, e1138017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleau, C.; Filliol, A.; Samson, M.; Lamontagne, L. Brain invasion by mouse hepatitis virus depends on impairment of tight junctions and beta interferon production in brain microvascular endothelial cells. J. Virol. 2015, 89, 9896–9908. [Google Scholar]
- Alquisiras-Burgos, I.; Peralta-Arrieta, I.; Alonso-Palomares, L.A.; Zacapala-Gomez, A.E.; Salmeron-Barcenas, E.G.; Aguilera, P. Neurological complications associated with the blood-brain barrier damage induced by the inflammatory response during SARS-CoV-2 infection. Mol. Neurobiol. 2021, 58, 520–535. [Google Scholar]
- Yang, R.-C.; Huang, K.; Zhang, H.-P.; Li, L.; Zhang, Y.-F.; Tan, C.; Chen, H.-C.; Jin, M.-L.; Wang, X.-R. SARS-CoV-2 productively infects human brain microvascular endothelial cells. J. Neuroinflamm. 2022, 19, 149. [Google Scholar] [CrossRef]
- Almutairi, M.M.; Gong, C.; Xu, Y.G.; Chang, Y.; Shi, H. Factors controlling permeability of the blood–brain barrier. Cell. Mol. Life Sci. 2016, 73, 57–77. [Google Scholar] [CrossRef]
- Ranaivo, H.R.; Zunich, S.M.; Choi, N.; Hodge, J.N.; Wainwright, M.S. Mild stretch-induced injury increases susceptibility to interleukin-1β-induced release of matrix metalloproteinase-9 from astrocytes. J. Neurotrauma 2011, 28, 1757–1766. [Google Scholar] [CrossRef]
- Erickson, M.A.; Rhea, E.M.; Knopp, R.C.; Banks, W.A. Interactions of SARS-CoV-2 with the blood–brain barrier. Int. J. Mol. Sci. 2021, 22, 2681. [Google Scholar] [CrossRef]
- Rauti, R.; Shahoha, M.; Leichtmann-Bardoogo, Y.; Nasser, R.; Paz, E.; Tamir, R.; Miller, V.; Babich, T.; Shaked, K.; Ehrlich, A. Effect of SARS-CoV-2 proteins on vascular permeability. Elife 2021, 10, e69314. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.A.; Banks, W.A. Neuroimmune axes of the blood–brain barriers and blood–brain interfaces: Bases for physiological regulation, disease states, and pharmacological interventions. Pharmacol. Rev. 2018, 70, 278–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilic, E.; Kilic, U.; Wang, Y.; Bassetti, C.L.; Marti, H.H.; Hermann, D.M. The phosphatidylinositol-3 kinase/Akt pathway mediates VEGF’s neuroprotective activity and induces blood brain barrier permeability after focal cerebral ischemia. FASEB J. 2006, 20, 1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, S.J.; Feigen, C.M.; Vazquez, J.P.; Kobets, A.J.; Altschul, D.J. Neurological sequelae of COVID-19. J. Integr. Neurosci. 2022, 21, 77. [Google Scholar] [CrossRef]
- Staekenborg, S.S.; Van der Flier, W.M.; Van Straaten, E.C.; Lane, R.; Barkhof, F.; Scheltens, P. Neurological signs in relation to type of cerebrovascular disease in vascular dementia. Stroke 2008, 39, 317–322. [Google Scholar] [CrossRef]
- Miners, S.; Kehoe, P.G.; Love, S. Cognitive impact of COVID-19: Looking beyond the short term. Alzheimer’s Res. Ther. 2020, 12, 1–16. [Google Scholar]
- Loo, J.; Spittle, D.A.; Newnham, M. COVID-19, immunothrombosis and venous thromboembolism: Biological mechanisms. Thorax 2021, 76, 412–420. [Google Scholar]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 170. [Google Scholar] [CrossRef]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar]
- Harris, A.G.; Skalak, T.C. Leukocyte cytoskeletal structure determines capillary plugging and network resistance. Am. J. Physiol.-Heart Circ. Physiol. 1993, 265, H1670–H1675. [Google Scholar] [CrossRef]
- Cruz Hernández, J.C.; Bracko, O.; Kersbergen, C.J.; Muse, V.; Haft-Javaherian, M.; Berg, M.; Park, L.; Vinarcsik, L.K.; Ivasyk, I.; Rivera, D.A. Neutrophil adhesion in brain capillaries reduces cortical blood flow and impairs memory function in Alzheimer’s disease mouse models. Nat. Neurosci. 2019, 22, 413–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Cao, C.; Chen, Z.; Bankaitis, V.; Tzima, E.; Sheibani, N.; Burridge, K. Pericytes regulate vascular basement membrane remodeling and govern neutrophil extravasation during inflammation. PLoS ONE 2012, 7, e45499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, R.; Zhang, Z.G.; Eliceiri, B.P.; Jiang, Q.; Boccia, A.D.; Zhang, R.L.; Chopp, M.; Cheresh, D.A. Src deficiency or blockade of Src activity in mice provides cerebral protection following stroke. Nat. Med. 2001, 7, 222–227. [Google Scholar] [CrossRef]
- Liu, D.Z.; Ander, B.P.; Xu, H.; Shen, Y.; Kaur, P.; Deng, W.; Sharp, F.R. Blood–brain barrier breakdown and repair by Src after thrombin-induced injury. Ann. Neurol. 2010, 67, 526–533. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, N.; Roberts, A.M.; Dean, W.L.; Tyagi, S.C.; Lominadze, D. Fibrinogen induces endothelial cell permeability. Mol. Cell. Biochem. 2008, 307, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Trepat, X.; Grabulosa, M.; Buscemi, L.; Rico, F.; Farré, R.; Navajas, D. Thrombin and histamine induce stiffening of alveolar epithelial cells. J. Appl. Physiol. 2005, 98, 1567–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yepes, M.; Sandkvist, M.; Moore, E.G.; Bugge, T.H.; Strickland, D.K.; Lawrence, D.A. Tissue-type plasminogen activator induces opening of the blood-brain barrier via the LDL receptor–related protein. J. Clin. Investig. 2003, 112, 1533–1540. [Google Scholar] [CrossRef]
- Grammas, P.; Samany, P.G.; Thirumangalakudi, L. Thrombin and inflammatory proteins are elevated in Alzheimer’s disease microvessels: Implications for disease pathogenesis. J. Alzheimer’s Dis. 2006, 9, 51–58. [Google Scholar] [CrossRef]
- Shah, F.A.; Pike, F.; Alvarez, K.; Angus, D.; Newman, A.B.; Lopez, O.; Tate, J.; Kapur, V.; Wilsdon, A.; Krishnan, J.A. Bidirectional relationship between cognitive function and pneumonia. Am. J. Respir. Crit. Care Med. 2013, 188, 586–592. [Google Scholar] [CrossRef] [Green Version]
- Salive, M.E.; Satterfield, S.; Ostfeld, A.M.; Wallace, R.B.; Havlik, R.J. Disability and cognitive impairment are risk factors for pneumonia-related mortality in older adults. Public Health Rep. 1993, 108, 314. [Google Scholar]
- Herridge, M.S.; Moss, M.; Hough, C.L.; Hopkins, R.O.; Rice, T.W.; Bienvenu, O.J.; Azoulay, E. Recovery and outcomes after the acute respiratory distress syndrome (ARDS) in patients and their family caregivers. Intensive Care Med. 2016, 42, 725–738. [Google Scholar] [CrossRef]
- Hopkins, R.O.; Jackson, J.C. Long-term neurocognitive function after critical illness. Chest 2006, 130, 869–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tate, J.A.; Snitz, B.E.; Alvarez, K.A.; Nahin, R.L.; Weissfeld, L.A.; Lopez, O.; Angus, D.C.; Shah, F.; Ives, D.G.; Fitzpatrick, A.L. Infection hospitalization increases risk of dementia in the elderly. Crit. Care Med. 2014, 42, 1037. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, M.E.; Brummel, N.E.; Archer, K.; Ely, E.W.; Jackson, J.C.; Hopkins, R.O. Cognitive dysfunction in ICU patients: Risk factors, predictors, and rehabilitation interventions. Crit. Care Med. 2013, 41, S81–S98. [Google Scholar] [CrossRef] [PubMed]
- Cervos-Navarro, J.; Diemer, N. Selective vulnerability in brain hypoxia. Crit. Rev. Neurobiol. 1991, 6, 149–182. [Google Scholar] [PubMed]
- Moskowitz, M.A.; Lo, E.H.; Iadecola, C. The science of stroke: Mechanisms in search of treatments. Neuron 2010, 67, 181–198. [Google Scholar] [CrossRef] [Green Version]
- Fernando, M.S.; Simpson, J.E.; Matthews, F.; Brayne, C.; Lewis, C.E.; Barber, R.; Kalaria, R.N.; Forster, G.; Esteves, F.; Wharton, S.B. White matter lesions in an unselected cohort of the elderly: Molecular pathology suggests origin from chronic hypoperfusion injury. Stroke 2006, 37, 1391–1398. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Rosenberg, G.A. Blood–brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke 2011, 42, 3323–3328. [Google Scholar] [CrossRef] [Green Version]
- Esch, T.; Stefano, G.B.; Ptacek, R.; Kream, R.M. Emerging roles of blood-borne intact and respiring mitochondria as bidirectional mediators of pro-and anti-inflammatory processes. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e924331–e924337. [Google Scholar] [CrossRef]
- Shenoy, S. Coronavirus (Covid-19) sepsis: Revisiting mitochondrial dysfunction in pathogenesis, aging, inflammation, and mortality. Inflamm. Res. 2020, 69, 1077–1085. [Google Scholar] [CrossRef]
- Singh, K.K.; Chaubey, G.; Chen, J.Y.; Suravajhala, P. Decoding SARS-CoV-2 hijacking of host mitochondria in COVID-19 pathogenesis. Am. J. Physiol.-Cell Physiol. 2020, 319, C258–C267. [Google Scholar] [CrossRef] [PubMed]
- Ptacek, R.; Ptackova, H.; Martin, A.; Stefano, G.B. Psychiatric manifestations of COVID-19 and their social significance. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e930340–e930341. [Google Scholar] [CrossRef] [PubMed]
- Bostancıklıoğlu, M. SARS-CoV2 entry and spread in the lymphatic drainage system of the brain. Brain Behav. Immun. 2020, 87, 122. [Google Scholar] [CrossRef] [PubMed]
- Steardo, L.; Steardo Jr, L.; Zorec, R.; Verkhratsky, A. Neuroinfection may contribute to pathophysiology and clinical manifestations of COVID-19. Acta Physiol. 2020, 229, e13473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellon, M.; Schweblin, C.; Lambeng, N.; Cherpillod, P.; Vazquez, J.; Lalive, P.H.; Schibler, M.; Deffert, C. Cerebrospinal fluid features in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) reverse transcription polymerase chain reaction (RT-PCR) positive patients. Clin. Infect. Dis. 2021, 73, e3102–e3105. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Wang, L.; Zhang, L.; Qin, C.; Song, Y.; Ma, Y.; Chen, Y.; Chen, S.; Wang, Y.; Zhang, Z. Blood-brain barrier disruption induced cognitive impairment is associated with increase of inflammatory cytokine. Front. Aging Neurosci. 2018, 10, 129. [Google Scholar] [CrossRef] [Green Version]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 leads to blood–brain barrier dysfunction predicting cognitive decline. Nature 2020, 581, 71–76. [Google Scholar] [CrossRef]
- Insel, P.S.; Donohue, M.C.; Sperling, R.; Hansson, O.; Mattsson-Carlgren, N. The A4 study: β-amyloid and cognition in 4432 cognitively unimpaired adults. Ann. Clin. Transl. Neurol. 2020, 7, 776–785. [Google Scholar] [CrossRef] [Green Version]
- Ma, G.; Zhang, D.-F.; Zou, Q.-C.; Xie, X.; Xu, L.; Feng, X.-L.; Li, X.; Han, J.-B.; Yu, D.; Deng, Z.-H.; et al. SARS-CoV-2 Spike protein S2 subunit modulates γ-secretase and enhances amyloid-β production in COVID-19 neuropathy. Cell Discov. 2022, 8, 99. [Google Scholar] [CrossRef]
- Shen, W.-B.; Elahi, M.; Logue, J.; Yang, P.; Baracco, L.; Reece, E.A.; Wang, B.; Li, L.; Blanchard, T.G.; Han, Z.; et al. SARS-CoV-2 invades cognitive centers of the brain and induces Alzheimer’s-like neuropathology. BioRxiv 2022. [Google Scholar] [CrossRef]
- Vasek, M.J.; Garber, C.; Dorsey, D.; Durrant, D.M.; Bollman, B.; Soung, A.; Yu, J.; Perez-Torres, C.; Frouin, A.; Wilton, D.K.; et al. A complement–microglial axis drives synapse loss during virus-induced memory impairment. Nature 2016, 534, 538–543. [Google Scholar] [CrossRef] [Green Version]
- Abate, G.; Memo, M.; Uberti, D. Impact of COVID-19 on Alzheimer’s disease risk: Viewpoint for research action. MDPI 2020, 8, 286. [Google Scholar] [CrossRef]
- Patel, A.B.; Tsilioni, I.; Leeman, S.E.; Theoharides, T.C. Neurotensin stimulates sortilin and mTOR in human microglia inhibitable by methoxyluteolin, a potential therapeutic target for autism. Proc. Natl. Acad. Sci. USA 2016, 113, E7049–E7058. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Kempuraj, D. Role of SARS-CoV-2 Spike-Protein-Induced Activation of Microglia and Mast Cells in the Pathogenesis of Neuro-COVID. Cells 2023, 12, 688. [Google Scholar] [CrossRef]
- Wu, Y.; Xu, X.; Chen, Z.; Duan, J.; Hashimoto, K.; Yang, L.; Liu, C.; Yang, C. Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav. Immun. 2020, 87, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.M. Immunotherapies and COVID-19 related neurological manifestations: A comprehensive review article. J. Immunoass. Immunochem. 2020, 41, 960–975. [Google Scholar] [CrossRef] [PubMed]
- Alnefeesi, Y.; Siegel, A.; Lui, L.M.; Teopiz, K.M.; Ho, R.; Lee, Y.; Nasri, F.; Gill, H.; Lin, K.; Cao, B. Impact of SARS-CoV-2 infection on cognitive function: A systematic review. Front. Psychiatry 2021, 11, 1629. [Google Scholar] [CrossRef]
- Gao, W.; Li, Y.; Zhang, T.; Lu, J.; Pan, J.; Qi, Q.; Dong, S.; Chen, X.; Su, Z.; Li, J. Systematic analysis of chemokines reveals CCL18 is a prognostic biomarker in glioblastoma. J. Inflamm. Res. 2022, 15, 2731–2743. [Google Scholar] [CrossRef]
- Kolodziej, A.; Schulz, S.; Guyon, A.; Wu, D.-F.; Pfeiffer, M.; Odemis, V.; Höllt, V.; Stumm, R. Tonic activation of CXC chemokine receptor 4 in immature granule cells supports neurogenesis in the adult dentate gyrus. J. Neurosci. 2008, 28, 4488–4500. [Google Scholar] [CrossRef] [PubMed]
- Senf, K.; Karius, J.; Stumm, R.; Neuhaus, E.M. Chemokine signaling is required for homeostatic and injury-induced neurogenesis in the olfactory epithelium. Stem Cells 2021, 39, 617–635. [Google Scholar] [CrossRef] [PubMed]
- Karimabad, M.N.; Hassanshahi, G.; Kounis, N.G.; Mplani, V.; Roditis, P.; Gogos, C.; Lagadinou, M.; Assimakopoulos, S.F.; Dousdampanis, P.; Koniari, I. The Chemokines CXC, CC and C in the Pathogenesis of COVID-19 Disease and as Surrogates of Vaccine-Induced Innate and Adaptive Protective Responses. Vaccines 2022, 10, 1299. [Google Scholar] [CrossRef]
- Yasui, H.; Kajiyama, H.; Tamauchi, S.; Suzuki, S.; Peng, Y.; Yoshikawa, N.; Sugiyama, M.; Nakamura, K.; Kikkawa, F. CCL2 secreted from cancer-associated mesothelial cells promotes peritoneal metastasis of ovarian cancer cells through the P38-MAPK pathway. Clin. Exp. Metastasis 2020, 37, 145–158. [Google Scholar] [CrossRef]
- Parajuli, B.; Horiuchi, H.; Mizuno, T.; Takeuchi, H.; Suzumura, A. CCL11 enhances excitotoxic neuronal death by producing reactive oxygen species in microglia. Glia 2015, 63, 2274–2284. [Google Scholar] [CrossRef] [PubMed]
- Villeda, S.A.; Luo, J.; Mosher, K.I.; Zou, B.; Britschgi, M.; Bieri, G.; Stan, T.M.; Fainberg, N.; Ding, Z.; Eggel, A. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 2011, 477, 90–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minton, K. Mechanistic insights into Long COVID in hamsters. Nat. Rev. Immunol. 2022, 22, 463. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Lee, E.E.; Martin, A.S.; Soontornniyomkij, B.; Soontornniyomkij, V.; Achim, C.L.; Reuter, C.; Irwin, M.R.; Eyler, L.T.; Jeste, D.V. Abnormalities in chemokine levels in schizophrenia and their clinical correlates. Schizophr. Res. 2017, 181, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Castañeda, A.; Lu, P.; Geraghty, A.C.; Song, E.; Lee, M.-H.; Wood, J.; Yalçın, B.; Taylor, K.R.; Dutton, S.; Acosta-Alvarez, L. Mild respiratory SARS-CoV-2 infection can cause multi-lineage cellular dysregulation and myelin loss in the brain. bioRxiv 2022. [Google Scholar] [CrossRef]
- Xu, J.; Dong, H.; Qian, Q.; Zhang, X.; Wang, Y.; Jin, W.; Qian, Y. Astrocyte-derived CCL2 participates in surgery-induced cognitive dysfunction and neuroinflammation via evoking microglia activation. Behav. Brain Res. 2017, 332, 145–153. [Google Scholar] [CrossRef]
- Thirumangalakudi, L.; Samany, P.G.; Owoso, A.; Wiskar, B.; Grammas, P. Angiogenic proteins are expressed by brain blood vessels in Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 10, 111–118. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Teuwen, L.-A.; Geldhof, V.; Pasut, A.; Carmeliet, P. COVID-19: The vasculature unleashed. Nat. Rev. Immunol. 2020, 20, 389–391. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of SARS-CoV-2 infection causing vascular dysfunction and BBB disruption. Proinflammatory cytokines result in VEGF production in astrocytes via allowing immune cells to pass through the BBB. VEGF is able to disrupt TJ proteins in brain capillary endothelial cells by triggering the phosphoinositide 3 (PI3)-kinase and AKT signaling pathways and MMP-9 upregulation, leading to BBB breakdown.
Figure 1.
Schematic representation of SARS-CoV-2 infection causing vascular dysfunction and BBB disruption. Proinflammatory cytokines result in VEGF production in astrocytes via allowing immune cells to pass through the BBB. VEGF is able to disrupt TJ proteins in brain capillary endothelial cells by triggering the phosphoinositide 3 (PI3)-kinase and AKT signaling pathways and MMP-9 upregulation, leading to BBB breakdown.
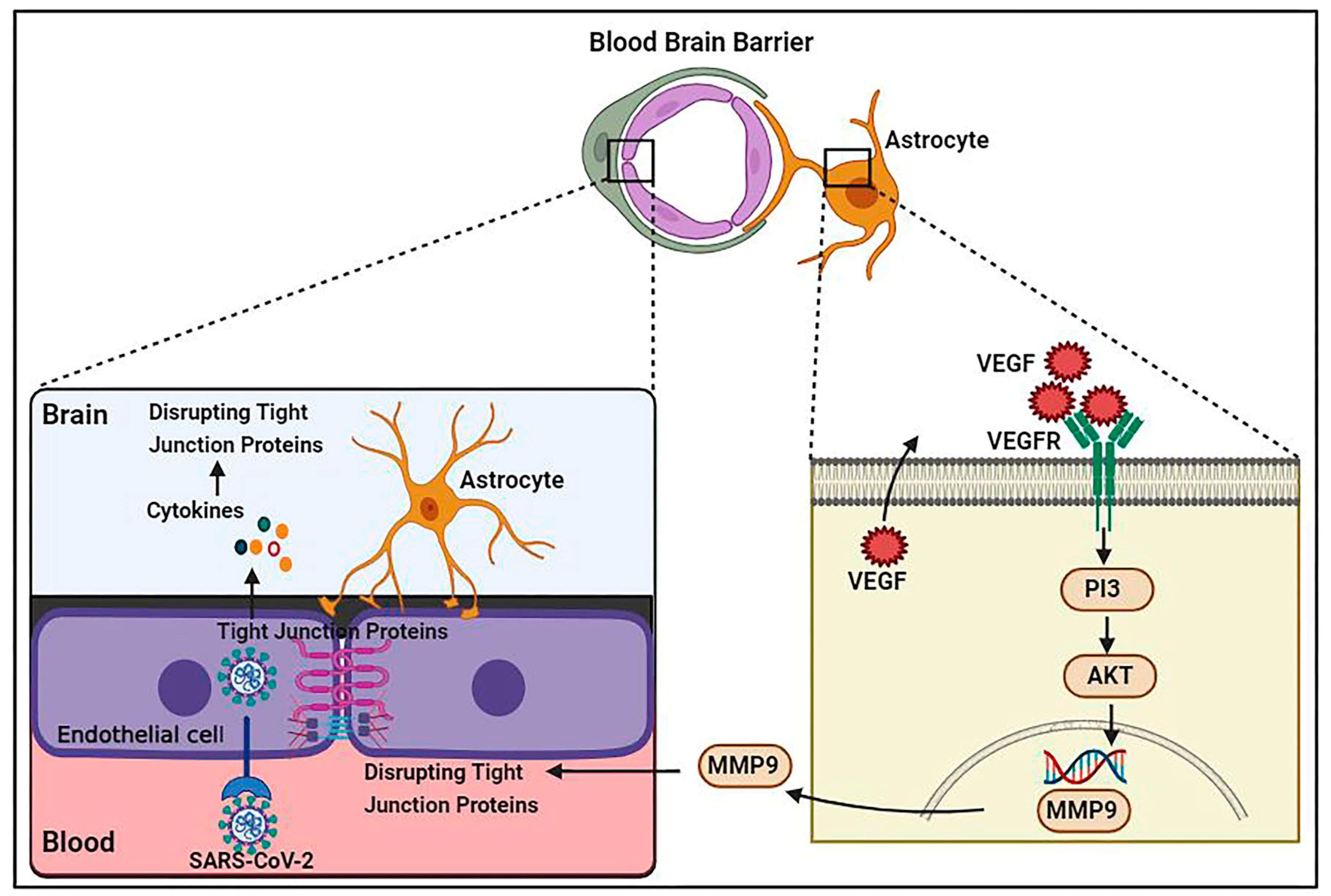
Figure 2.
Schematic representation of indirect BBB disruption resulting from vascular dysfunction in COVID-19. The virus acts via binding of the spike glycoprotein of the virus to the angiotensin converting enzyme 2 (ACE2) on the cell surface. Disharmonic and disturbed immune reaction in COVID-19 patients lead to wide local and systemic inflammation by generating cytokine storm, leading to ARDS and vascular coagulation. Alternatively, it may lead to the formation of neutrophil extracellular traps (NETs), composed of chromatin and microbicidal proteins, which participate in the pathobiology of thrombosis and platelets aggregation. Activation of the complement system results in the generation of C5a and C5b, which are involved in the inflammation and activation of platelets. This C5b forms the membrane attack complex (MAC) to activate microvascular coagulation and inflammation.
Figure 2.
Schematic representation of indirect BBB disruption resulting from vascular dysfunction in COVID-19. The virus acts via binding of the spike glycoprotein of the virus to the angiotensin converting enzyme 2 (ACE2) on the cell surface. Disharmonic and disturbed immune reaction in COVID-19 patients lead to wide local and systemic inflammation by generating cytokine storm, leading to ARDS and vascular coagulation. Alternatively, it may lead to the formation of neutrophil extracellular traps (NETs), composed of chromatin and microbicidal proteins, which participate in the pathobiology of thrombosis and platelets aggregation. Activation of the complement system results in the generation of C5a and C5b, which are involved in the inflammation and activation of platelets. This C5b forms the membrane attack complex (MAC) to activate microvascular coagulation and inflammation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Shabani, Z.; Liu, J.; Su, H. Vascular Dysfunctions Contribute to the Long-Term Cognitive Deficits Following COVID-19. Biology 2023, 12, 1106. https://doi.org/10.3390/biology12081106
AMA Style
Shabani Z, Liu J, Su H. Vascular Dysfunctions Contribute to the Long-Term Cognitive Deficits Following COVID-19. Biology. 2023; 12(8):1106. https://doi.org/10.3390/biology12081106
Chicago/Turabian StyleShabani, Zahra, Jialing Liu, and Hua Su. 2023. "Vascular Dysfunctions Contribute to the Long-Term Cognitive Deficits Following COVID-19" Biology 12, no. 8: 1106. https://doi.org/10.3390/biology12081106
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.