Abstract
The development of multicellular eukaryotes, according to their body plan, is often directed by members of multigene families that encode transcription factors. MADS (for MINICHROMOSOME MAINTENANCE1, AGAMOUS, DEFICIENS and SERUM RESPONSE FACTOR)-box genes form one of those families controlling nearly all major aspects of plant development. Knowing the complete complement of MADS-box genes in sequenced plant genomes will allow a better understanding of the evolutionary patterns of these genes and the association of their evolution with the evolution of plant morphologies. Here, we have applied a combination of automatic and manual annotations to identify the complete set of MADS-box genes in 17 plant genomes. Furthermore, three plant genomes were reanalyzed and published datasets were used for four genomes such that more than 2,600 genes from 24 species were classified into the two types of MADS-box genes, Type I and Type II. Our results extend previous studies, highlighting the remarkably different evolutionary patterns of Type I and Type II genes and provide a basis for further studies on the evolution and function of MADS-box genes.
1. Introduction
MADS-box genes encode transcription factors and have been identified in nearly all groups of eukaryotes [1]. They are major regulators of development [2,3]. The acronym MADS is derived from the four founding members of this gene family: MINICHROMOSOME MAINTENANCE1 from Saccharomyces cerevisiae, AGAMOUS from Arabidopsis thaliana, DEFICIENS from Antirrhinum majus, and SERUM RESPONSE FACTOR from Homo sapiens [4]. Whereas only two to six MADS-box genes are present in the genomes of animals and fungi, the number of these genes has greatly expanded in plants [2,5,6,7]. Roughly, about 100 MADS-box genes are typically found in flowering plant genomes [8,9]. The expansion and diversification of MADS-box genes in plants is closely linked to the evolution of novel structures, such as seeds, flowers, and fruits [5]. Consequently, MADS-box genes are key objects in studies of evolutionary developmental genetics in plants. Knowledge of the full complement of MADS-box genes along the plant lineage will enable a more profound comprehension of the evolution of the major land plant groups and the structures which define them.
MADS-box genes encode MADS-domain proteins. These are characterized by the highly conserved DNA-binding MADS domain which has a length of 56–60 amino acids [4]. Two types of MADS-box genes are distinguished throughout the eukaryotes, Type I and Type II [1,10]. Type II MADS-box genes of plants are also termed MIKC-type genes, referring to the typical domain structure of the encoded proteins. In MIKC-domain proteins, the MADS (M) domain is followed by an Intervening (I), a Keratin-like (K), and a C-terminal (C) domain [11]. The K domain is the second most conserved domain after the MADS domain. The two types of MADS-box genes are further subdivided into the groups Mα, Mβ, and Mγ (Type I), and MIKCC and MIKC* (Type II) based on phylogenetic and structural features [9,11]. More than a dozen ancient clades of MIKCC-group genes are distinguished by different motifs in their C-terminal regions [2].
While only a few Type I MADS-box genes have been functionally characterized [12,13,14,15], the Type II, especially the MIKCC-group MADS-box genes have been well studied (e.g., [4,16,17,18,19]). In flowering plants, their functions range from root development via floral organ specification to fruit development. Interestingly, genes with similar functions and similar expression patterns in different species are generally closely related and belong to the same clade of MADS-box genes [2,5,20]. Hence, identification and determination of the phylogenetic position of MADS-box genes in plant species allows the formulation of hypotheses about both, the morphological development of these species and the function of respective genes.
The complete set of MADS-box genes has been studied for the plants Arabidopsis thaliana, Glycine max, Cucumis sativus, Oryza sativa, Populus trichocarpa, Selaginella moellendorffii, and Physcomitrella patens [6,8,9,21,22,23,24]. Type II MADS-box genes have also been studied in Vitis vinifera [25]. As of June 2013, Phytozome offers access to the genomes of 28 additional land plant species (Figure 1). Many more whole-genome sequencing projects are ongoing.
Here, we use available whole-genome information to study the evolution of MADS-box genes in land plants. We have developed a pipeline which enables the fast annotation of MADS-box genes. Our pipeline first gathers MADS-box genes from gene prediction sets provided by the genome-sequencing projects. It then uses Hidden Markov Model (HMM) searches [26] of the whole genomes and the gene prediction programs GlimmerHMM [27] and FgenesH to identify MADS-box genes that have escaped the automatic annotation. Using our pipeline, we have newly identified 2,060 MADS-box genes in 17 plant species. Reanalyzing three, and using published datasets for four plant species, we analyzed a total of 2,603 MADS-box genes from 24 plant species. Approximately equal numbers of these genes were classified as Type I and Type II genes, respectively. Our phylogenies emphasize differences in the evolution of Type I and Type II genes and hence corroborate previous findings [28]. Our results will facilitate more in-depth studies on the mechanisms leading to these differences in evolutionary rates of the two types of MADS-box genes in flowering plants.

Figure 1.
Phylogeny of land plants for which whole-genome information is now available. Chlamydomonas reinhardtii and Volvox carteri are green algae and used as representatives of the outgroup. Species for which MADS-box genes are studied here are indicated by an asterisk. The topology of the tree was taken from [29].
2. Experimental Section
2.1. Sequence Data
MADS-box genes were analyzed in 24 plant genomes. Of these, MADS-box genes were newly predicted in 17 genomes, reanalyzed in three genomes (G.max, C. sativus, and V. vinifera) and taken from the literature for four genomes (A. thaliana, O. sativa, S. moellendorffii, and P. patens). To do so, whole-genome assemblies of all but the last four mentioned plant species were downloaded from the genome websites indicated in Table 1 or from Phytozome [29]. The coding sequences (CDS) for these species were retrieved from Phytozome.

Table 1.
List of some species and websites from which whole-genome data were retrieved.
| Species | Website |
|---|---|
| Ricinus communis | http://castorbean.jcvi.org/ |
| Medicago truncatula | http://medicagohapmap.org/ |
| Carica papaya | http://asgpb.mhpcc.hawaii.edu/papaya/ |
| Sorghum bicolor | http://genome.jgi-psf.org/Sorbi1/ |
| Brachypodium distachyon | http://www.brachypodium.org/ |
2.2. Annotation Pipeline
Customized perl scripts (available as Supplementary Data 1) were written to semi-automatically predict MADS-box genes in 17 plant genomes (Figure 2). Predicted CDS made available by the genome projects were translated into protein sequences and searched using the HMMer program [26] and a customized HMM of the MADS domain (Figure 3). This way, we aimed at identifying MADS‑box genes, which were already predicted by standard methods of the genome projects. The HMM is based on an alignment of exemplary MADS domains from all types and groups of MADS‑domain proteins from the species A. thaliana, O. sativa, P. trichocarpa, and Gnetum gnemon (Figure 3). The presence of the MADS domain in thereby detected sequences was confirmed by searches of the National Centre for Biotechnology Information (NCBI) conserved domains database (CDD) [30].
To detect additional MADS-box genes, which may have been missed during the automatic annotation procedure, whole-genome sequences were translated in all six reading frames. The translated genomes were again searched using the customized HMM of the MADS domain and presence of the MADS domain was counterchecked using the CDD. To annotate MADS-box genes encoding for the identified MADS domains, the corresponding genomic regions were retrieved including 300 bp upstream and 19,700 bp downstream of the start of the detected MADS box. Automatic gene predictions on these genome fragments were conducted using GlimmerHMM with the training sets of A. thaliana and O. sativa [27]. The thereby predicted genes were manually inspected and where gene predictions were short or the MADS- or K-domains were only partially included, improvements of gene predictions were attempted using FgenesH [31] with different training sets. The pipeline was tested using the genomes of A. thaliana and O. sativa where the full complements of MADS-box genes have been studied [8,9].
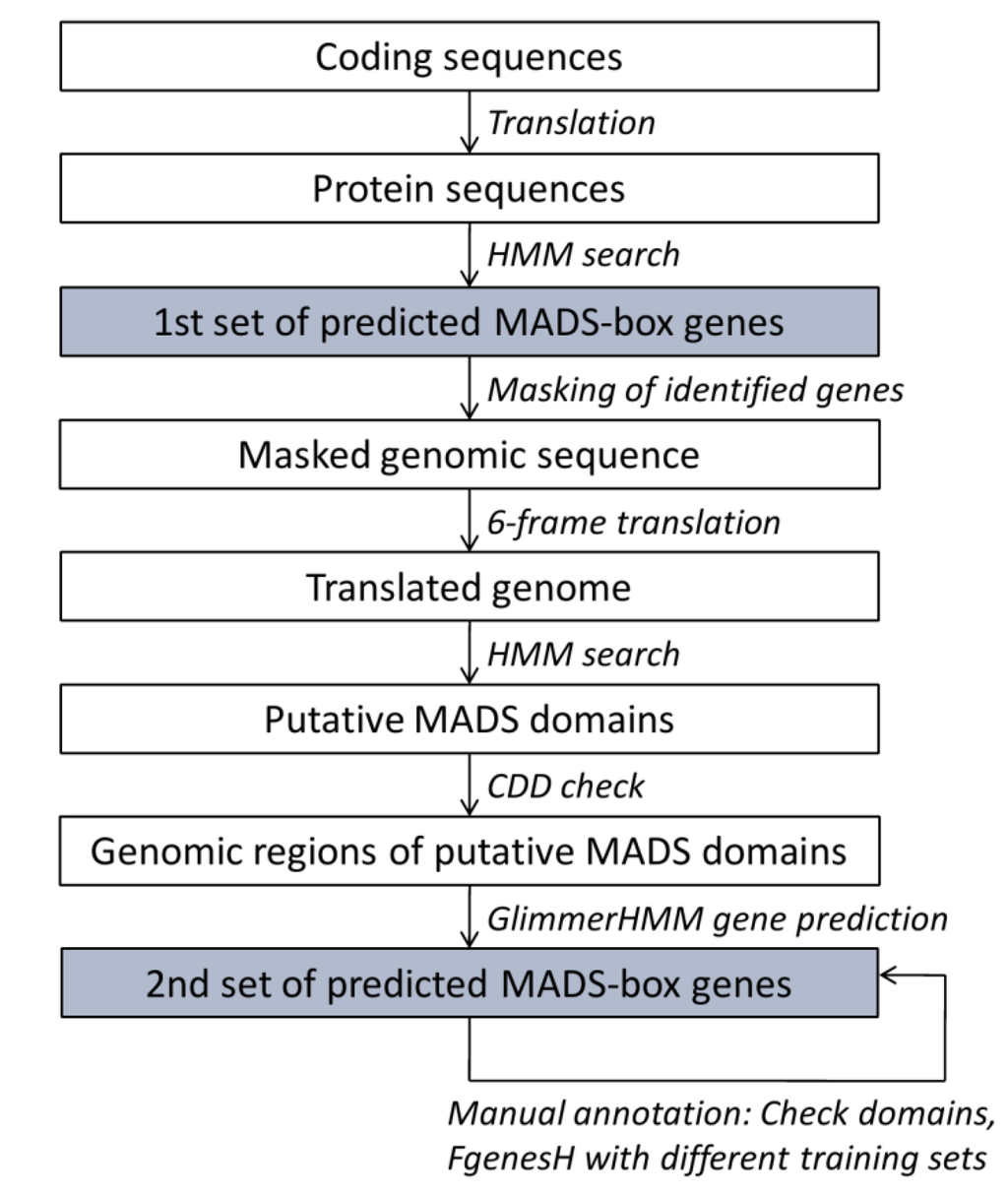
Figure 2.
Pipeline for annotation of MADS-box genes in plants. The two sets of predicted MADS-box genes are combined to obtain what is presumably the full complement of MADS-box genes in a species.
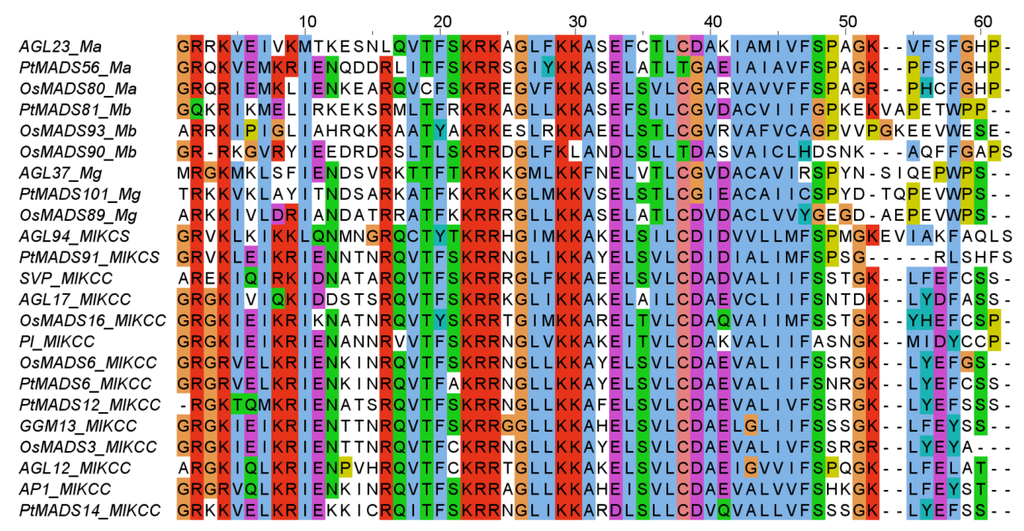
Figure 3.
Alignment of exemplary MADS domains from all types and groups of MADS‑domain proteins from the species Oryza sativa (OsMADS), Populus trichocarpa (PtMADS), Gnetum gnemon (GGM), and Arabidopsis thaliana (all other gene names). The labelling after the underscore denotes the group: Ma, Mα; Mb, Mβ; Mg, Mγ; MIKCS, MIKC*, and MIKCC, MIKCC. The alignment is colored according to the Clustal X color scheme [32].
2.3. Phylogeny Reconstructions
Identified MADS-domain sequences were aligned with known MADS domains of A. thaliana, O. sativa, P. trichocarpa, C. sativus, G. max, and V. vinifera using MAFFT [33]. An unrooted Maximum Likelihood tree was constructed using RAxML [34]. The different types of MADS-domain proteins were identified from the phylogeny based on the position of the characterized proteins of A. thaliana, P. trichocarpa, and O. sativa [8,9,21]. Separate phylogenies were then reconstructed for Type I and Type II genes, again using MAFFT for alignments and RAxML to obtain Maximum Likelihood phylogenies.
3. Results and Discussion
3.1. Identified MADS-Box Genes
We tested our pipeline using the genomes of A. thaliana and O. sativa. All but one and four of the known MADS-box genes of A. thaliana and O. sativa, respectively, were retrieved from their predicted CDS. In case of A. thaliana, five additional potential MADS-box genes have then been identified from the genome while, for O. sativa, one additional gene was detected.
From the 17 plant genomes we newly identified 2,060 MADS-box genes, of which 1,057 were classified as Type I and 990 were classified as Type II (Table 2; all identified as well as previously known MADS-box genes used in this study are available as Supplementary Data 2–4). The largest number of MADS-box genes was identified in S. tuberosum (265) of which 173 genes belong to Type I. This is the second highest number of Type I genes found in all species. The highest number of Type I genes was found in C. papaya where 229 of 262 MADS-box genes are Type I. Papaya was shown to have a comparably low number of genes, so the high number of MADS-box genes is surprising [35]. The highest number of Type II genes was identified in M. domestica with 113 Type II MADS-box genes. R. communis has the lowest number of MADS-box genes in the flowering plants studied at 60 genes. However, this number is still higher than the number of MADS-box genes previously identified in C. sativus with 50 genes [24]. Low numbers of Type I genes in flowering plants were observed in C. sativus (11 genes) and Z. mays (14 genes). The lowest number of Type II genes in a flowering plant was found in M. truncatula with 24 genes. Thus, the range of MADS-box genes in flowering plants is greater for Type I genes (11 to 229 genes) than for Type II genes (24 to 113 genes).
3.2. Phylogenies of MADS-Box Genes
We have reconstructed separate maximum likelihood phylogenies of the Type I and Type II MADS-box genes (Figure 4, Figure 5; Supplementary Figures 1 and 2). The newly identified genes were assigned to the different groups of MADS-box genes according to their position in the phylogenies relative to the previously classified genes of A. thaliana and O. sativa [8,9]. Unlike a previous classification, OsMADS90 and OsMADS91 fall into the Mα group rather than into the Mβ group in our phylogeny. Furthermore, OsMADS86 is adjacent to the Mβ and Mγ groups rather than in the Mγ group in our phylogeny. Apart from this, the three groups may well be monophyletic.
Table 2.
Total number of MADS-box genes in 24 plant species and classification into Type I and Type II genes. The complete set of MADS-box genes was previously known for P. trichocarpa, G. max, C. sativus, A. thaliana, O. sativa, S. moellendorffii, and P. patens (indicated by bold writing) [6,8,9,21,22,23,24]. * The remaining MADS-box genes could not be classified into Type I or Type II.
| Species [Genome reference] | Total | Type I | Type II |
|---|---|---|---|
| Manihot esculenta[36] | 85 | 28 | 57 |
| Ricinus communis [37] | 60 | 23 | 37 |
| Linum usitatissimum [38] | 123 | 45 | 78 |
| Populus trichocarpa [39] | 95 | 35 | 60 |
| Medicago truncatula [40] | 81 | 57 | 24 |
| Glycine max* [41] | 180 | 82 | 96 |
| Cucumis sativus [42] | 50 | 11 | 39 |
| Prunus persica [43] | 85 | 41 | 44 |
| Malus domestica [44] | 179 | 66 | 113 |
| Arabidopsis thaliana [45] | 105 | 59 | 46 |
| Arabidopsis lyrata [46] | 110 | 61 | 49 |
| Carica papaya * [35] | 262 | 229 | 30 |
| Citrus sinensis * [47] | 89 | 24 | 64 |
| Vitis vinifera * [48] | 90 | 44 | 44 |
| Solanum lycopersicum * [49] | 190 | 127 | 62 |
| Solanum tuberosum * [50] | 265 | 173 | 86 |
| Sorghum bicolor [51] | 67 | 24 | 43 |
| Zea mays [52] | 69 | 14 | 55 |
| Setaria italica [53] | 137 | 41 | 96 |
| Oryza sativa [54] | 71 | 29 | 42 |
| Brachypodium distachyon [55] | 75 | 35 | 40 |
| Musa accuminata [56] | 93 | 25 | 68 |
| Selaginella moellendorffii [57] | 19 | 13 | 6 |
| Physcomitrella patens [58] | 23 | 7 | 16 |
| Total* | 2,603 | 1,293 | 1,295 |
In the phylogeny of Type II genes, the group of MIKC* genes and the different groups of MIKCC‑group genes are well identifiable (Figure 5; Supplementary Figure 2). All of the previously defined groups may well be monophyletic also in our phylogeny. All of the groups contain a similar number of genes from nearly all species analyzed indicating that Type II MADS-box genes are rarely duplicated or lost or are about as frequently duplicated as lost after the establishment of the different groups.
Type I genes have been recognized to have faster birth-and-death rates than Type II genes before, when MADS-box genes in A. thaliana and O. sativa were studied [28]. The lineages that led to A. thaliana and O. sativa diverged around 150 million years ago [59]. Here, we show that, even when examining a larger number and more closely related species, still a large number of lineage-specific expansions can be seen for Type I genes. For example, when comparing the number of independent duplications in A. thaliana which are not found in A. lyrata and vice versa (Table 3), it becomes apparent that even within the last five million years after the divergence of these two Arabidopsis species [60], many duplications happened for Type I but not for Type II MADS-box genes.
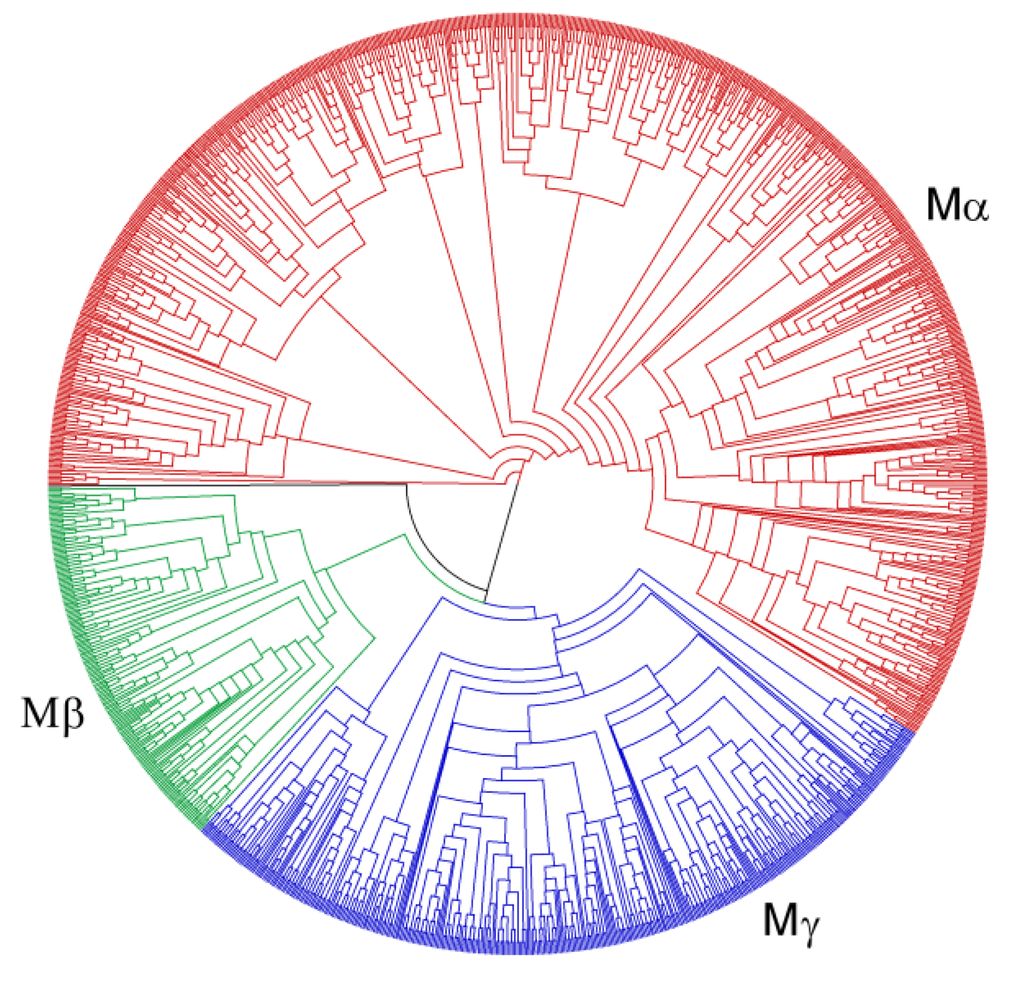
Figure 4.
Phylogeny of Type I MADS-box genes from 24 plant species including known genes from A. thaliana, P. trichocarpa, G. max, C. sativus, O. sativa, S. moellendorffii, and P. patens and newly annotated genes. Groups of Type I genes are indicated by different colours and their names are shown. Black indicates genes that could not be assigned unambiguously to one of the groups.
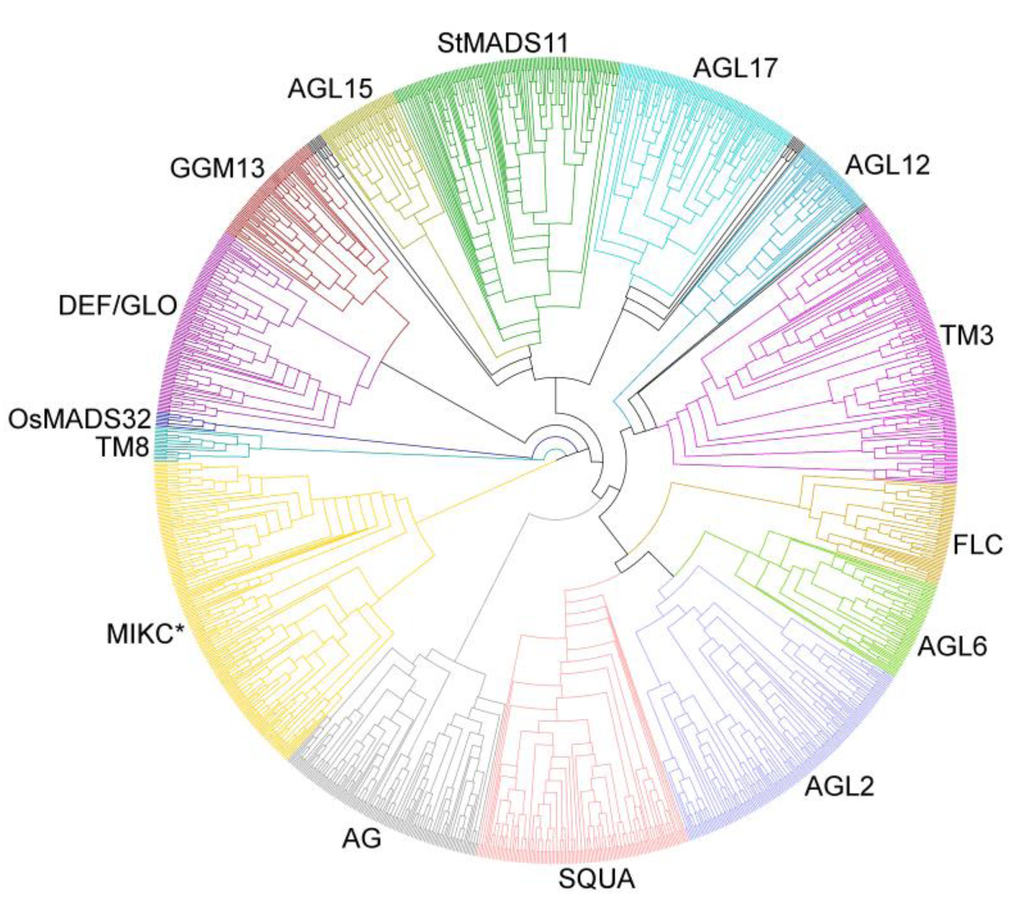
Figure 5.
Phylogeny of Type II MADS-box genes from 24 plant species including known genes from A. thaliana, P. trichocarpa, G. max, C. sativus, O. sativa, S. moellendorffii, and P. patens and newly annotated genes. Groups of Type II genes are indicated by different colours and their names are shown. Black indicates genes that could not be assigned unambiguously to one of the groups.
Table 3.
Number of independent duplications of Type I and Type II MADS-box genes in A. thaliana as compared to A. lyrata.
| Species | Type I | Type II |
|---|---|---|
| A. thaliana | 6 | 0 |
| A. lyrata | 9 | 2 |
3.3. Different Evolutionary Patterns of Type I and Type II MADS-Box Genes
Comparing the phylogenies of Type I and Type II MADS-box genes, it becomes apparent that there are more large species-specific groups of Type I genes than of Type II genes (Table 4). While there are 10 groups containing 10 or more genes from the same species for Type I genes, only three such groups exist for Type II genes. Also, the number of groups containing five to nine genes from the same species is larger for Type I genes at 27 groups as compared to eight groups for Type II genes. This bias is not due to a bias in gene number as the number of Type I (1,293) and Type II genes (1,295) is nearly identical in the 24 flowering plant species analyzed. Most prominent are expansions of Type I genes for C. papaya. There is one group comprised of 36 genes and another group of 58 genes from C. papaya. Why these genes have undergone such massive expansions in papaya in particular, is unknown. We did not include gymnosperm genomes in this study. However, the genome of Picea abies reveals that the situation might be different in gymnosperms where Type I MADS-box genes might be rare [61].
Table 4.
Number of species-specific groups of a certain size in our dataset for Type I and Type II genes.
| Size | Type I | Type II |
|---|---|---|
| ≥10 | 10 | 3 |
| 9 | 1 | 0 |
| 8 | 5 | 2 |
| 7 | 4 | 1 |
| 6 | 8 | 2 |
| 5 | 9 | 3 |
Which mechanisms lead to the higher rate of duplications or the higher rate of ‘short-term’ retention of Type I genes as compared to Type II genes, and to the different evolutionary dynamics of both gene types in general, is largely unknown.
According to the gene balance hypothesis genes that are more connected, e.g., due to protein-protein interactions of their gene products, are more likely retained after whole genome or large-scale duplications than after tandem or other small-scale duplications [62,63]. Type II MADS-domain proteins are probably involved in a higher number of complexes and in more complex multimers than Type I proteins [64,65], possibly due to the presence of the K domain that facilitates protein-protein interactions [66]. Therefore, Type II genes may have higher probabilities than Type I genes to be maintained after whole genome duplications and to be lost after small-scale duplications due to purifying selection that tries to keep genes in balance. Thus, the gene balance hypothesis might at least partially explain the differences in evolutionary patterns of Type I and Type II genes.
Secondly, there may be positive selection on the duplication and retention of certain genes after small scale duplications. This has been hypothesized for genes which are involved in coevolutionary scenarios, e.g., in some reproductive processes [67]. Compared to Type II genes Type I genes are less well studied, but the few Type I genes for which functions are known are involved in female gametophyte, embryo, and seed development and hence quite directly in reproduction [7,68]. There is evidence that some Type I genes in A. thaliana are involved in the development of interspecific barriers and hence may promote speciation [69]. Thus, the difference in the evolutionary patterns between Type I and Type II genes may also be explained by selection for diversity and neofunctionalization of Type I genes.
However, in addition to selection regimes also mutation patterns could be different for Type I and Type II genes. It has been hypothesized that Type I MADS-box genes have been highjacked by transposons, or have an intrinsic transposon activity [70], and, thus, might duplicate more often than ‘ordinary’ genes including Type II MADS-box genes. Furthermore, Type I genes are shorter and have less exons than Type II genes. Hence, the chance of Type I genes being duplicated as functional units might be higher than that of the larger Type II genes.
To find out more about the mutational mechanisms by which Type I genes are duplicated, we investigated the chromosomal position of the Type I genes with lineage-specific duplications in A. thaliana and A. lyrata. We found two tandem, three proximal, and ten distal duplications (as defined in [71]). The distal duplications might be caused by transposon activity. Hence, the investigated Type I duplications indicate both easy duplicatability and dispersal by transposon activity. Further studies are needed to gain more insights into the reasons for the different evolutionary rates of Type I and Type II genes.
4. Conclusions
Here we have identified the complete set of MADS-box genes in 17 plant species (some minor refinements in the future notwithstanding). We have analyzed more than 2,600 MADS-box genes and classified them into the known types. Our results corroborate a previous finding that Type I genes have a faster evolutionary rate than Type II genes and show that the number of duplications of Type I genes is high even in short time frames as can be recognized from the comparison of the closely related species A. thaliana and A. lyrata. Our study provides the basis for more elaborate studies on the striking differences in the evolutionary patterns of Type I and Type II genes and on the function of MADS-box genes in different plant species.
Acknowledgments
We would like to thank all members of the Theißen lab for stimulating discussions and Thomas Winterberg for his help in programming the pipeline. We are also grateful to two anonymous reviewers for helpful comments on a previous version of the manuscript and the Friedrich Schiller University Jena for general support.
Conflicts of Interest
The authors declare no conflict of interest
Supplementary Material
Figure 1. Phylogeny of Type I MADS-box genes from 24 plant species.
Figure 2. Phylogeny of Type II MADS-box genes from 24 plant species.
Data 1. pipeline.zip, Perl scripts to predict MADS-box genes in sequences plant genomes.
Data 2. TypeI.fa, Type I MADS-box genes in angiosperms as identified before and in this study.
Data 3. TypeII.fa, Type II MADS-box genes in angiosperms as identified before and in this study.
Data 4. unassigned.fa, MADS-box genes in angiosperms identified in this study which could not be assigned to Type I or Type II unambiguously.
References
- Gramzow, L.; Ritz, M.S.; Theissen, G. On the origin of MADS-domain transcription factors. Trends Genet. 2010, 26, 149–153. [Google Scholar] [CrossRef]
- Becker, A.; Theissen, G. The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol. Phylogenet. Evol. 2003, 29, 464–489. [Google Scholar] [CrossRef]
- Messenguy, F.; Dubois, E. Role of MADS box proteins and their cofactors in combinatorial control of gene expression and cell development. Gene 2003, 316, 1–21. [Google Scholar] [CrossRef]
- Schwarz-Sommer, Z.; Huijser, P.; Nacken, W.; Saedler, H.; Sommer, H. Genetic control of flower development by homeotic genes in Antirrhinum majus. Science 1990, 250, 931–936. [Google Scholar]
- Theissen, G.; Becker, A.; di Rosa, A.; Kanno, A.; Kim, J.T.; Munster, T.; Winter, K.U.; Saedler, H. A short history of MADS-box genes in plants. Plant Mol. Biol. 2000, 42, 115–149. [Google Scholar] [CrossRef]
- Gramzow, L.; Barker, E.; Schulz, C.; Ambrose, B.; Ashton, N.; Theissen, G.; Litt, A. Selaginella Genome Analysis—Entering the “Homoplasy Heaven” of the MADS World. Front. Plant Sci. 2012, 3, 214. [Google Scholar]
- Gramzow, L.; Theissen, G. A hitchhiker’s guide to the MADS world of plants. Genome Biol. 2010, 11, 214. [Google Scholar] [CrossRef]
- Arora, R.; Agarwal, P.; Ray, S.; Singh, A.K.; Singh, V.P.; Tyagi, A.K.; Kapoor, S. MADS-box gene family in rice: genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genomics 2007, 8, 242. [Google Scholar] [CrossRef]
- Parenicova, L.; de Folter, S.; Kieffer, M.; Horner, D.S.; Favalli, C.; Busscher, J.; Cook, H.E.; Ingram, R.M.; Kater, M.M.; Davies, B.; et al. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: new openings to the MADS world. Plant Cell 2003, 15, 1538–1551. [Google Scholar] [CrossRef]
- Alvarez-Buylla, E.R.; Pelaz, S.; Liljegren, S.J.; Gold, S.E.; Burgeff, C.; Ditta, G.S.; de Pouplana, L.R.; Martinez-Castilla, L.; Yanofsky, M.F. An ancestral MADS-box gene duplication occurred before the divergence of plants and animals. Proc. Natl. Acad. Sci. USA 2000, 97, 5328–5333. [Google Scholar] [CrossRef]
- Henschel, K.; Kofuji, R.; Hasebe, M.; Saedler, H.; Munster, T.; Theissen, G. Two ancient classes of MIKC-type MADS-box genes are present in the moss Physcomitrella patens. Mol. Biol. Evol. 2002, 19, 801–814. [Google Scholar] [CrossRef]
- Bemer, M.; Wolters-Arts, M.; Grossniklaus, U.; Angenent, G.C. The MADS domain protein DIANA acts together with AGAMOUS-LIKE80 to specify the central cell in Arabidopsis ovules. Plant Cell 2008, 20, 2088–2101. [Google Scholar] [CrossRef]
- Colombo, M.; Masiero, S.; Vanzulli, S.; Lardelli, P.; Kater, M.M.; Colombo, L. AGL23, a type I MADS-box gene that controls female gametophyte and embryo development in Arabidopsis. Plant J. 2008, 54, 1037–1048. [Google Scholar] [CrossRef]
- Kang, I.H.; Steffen, J.G.; Portereiko, M.F.; Lloyd, A.; Drews, G.N. The AGL62 MADS domain protein regulates cellularization during endosperm development in Arabidopsis. Plant Cell 2008, 20, 635–647. [Google Scholar] [CrossRef]
- Steffen, J.G.; Kang, I.H.; Portereiko, M.F.; Lloyd, A.; Drews, G.N. AGL61 interacts with AGL80 and is required for central cell development in Arabidopsis. Plant Physiol. 2008, 148, 259–268. [Google Scholar] [CrossRef]
- Mandel, M.A.; Gustafson-Brown, C.; Savidge, B.; Yanofsky, M.F. Molecular characterization of the Arabidopsis floral homeotic gene APETALA1. Nature 1992, 360, 273–277. [Google Scholar] [CrossRef]
- Pelaz, S.; Tapia-Lopez, R.; Alvarez-Buylla, E.R.; Yanofsky, M.F. Conversion of leaves into petals in Arabidopsis. Curr. Biol. 2001, 11, 182–184. [Google Scholar]
- Trobner, W.; Ramirez, L.; Motte, P.; Hue, I.; Huijser, P.; Lonnig, W.E.; Saedler, H.; Sommer, H.; Schwarz-Sommer, Z. GLOBOSA—A homeotic gene which interacts with DEFICIENS in the control of antirrhinum floral organogenesis. EMBO J. 1992, 11, 4693–4704. [Google Scholar]
- Yanofsky, M.F.; Ma, H.; Bowman, J.L.; Drews, G.N.; Feldmann, K.A.; Meyerowitz, E.M. The protein encoded by the Arabidopsis homeotic gene agamous resembles transcription factors. Nature 1990, 346, 35–39. [Google Scholar] [CrossRef]
- Theissen, G.; Kim, J.T.; Saedler, H. Classification and phylogeny of the MADS-box multigene family suggest defined roles of MADS-box gene subfamilies in the morphological evolution of eukaryotes. J. Mol. Evol. 1996, 43, 484–516. [Google Scholar] [CrossRef]
- Leseberg, C.H.; Li, A.; Kang, H.; Duvall, M.; Mao, L. Genome-wide analysis of the MADS-box gene family in Populus trichocarpa. Gene 2006, 378, 84–94. [Google Scholar] [CrossRef]
- Barker, E.I.; Ashton, N.W. A parsimonious model of lineage-specific expansion of MADS-box genes in Physcomitrella patens. Plant Cell Rep. 2013, 32, 1161–1177. [Google Scholar] [CrossRef]
- Shu, Y.; Yu, D.; Wang, D.; Guo, D.; Guo, C. Genome-wide survey and expression analysis of the MADS-box gene family in soybean. Mol. Biol. Rep. 2013, 40, 3901–3911. [Google Scholar] [CrossRef]
- Hu, L.; Liu, S. Genome-wide analysis of the MADS-box gene family in cucumber. Genome 2012, 55, 245–256. [Google Scholar] [CrossRef]
- Diaz-Riquelme, J.; Lijavetzky, D.; Martinez-Zapater, J.M.; Carmona, M.J. Genome-wide analysis of MIKCC-type MADS box genes in grapevine. Plant Physiol. 2009, 149, 354–369. [Google Scholar] [CrossRef]
- Eddy, S.R. Hidden Markov models. Curr. Opin. Struct. Biol. 1996, 6, 361–365. [Google Scholar] [CrossRef]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef]
- Nam, J.; Kim, J.; Lee, S.; An, G.; Ma, H.; Nei, M. Type I MADS-box genes have experienced faster birth-and-death evolution than type II MADS-box genes in angiosperms. Proc. Natl. Acad. Sci. USA 2004, 101, 1910–1915. [Google Scholar] [CrossRef]
- Phytozome. Available online: http://www.phytozome.net (accessed on 29 April 2013).
- Marchler-Bauer, A.; Anderson, J.B.; Derbyshire, M.K.; DeWeese-Scott, C.; Gonzales, N.R.; Gwadz, M.; Hao, L.N.; He, S.Q.; Hurwitz, D.I.; Jackson, J.D.; et al. CDD: A conserved domain database for interactive domain family analysis. Nucleic Acids Res. 2007, 35, D237–D240. [Google Scholar] [CrossRef]
- Salamov, A.A.; Solovyev, V.V. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 2000, 10, 516–522. [Google Scholar] [CrossRef]
- Chenna, R.; Sugawara, H.; Koike, T.; Lopez, R.; Gibson, T.J.; Higgins, D.G.; Thompson, J.D. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003, 31, 3497–3500. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef]
- Ming, R.; Hou, S.; Feng, Y.; Yu, Q.; Dionne-Laporte, A.; Saw, J.H.; Senin, P.; Wang, W.; Ly, B.V.; Lewis, K.L.; et al. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 2008, 452, 991–996. [Google Scholar] [CrossRef]
- Prochnik, S.; Marri, P.R.; Desany, B.; Rabinowicz, P.D.; Kodira, C.; Mohiuddin, M.; Rodriguez, F.; Fauquet, C.; Tohme, J.; Harkins, T.; et al. The Cassava Genome: Current Progress, Future Directions. Trop. Plant Biol. 2012, 5, 88–94. [Google Scholar] [CrossRef]
- Chan, A.P.; Crabtree, J.; Zhao, Q.; Lorenzi, H.; Orvis, J.; Puiu, D.; Melake-Berhan, A.; Jones, K.M.; Redman, J.; Chen, G.; et al. Draft genome sequence of the oilseed species Ricinus communis. Nat. Biotechnol. 2010, 28, 951–956. [Google Scholar] [CrossRef]
- Wang, Z.; Hobson, N.; Galindo, L.; Zhu, S.; Shi, D.; McDill, J.; Yang, L.; Hawkins, S.; Neutelings, G.; Datla, R.; et al. The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. Plant J. 2012, 72, 461–473. [Google Scholar] [CrossRef]
- Tuskan, G.A.; Difazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar] [CrossRef]
- Cannon, S.B.; Sterck, L.; Rombauts, S.; Sato, S.; Cheung, F.; Gouzy, J.; Wang, X.; Mudge, J.; Vasdewani, J.; Schiex, T.; et al. Legume genome evolution viewed through the Medicago truncatula and Lotus japonicus genomes. Proc. Natl. Acad. Sci. USA 2006, 103, 14959–14964. [Google Scholar] [CrossRef]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.X.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.J.; Thelen, J.J.; Cheng, J.L.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef]
- Huang, S.; Li, R.; Zhang, Z.; Li, L.; Gu, X.; Fan, W.; Lucas, W.J.; Wang, X.; Xie, B.; Ni, P.; et al. The genome of the cucumber, Cucumis sativus L. Nat. Genet. 2009, 41, 1275–1281. [Google Scholar] [CrossRef]
- Verde, I.; Abbott, A.G.; Scalabrin, S.; Jung, S.; Shu, S.; Marroni, F.; Zhebentyayeva, T.; Dettori, M.T.; Grimwood, J.; Cattonaro, F.; et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 2013, 45, 487–494. [Google Scholar] [CrossRef]
- Velasco, R.; Zharkikh, A.; Affourtit, J.; Dhingra, A.; Cestaro, A.; Kalyanaraman, A.; Fontana, P.; Bhatnagar, S.K.; Troggio, M.; Pruss, D.; et al. The genome of the domesticated apple (Malus x domestica Borkh.). Nat. Genet. 2010, 42, 833–839. [Google Scholar] [CrossRef]
- The Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796–815. [CrossRef]
- Hu, T.T.; Pattyn, P.; Bakker, E.G.; Cao, J.; Cheng, J.F.; Clark, R.M.; Fahlgren, N.; Fawcett, J.A.; Grimwood, J.; Gundlach, H.; et al. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat. Genet. 2011, 43, 476–481. [Google Scholar] [CrossRef]
- Xu, Q.; Chen, L.L.; Ruan, X.; Chen, D.; Zhu, A.; Chen, C.; Bertrand, D.; Jiao, W.B.; Hao, B.H.; Lyon, M.P.; et al. The draft genome of sweet orange (Citrus sinensis). Nat. Genet. 2013, 45, 59–66. [Google Scholar]
- Jaillon, O.; Aury, J.M.; Noel, B.; Policriti, A.; Clepet, C.; Casagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C.; et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449, 463–467. [Google Scholar] [CrossRef]
- Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 2012, 485, 635–641. [CrossRef]
- Xu, X.; Pan, S.; Cheng, S.; Zhang, B.; Mu, D.; Ni, P.; Zhang, G.; Yang, S.; Li, R.; Wang, J.; et al. Genome sequence and analysis of the tuber crop potato. Nature 2011, 475, 189–195. [Google Scholar] [CrossRef]
- Paterson, A.H.; Bowers, J.E.; Bruggmann, R.; Dubchak, I.; Grimwood, J.; Gundlach, H.; Haberer, G.; Hellsten, U.; Mitros, T.; Poliakov, A.; et al. The Sorghum bicolor genome and the diversification of grasses. Nature 2009, 457, 551–556. [Google Scholar] [CrossRef]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, X.; Quan, Z.; Cheng, S.; Xu, X.; Pan, S.; Xie, M.; Zeng, P.; Yue, Z.; Wang, W.; et al. Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat. Biotechnol. 2012, 30, 549–554. [Google Scholar] [CrossRef]
- Goff, S.A.; Ricke, D.; Lan, T.H.; Presting, G.; Wang, R.L.; Dunn, M.; Glazebrook, J.; Sessions, A.; Oeller, P.; Varma, H.; et al. A draft sequence of the rice genome (Oryza sativa L. ssp japonica). Science 2000, 296, 92–100. [Google Scholar]
- International Brachypodium Initiative. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 2010, 463, 763–768. [CrossRef]
- D'Hont, A.; Denoeud, F.; Aury, J.M.; Baurens, F.C.; Carreel, F.; Garsmeur, O.; Noel, B.; Bocs, S.; Droc, G.; Rouard, M.; et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 2012, 488, 213–217. [Google Scholar] [CrossRef]
- Banks, J.A.; Nishiyama, T.; Hasebe, M.; Bowman, J.L.; Gribskov, M.; dePamphilis, C.; Albert, V.A.; Aono, N.; Aoyama, T.; Ambrose, B.A.; et al. The Selaginella genome identifies genetic changes associated with the evolution of vascular plants. Science 2011, 332, 960–963. [Google Scholar] [CrossRef]
- Rensing, S.A.; Lang, D.; Zimmer, A.D.; Terry, A.; Salamov, A.; Shapiro, H.; Nishiyama, T.; Perroud, P.F.; Lindquist, E.A.; Kamisugi, Y.; et al. The Physcomitrella genome reveals evolutionary insights into the conquest of land by plants. Science 2008, 319, 64–69. [Google Scholar] [CrossRef]
- Fawcett, J.A.; Maere, S.; van de Peer, Y. Plants with double genomes might have had a better chance to survive the Cretaceous-Tertiary extinction event. Proc. Natl. Acad. Sci. USA 2009, 106, 5737–5742. [Google Scholar] [CrossRef]
- Koch, M.A.; Haubold, B.; Mitchell-Olds, T. Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis, and related genera (Brassicaceae). Mol. Biol. Evol. 2000, 17, 1483–1498. [Google Scholar] [CrossRef]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef]
- Freeling, M. Bias in plant gene content following different sorts of duplication: Tandem, whole-genome, segmental, or by transposition. Annu. Rev. Plant Biol. 2009, 60, 433–453. [Google Scholar] [CrossRef]
- Edger, P.P.; Pires, J.C. Gene and genome duplications: The impact of dosage-sensitivity on the fate of nuclear genes. Chromosome Res. 2009, 17, 699–717. [Google Scholar] [CrossRef]
- Melzer, R.; Theissen, G. Reconstitution of 'floral quartets’ in vitro involving class B and class E floral homeotic proteins. Nucleic Acids Res. 2009, 37, 2723–2736. [Google Scholar] [CrossRef]
- De Folter, S.; Immink, R.G.; Kieffer, M.; Parenicova, L.; Henz, S.R.; Weigel, D.; Busscher, M.; Kooiker, M.; Colombo, L.; Kater, M.M.; et al. Comprehensive interaction map of the Arabidopsis MADS Box transcription factors. Plant Cell 2005, 17, 1424–1433. [Google Scholar] [CrossRef]
- Kaufmann, K.; Melzer, R.; Theissen, G. MIKC-type MADS-domain proteins: Structural modularity, protein interactions and network evolution in land plants. Gene 2005, 347, 183–198. [Google Scholar] [CrossRef]
- Freeling, M.; Lyons, E.; Pedersen, B.; Alam, M.; Ming, R.; Lisch, D. Many or most genes in Arabidopsis transposed after the origin of the order Brassicales. Genome Res. 2008, 18, 1924–1937. [Google Scholar] [CrossRef]
- Masiero, S.; Colombo, L.; Grini, P.E.; Schnittger, A.; Kater, M.M. The emerging importance of type I MADS box transcription factors for plant reproduction. Plant Cell 2011, 23, 865–872. [Google Scholar] [CrossRef]
- Walia, H.; Josefsson, C.; Dilkes, B.; Kirkbride, R.; Harada, J.; Comai, L. Dosage-dependent deregulation of an AGAMOUS-LIKE gene cluster contributes to interspecific incompatibility. Curr. Biol. 2009, 19, 1128–1132. [Google Scholar] [CrossRef]
- De Bodt, S.; Raes, J.; Van de Peer, Y.; Theissen, G. And then there were many: MADS goes genomic. Trends Plant Sci. 2003, 8, 475–483. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Tang, H.; Tan, X.; Ficklin, S.P.; Feltus, F.A.; Paterson, A.H. Modes of gene duplication contribute differently to genetic novelty and redundancy, but show parallels across divergent angiosperms. PLoS One 2011, 6, e28150. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).