Phylogenomics of MADS-Box Genes in Plants — Two Opposing Life Styles in One Gene Family

Abstract
:1. Introduction

2. Experimental Section
2.1. Sequence Data

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Website |
|---|---|
| Ricinus communis | http://castorbean.jcvi.org/ |
| Medicago truncatula | http://medicagohapmap.org/ |
| Carica papaya | http://asgpb.mhpcc.hawaii.edu/papaya/ |
| Sorghum bicolor | http://genome.jgi-psf.org/Sorbi1/ |
| Brachypodium distachyon | http://www.brachypodium.org/ |
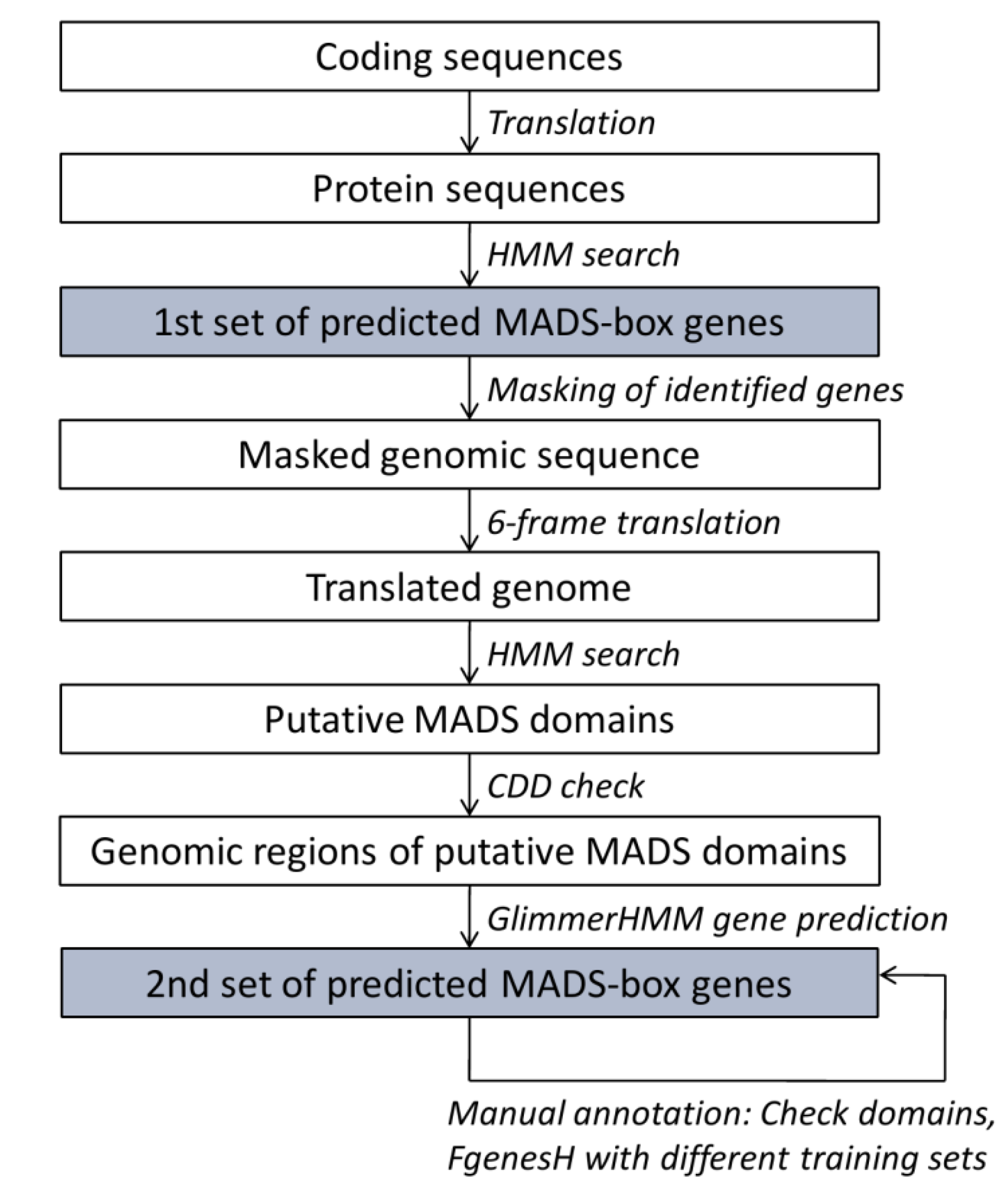
2.2. Annotation Pipeline
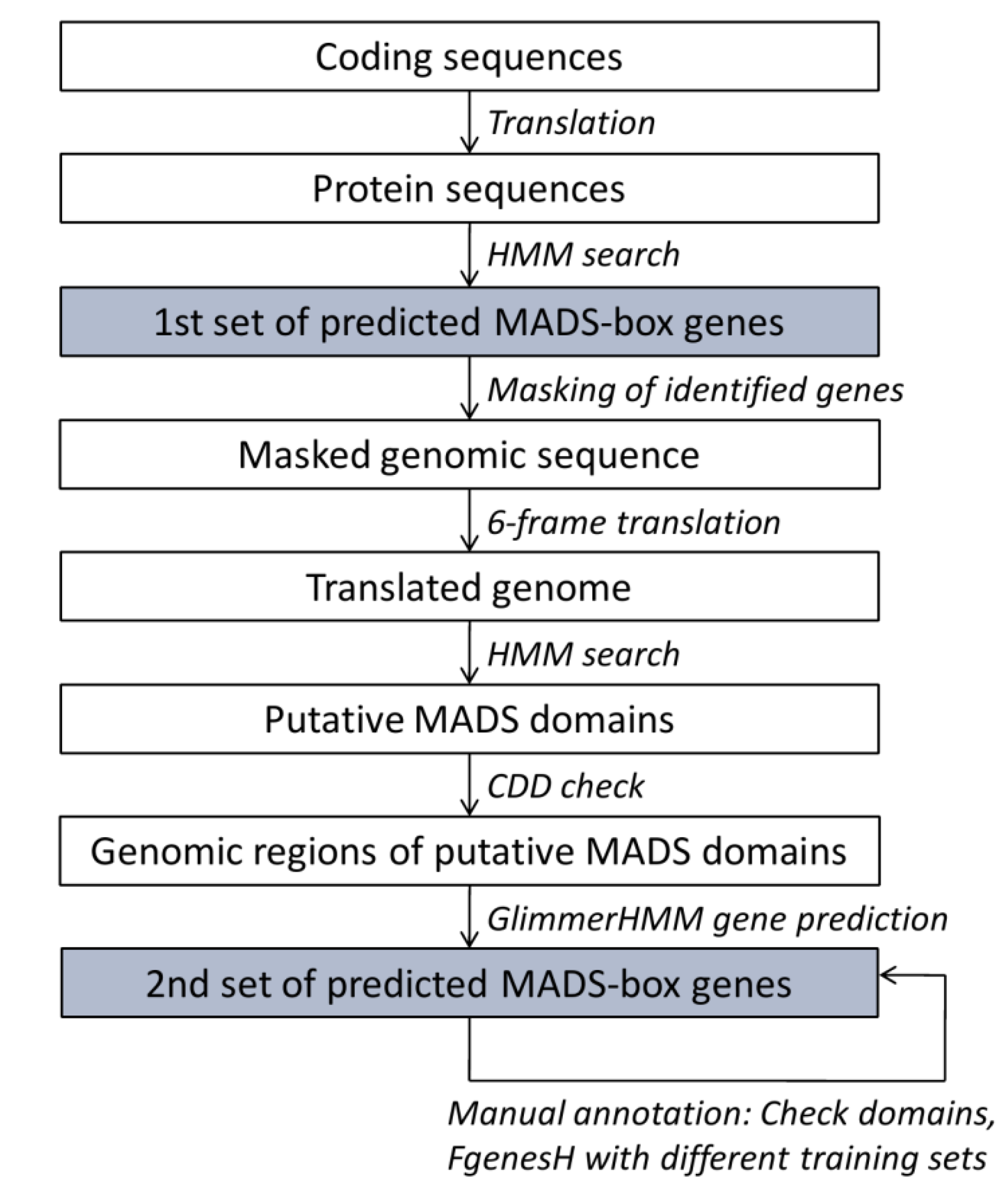
2.3. Phylogeny Reconstructions
3. Results and Discussion
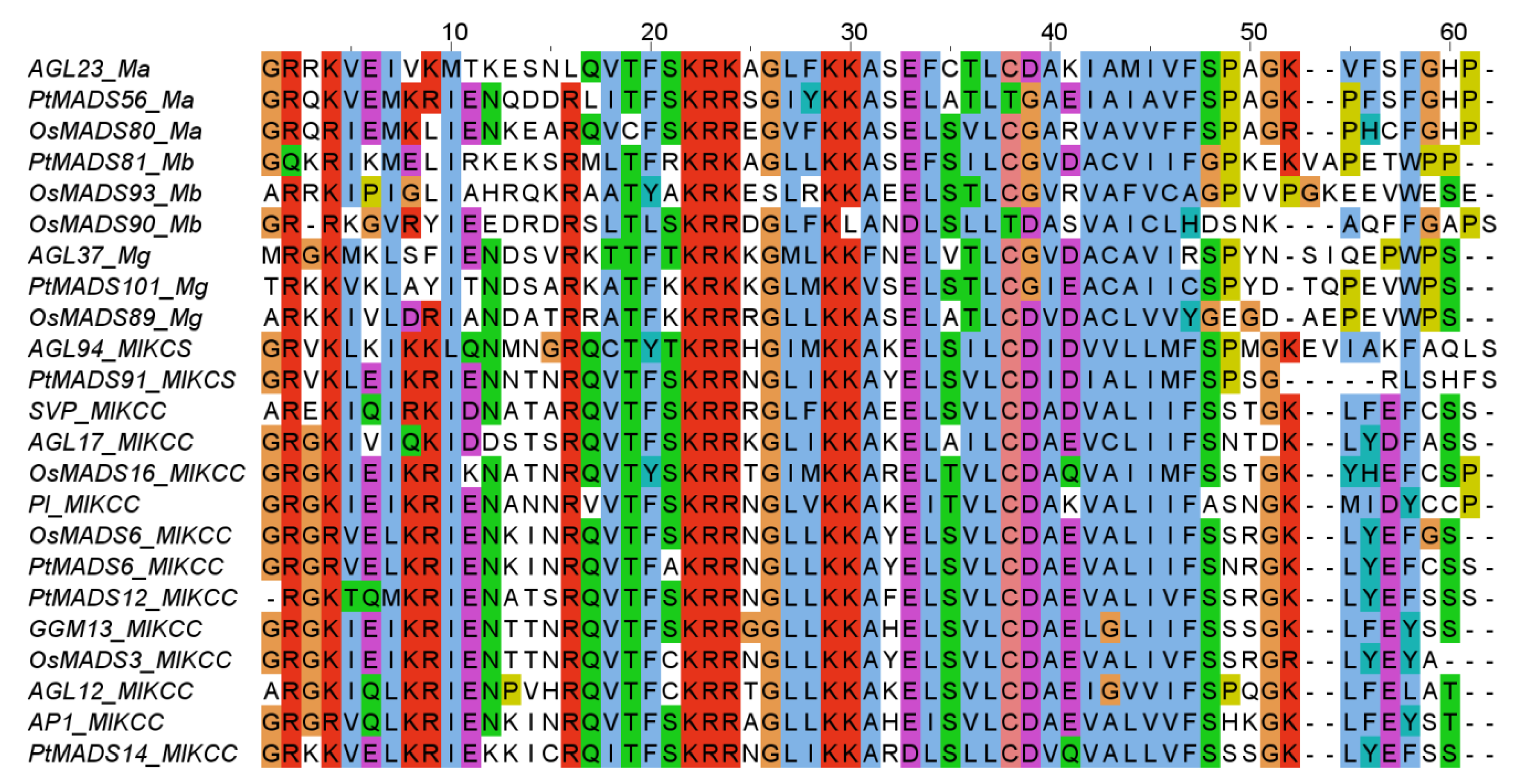
3.1. Identified MADS-Box Genes
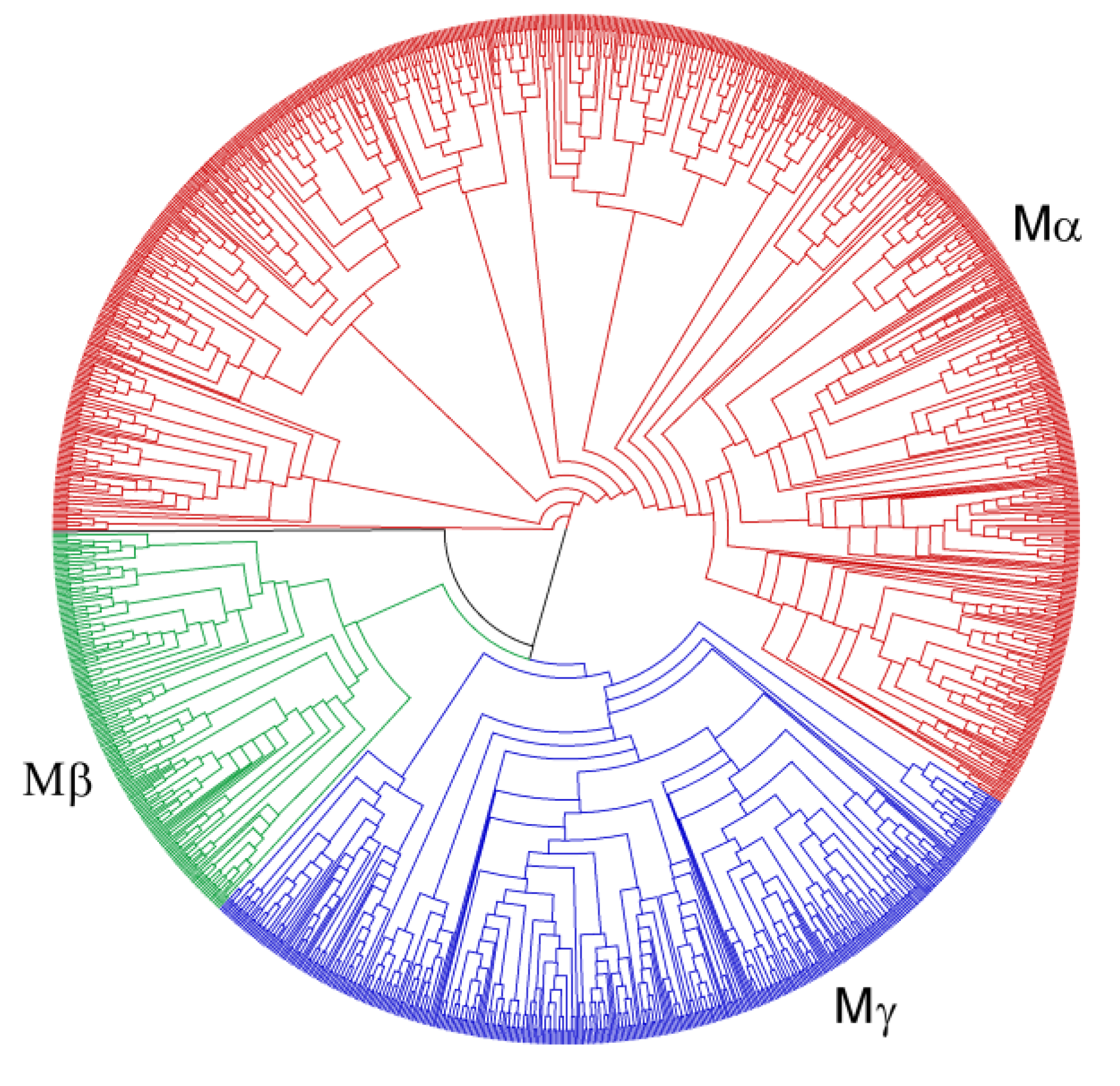
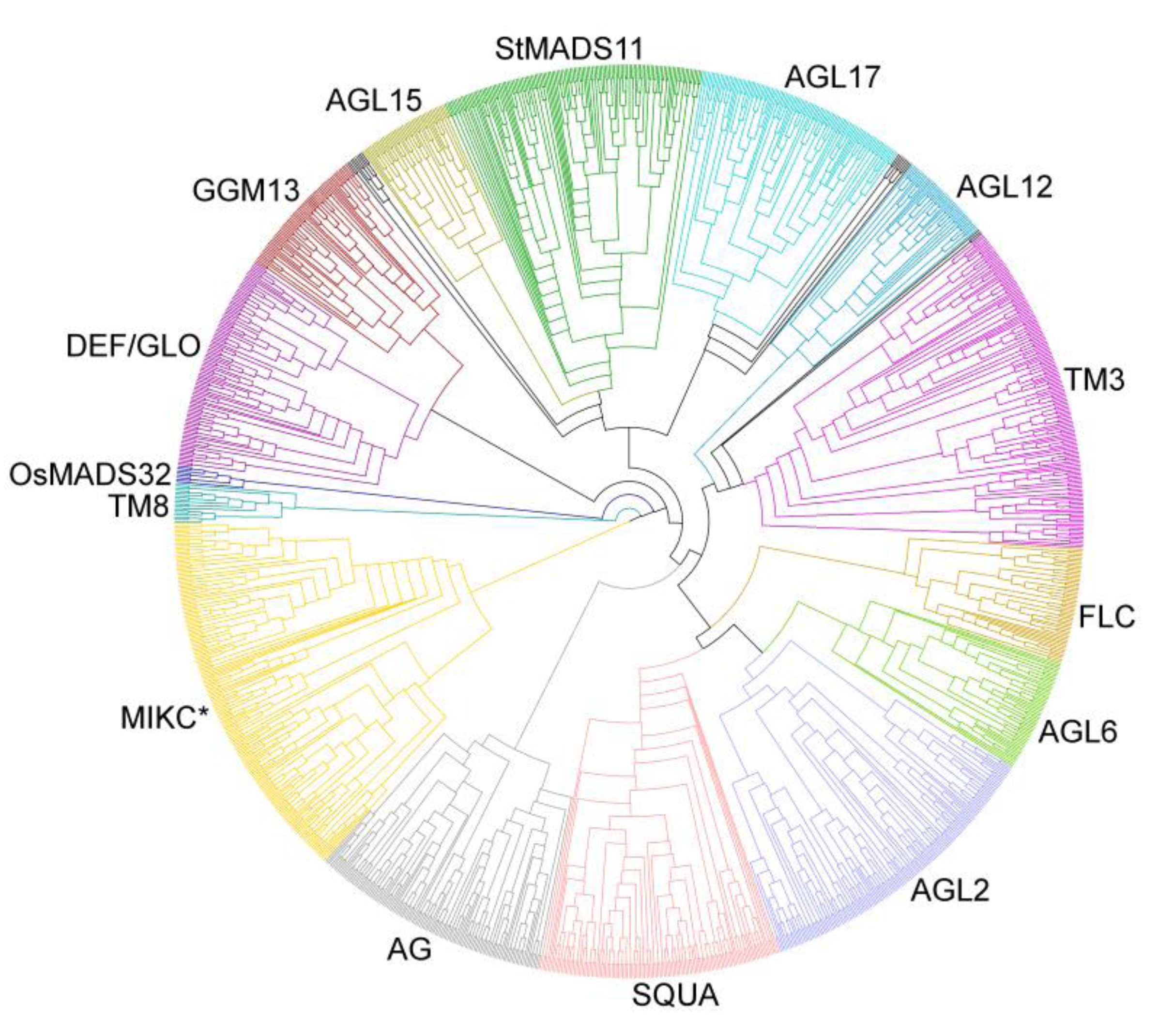
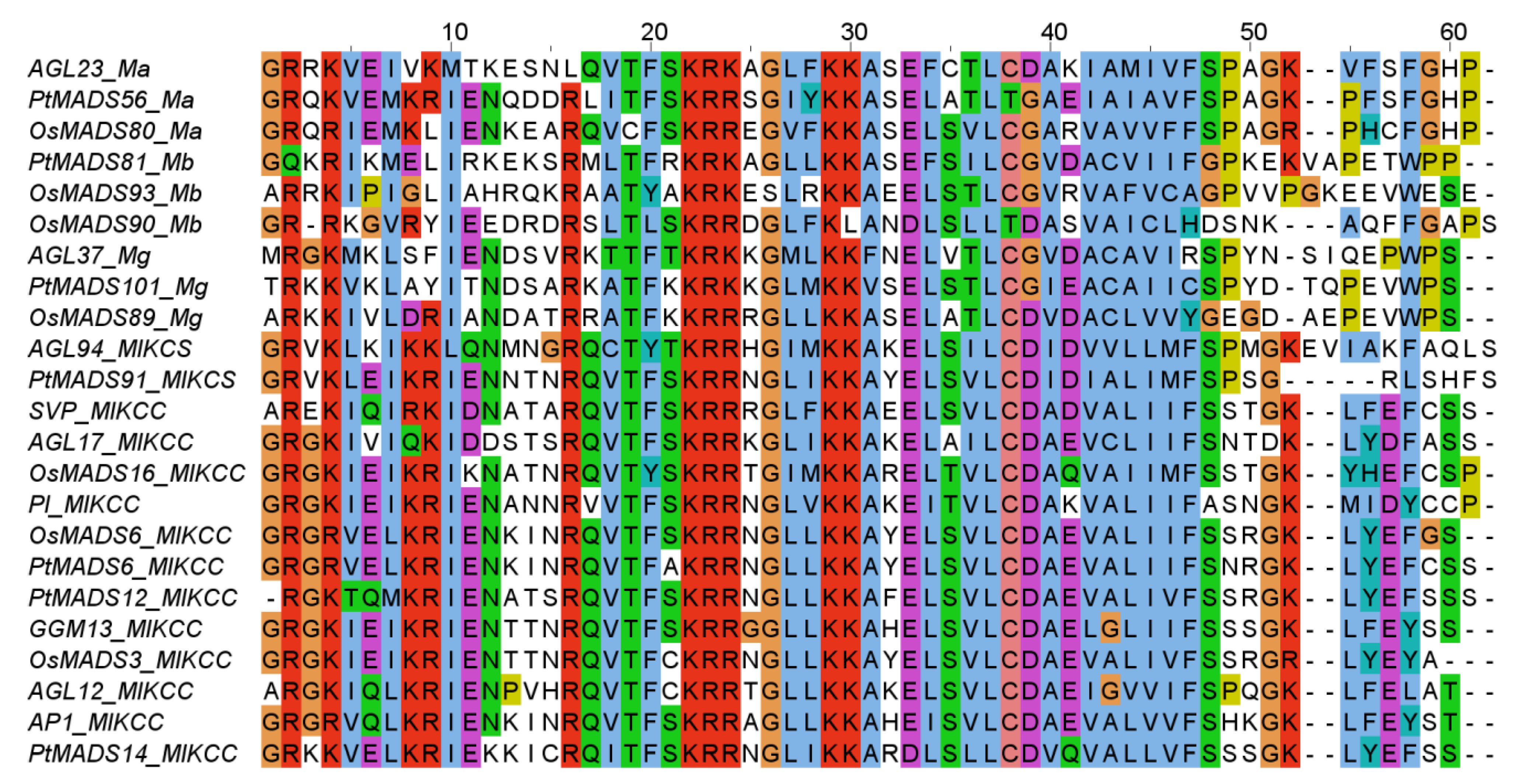
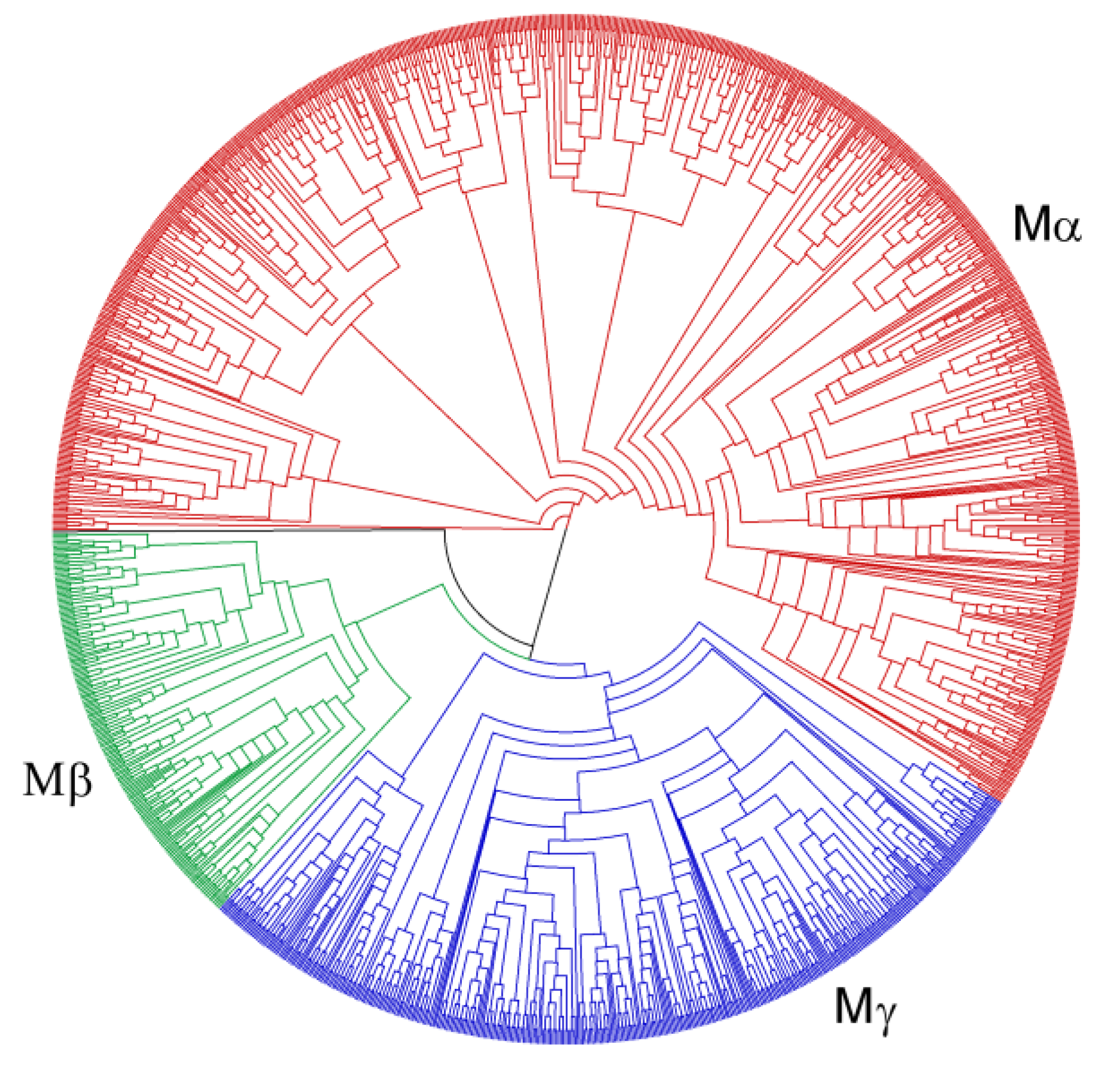
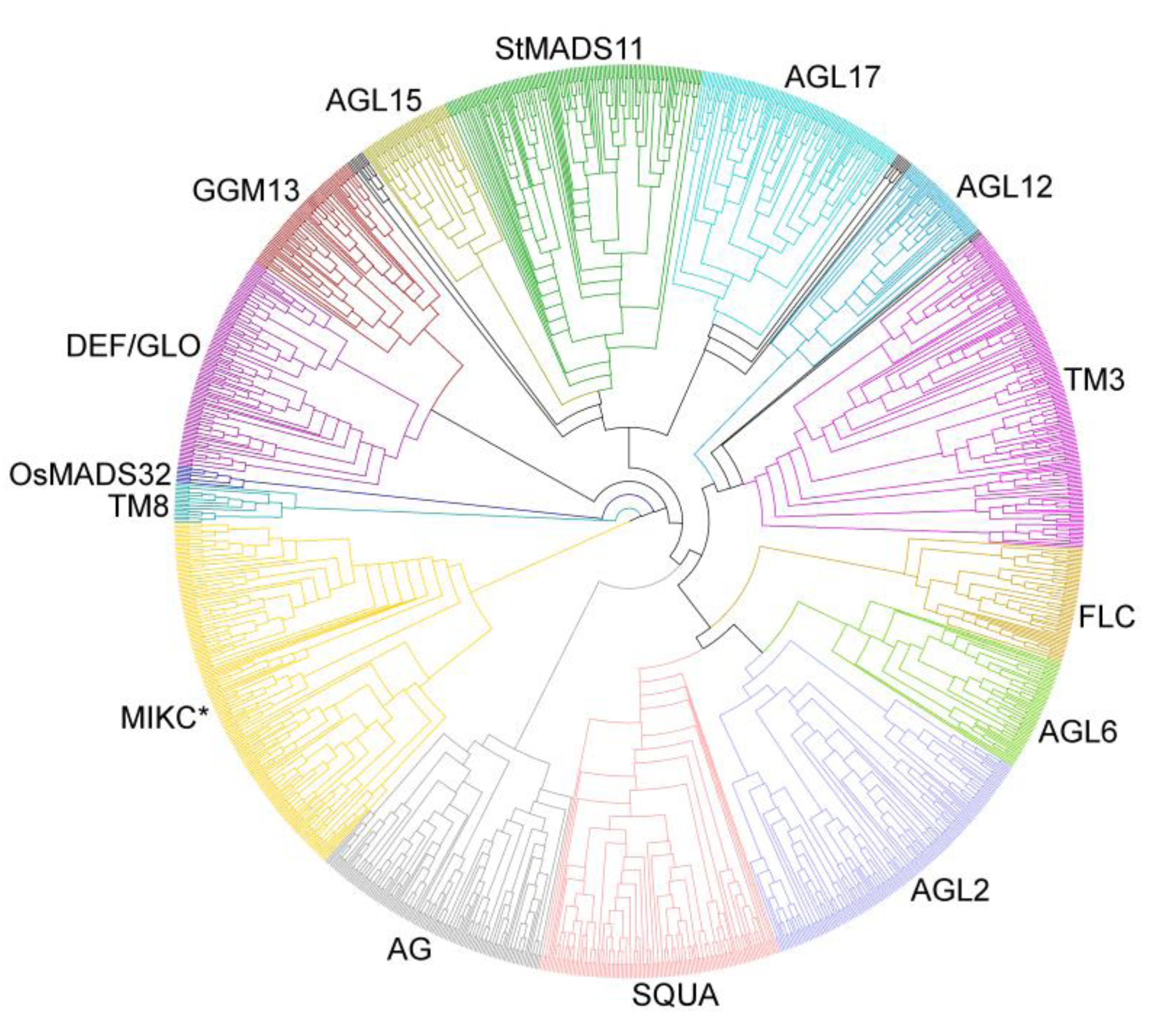
3.2. Phylogenies of MADS-Box Genes
| Species [Genome reference] | Total | Type I | Type II |
|---|---|---|---|
| Manihot esculenta[36] | 85 | 28 | 57 |
| Ricinus communis [37] | 60 | 23 | 37 |
| Linum usitatissimum [38] | 123 | 45 | 78 |
| Populus trichocarpa [39] | 95 | 35 | 60 |
| Medicago truncatula [40] | 81 | 57 | 24 |
| Glycine max* [41] | 180 | 82 | 96 |
| Cucumis sativus [42] | 50 | 11 | 39 |
| Prunus persica [43] | 85 | 41 | 44 |
| Malus domestica [44] | 179 | 66 | 113 |
| Arabidopsis thaliana [45] | 105 | 59 | 46 |
| Arabidopsis lyrata [46] | 110 | 61 | 49 |
| Carica papaya * [35] | 262 | 229 | 30 |
| Citrus sinensis * [47] | 89 | 24 | 64 |
| Vitis vinifera * [48] | 90 | 44 | 44 |
| Solanum lycopersicum * [49] | 190 | 127 | 62 |
| Solanum tuberosum * [50] | 265 | 173 | 86 |
| Sorghum bicolor [51] | 67 | 24 | 43 |
| Zea mays [52] | 69 | 14 | 55 |
| Setaria italica [53] | 137 | 41 | 96 |
| Oryza sativa [54] | 71 | 29 | 42 |
| Brachypodium distachyon [55] | 75 | 35 | 40 |
| Musa accuminata [56] | 93 | 25 | 68 |
| Selaginella moellendorffii [57] | 19 | 13 | 6 |
| Physcomitrella patens [58] | 23 | 7 | 16 |
| Total* | 2,603 | 1,293 | 1,295 |
| Species | Type I | Type II |
|---|---|---|
| A. thaliana | 6 | 0 |
| A. lyrata | 9 | 2 |
3.3. Different Evolutionary Patterns of Type I and Type II MADS-Box Genes
| Size | Type I | Type II |
|---|---|---|
| ≥10 | 10 | 3 |
| 9 | 1 | 0 |
| 8 | 5 | 2 |
| 7 | 4 | 1 |
| 6 | 8 | 2 |
| 5 | 9 | 3 |
4. Conclusions
Acknowledgments
Conflicts of Interest
Supplementary Material
References
- Gramzow, L.; Ritz, M.S.; Theissen, G. On the origin of MADS-domain transcription factors. Trends Genet. 2010, 26, 149–153. [Google Scholar] [CrossRef]
- Becker, A.; Theissen, G. The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol. Phylogenet. Evol. 2003, 29, 464–489. [Google Scholar] [CrossRef]
- Messenguy, F.; Dubois, E. Role of MADS box proteins and their cofactors in combinatorial control of gene expression and cell development. Gene 2003, 316, 1–21. [Google Scholar] [CrossRef]
- Schwarz-Sommer, Z.; Huijser, P.; Nacken, W.; Saedler, H.; Sommer, H. Genetic control of flower development by homeotic genes in Antirrhinum majus. Science 1990, 250, 931–936. [Google Scholar]
- Theissen, G.; Becker, A.; di Rosa, A.; Kanno, A.; Kim, J.T.; Munster, T.; Winter, K.U.; Saedler, H. A short history of MADS-box genes in plants. Plant Mol. Biol. 2000, 42, 115–149. [Google Scholar] [CrossRef]
- Gramzow, L.; Barker, E.; Schulz, C.; Ambrose, B.; Ashton, N.; Theissen, G.; Litt, A. Selaginella Genome Analysis—Entering the “Homoplasy Heaven” of the MADS World. Front. Plant Sci. 2012, 3, 214. [Google Scholar]
- Gramzow, L.; Theissen, G. A hitchhiker’s guide to the MADS world of plants. Genome Biol. 2010, 11, 214. [Google Scholar] [CrossRef]
- Arora, R.; Agarwal, P.; Ray, S.; Singh, A.K.; Singh, V.P.; Tyagi, A.K.; Kapoor, S. MADS-box gene family in rice: genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genomics 2007, 8, 242. [Google Scholar] [CrossRef]
- Parenicova, L.; de Folter, S.; Kieffer, M.; Horner, D.S.; Favalli, C.; Busscher, J.; Cook, H.E.; Ingram, R.M.; Kater, M.M.; Davies, B.; et al. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: new openings to the MADS world. Plant Cell 2003, 15, 1538–1551. [Google Scholar] [CrossRef]
- Alvarez-Buylla, E.R.; Pelaz, S.; Liljegren, S.J.; Gold, S.E.; Burgeff, C.; Ditta, G.S.; de Pouplana, L.R.; Martinez-Castilla, L.; Yanofsky, M.F. An ancestral MADS-box gene duplication occurred before the divergence of plants and animals. Proc. Natl. Acad. Sci. USA 2000, 97, 5328–5333. [Google Scholar] [CrossRef]
- Henschel, K.; Kofuji, R.; Hasebe, M.; Saedler, H.; Munster, T.; Theissen, G. Two ancient classes of MIKC-type MADS-box genes are present in the moss Physcomitrella patens. Mol. Biol. Evol. 2002, 19, 801–814. [Google Scholar] [CrossRef]
- Bemer, M.; Wolters-Arts, M.; Grossniklaus, U.; Angenent, G.C. The MADS domain protein DIANA acts together with AGAMOUS-LIKE80 to specify the central cell in Arabidopsis ovules. Plant Cell 2008, 20, 2088–2101. [Google Scholar] [CrossRef]
- Colombo, M.; Masiero, S.; Vanzulli, S.; Lardelli, P.; Kater, M.M.; Colombo, L. AGL23, a type I MADS-box gene that controls female gametophyte and embryo development in Arabidopsis. Plant J. 2008, 54, 1037–1048. [Google Scholar] [CrossRef]
- Kang, I.H.; Steffen, J.G.; Portereiko, M.F.; Lloyd, A.; Drews, G.N. The AGL62 MADS domain protein regulates cellularization during endosperm development in Arabidopsis. Plant Cell 2008, 20, 635–647. [Google Scholar] [CrossRef]
- Steffen, J.G.; Kang, I.H.; Portereiko, M.F.; Lloyd, A.; Drews, G.N. AGL61 interacts with AGL80 and is required for central cell development in Arabidopsis. Plant Physiol. 2008, 148, 259–268. [Google Scholar] [CrossRef]
- Mandel, M.A.; Gustafson-Brown, C.; Savidge, B.; Yanofsky, M.F. Molecular characterization of the Arabidopsis floral homeotic gene APETALA1. Nature 1992, 360, 273–277. [Google Scholar] [CrossRef]
- Pelaz, S.; Tapia-Lopez, R.; Alvarez-Buylla, E.R.; Yanofsky, M.F. Conversion of leaves into petals in Arabidopsis. Curr. Biol. 2001, 11, 182–184. [Google Scholar]
- Trobner, W.; Ramirez, L.; Motte, P.; Hue, I.; Huijser, P.; Lonnig, W.E.; Saedler, H.; Sommer, H.; Schwarz-Sommer, Z. GLOBOSA—A homeotic gene which interacts with DEFICIENS in the control of antirrhinum floral organogenesis. EMBO J. 1992, 11, 4693–4704. [Google Scholar]
- Yanofsky, M.F.; Ma, H.; Bowman, J.L.; Drews, G.N.; Feldmann, K.A.; Meyerowitz, E.M. The protein encoded by the Arabidopsis homeotic gene agamous resembles transcription factors. Nature 1990, 346, 35–39. [Google Scholar] [CrossRef]
- Theissen, G.; Kim, J.T.; Saedler, H. Classification and phylogeny of the MADS-box multigene family suggest defined roles of MADS-box gene subfamilies in the morphological evolution of eukaryotes. J. Mol. Evol. 1996, 43, 484–516. [Google Scholar] [CrossRef]
- Leseberg, C.H.; Li, A.; Kang, H.; Duvall, M.; Mao, L. Genome-wide analysis of the MADS-box gene family in Populus trichocarpa. Gene 2006, 378, 84–94. [Google Scholar] [CrossRef]
- Barker, E.I.; Ashton, N.W. A parsimonious model of lineage-specific expansion of MADS-box genes in Physcomitrella patens. Plant Cell Rep. 2013, 32, 1161–1177. [Google Scholar] [CrossRef]
- Shu, Y.; Yu, D.; Wang, D.; Guo, D.; Guo, C. Genome-wide survey and expression analysis of the MADS-box gene family in soybean. Mol. Biol. Rep. 2013, 40, 3901–3911. [Google Scholar] [CrossRef]
- Hu, L.; Liu, S. Genome-wide analysis of the MADS-box gene family in cucumber. Genome 2012, 55, 245–256. [Google Scholar] [CrossRef]
- Diaz-Riquelme, J.; Lijavetzky, D.; Martinez-Zapater, J.M.; Carmona, M.J. Genome-wide analysis of MIKCC-type MADS box genes in grapevine. Plant Physiol. 2009, 149, 354–369. [Google Scholar] [CrossRef]
- Eddy, S.R. Hidden Markov models. Curr. Opin. Struct. Biol. 1996, 6, 361–365. [Google Scholar] [CrossRef]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef]
- Nam, J.; Kim, J.; Lee, S.; An, G.; Ma, H.; Nei, M. Type I MADS-box genes have experienced faster birth-and-death evolution than type II MADS-box genes in angiosperms. Proc. Natl. Acad. Sci. USA 2004, 101, 1910–1915. [Google Scholar] [CrossRef]
- Phytozome. Available online: http://www.phytozome.net (accessed on 29 April 2013).
- Marchler-Bauer, A.; Anderson, J.B.; Derbyshire, M.K.; DeWeese-Scott, C.; Gonzales, N.R.; Gwadz, M.; Hao, L.N.; He, S.Q.; Hurwitz, D.I.; Jackson, J.D.; et al. CDD: A conserved domain database for interactive domain family analysis. Nucleic Acids Res. 2007, 35, D237–D240. [Google Scholar] [CrossRef]
- Salamov, A.A.; Solovyev, V.V. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 2000, 10, 516–522. [Google Scholar] [CrossRef]
- Chenna, R.; Sugawara, H.; Koike, T.; Lopez, R.; Gibson, T.J.; Higgins, D.G.; Thompson, J.D. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003, 31, 3497–3500. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef]
- Ming, R.; Hou, S.; Feng, Y.; Yu, Q.; Dionne-Laporte, A.; Saw, J.H.; Senin, P.; Wang, W.; Ly, B.V.; Lewis, K.L.; et al. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 2008, 452, 991–996. [Google Scholar] [CrossRef]
- Prochnik, S.; Marri, P.R.; Desany, B.; Rabinowicz, P.D.; Kodira, C.; Mohiuddin, M.; Rodriguez, F.; Fauquet, C.; Tohme, J.; Harkins, T.; et al. The Cassava Genome: Current Progress, Future Directions. Trop. Plant Biol. 2012, 5, 88–94. [Google Scholar] [CrossRef]
- Chan, A.P.; Crabtree, J.; Zhao, Q.; Lorenzi, H.; Orvis, J.; Puiu, D.; Melake-Berhan, A.; Jones, K.M.; Redman, J.; Chen, G.; et al. Draft genome sequence of the oilseed species Ricinus communis. Nat. Biotechnol. 2010, 28, 951–956. [Google Scholar] [CrossRef]
- Wang, Z.; Hobson, N.; Galindo, L.; Zhu, S.; Shi, D.; McDill, J.; Yang, L.; Hawkins, S.; Neutelings, G.; Datla, R.; et al. The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. Plant J. 2012, 72, 461–473. [Google Scholar] [CrossRef]
- Tuskan, G.A.; Difazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar] [CrossRef]
- Cannon, S.B.; Sterck, L.; Rombauts, S.; Sato, S.; Cheung, F.; Gouzy, J.; Wang, X.; Mudge, J.; Vasdewani, J.; Schiex, T.; et al. Legume genome evolution viewed through the Medicago truncatula and Lotus japonicus genomes. Proc. Natl. Acad. Sci. USA 2006, 103, 14959–14964. [Google Scholar] [CrossRef]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.X.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.J.; Thelen, J.J.; Cheng, J.L.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef]
- Huang, S.; Li, R.; Zhang, Z.; Li, L.; Gu, X.; Fan, W.; Lucas, W.J.; Wang, X.; Xie, B.; Ni, P.; et al. The genome of the cucumber, Cucumis sativus L. Nat. Genet. 2009, 41, 1275–1281. [Google Scholar] [CrossRef] [Green Version]
- Verde, I.; Abbott, A.G.; Scalabrin, S.; Jung, S.; Shu, S.; Marroni, F.; Zhebentyayeva, T.; Dettori, M.T.; Grimwood, J.; Cattonaro, F.; et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 2013, 45, 487–494. [Google Scholar] [CrossRef]
- Velasco, R.; Zharkikh, A.; Affourtit, J.; Dhingra, A.; Cestaro, A.; Kalyanaraman, A.; Fontana, P.; Bhatnagar, S.K.; Troggio, M.; Pruss, D.; et al. The genome of the domesticated apple (Malus x domestica Borkh.). Nat. Genet. 2010, 42, 833–839. [Google Scholar] [CrossRef] [Green Version]
- The Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796–815. [CrossRef]
- Hu, T.T.; Pattyn, P.; Bakker, E.G.; Cao, J.; Cheng, J.F.; Clark, R.M.; Fahlgren, N.; Fawcett, J.A.; Grimwood, J.; Gundlach, H.; et al. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat. Genet. 2011, 43, 476–481. [Google Scholar] [CrossRef]
- Xu, Q.; Chen, L.L.; Ruan, X.; Chen, D.; Zhu, A.; Chen, C.; Bertrand, D.; Jiao, W.B.; Hao, B.H.; Lyon, M.P.; et al. The draft genome of sweet orange (Citrus sinensis). Nat. Genet. 2013, 45, 59–66. [Google Scholar]
- Jaillon, O.; Aury, J.M.; Noel, B.; Policriti, A.; Clepet, C.; Casagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C.; et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449, 463–467. [Google Scholar] [CrossRef]
- Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 2012, 485, 635–641. [CrossRef] [Green Version]
- Xu, X.; Pan, S.; Cheng, S.; Zhang, B.; Mu, D.; Ni, P.; Zhang, G.; Yang, S.; Li, R.; Wang, J.; et al. Genome sequence and analysis of the tuber crop potato. Nature 2011, 475, 189–195. [Google Scholar] [CrossRef]
- Paterson, A.H.; Bowers, J.E.; Bruggmann, R.; Dubchak, I.; Grimwood, J.; Gundlach, H.; Haberer, G.; Hellsten, U.; Mitros, T.; Poliakov, A.; et al. The Sorghum bicolor genome and the diversification of grasses. Nature 2009, 457, 551–556. [Google Scholar] [CrossRef] [Green Version]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, X.; Quan, Z.; Cheng, S.; Xu, X.; Pan, S.; Xie, M.; Zeng, P.; Yue, Z.; Wang, W.; et al. Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat. Biotechnol. 2012, 30, 549–554. [Google Scholar] [CrossRef]
- Goff, S.A.; Ricke, D.; Lan, T.H.; Presting, G.; Wang, R.L.; Dunn, M.; Glazebrook, J.; Sessions, A.; Oeller, P.; Varma, H.; et al. A draft sequence of the rice genome (Oryza sativa L. ssp japonica). Science 2000, 296, 92–100. [Google Scholar]
- International Brachypodium Initiative. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 2010, 463, 763–768. [CrossRef]
- D'Hont, A.; Denoeud, F.; Aury, J.M.; Baurens, F.C.; Carreel, F.; Garsmeur, O.; Noel, B.; Bocs, S.; Droc, G.; Rouard, M.; et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 2012, 488, 213–217. [Google Scholar] [CrossRef] [Green Version]
- Banks, J.A.; Nishiyama, T.; Hasebe, M.; Bowman, J.L.; Gribskov, M.; dePamphilis, C.; Albert, V.A.; Aono, N.; Aoyama, T.; Ambrose, B.A.; et al. The Selaginella genome identifies genetic changes associated with the evolution of vascular plants. Science 2011, 332, 960–963. [Google Scholar] [CrossRef]
- Rensing, S.A.; Lang, D.; Zimmer, A.D.; Terry, A.; Salamov, A.; Shapiro, H.; Nishiyama, T.; Perroud, P.F.; Lindquist, E.A.; Kamisugi, Y.; et al. The Physcomitrella genome reveals evolutionary insights into the conquest of land by plants. Science 2008, 319, 64–69. [Google Scholar] [CrossRef]
- Fawcett, J.A.; Maere, S.; van de Peer, Y. Plants with double genomes might have had a better chance to survive the Cretaceous-Tertiary extinction event. Proc. Natl. Acad. Sci. USA 2009, 106, 5737–5742. [Google Scholar] [CrossRef]
- Koch, M.A.; Haubold, B.; Mitchell-Olds, T. Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis, and related genera (Brassicaceae). Mol. Biol. Evol. 2000, 17, 1483–1498. [Google Scholar] [CrossRef]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [Green Version]
- Freeling, M. Bias in plant gene content following different sorts of duplication: Tandem, whole-genome, segmental, or by transposition. Annu. Rev. Plant Biol. 2009, 60, 433–453. [Google Scholar] [CrossRef]
- Edger, P.P.; Pires, J.C. Gene and genome duplications: The impact of dosage-sensitivity on the fate of nuclear genes. Chromosome Res. 2009, 17, 699–717. [Google Scholar] [CrossRef]
- Melzer, R.; Theissen, G. Reconstitution of 'floral quartets’ in vitro involving class B and class E floral homeotic proteins. Nucleic Acids Res. 2009, 37, 2723–2736. [Google Scholar] [CrossRef]
- De Folter, S.; Immink, R.G.; Kieffer, M.; Parenicova, L.; Henz, S.R.; Weigel, D.; Busscher, M.; Kooiker, M.; Colombo, L.; Kater, M.M.; et al. Comprehensive interaction map of the Arabidopsis MADS Box transcription factors. Plant Cell 2005, 17, 1424–1433. [Google Scholar] [CrossRef]
- Kaufmann, K.; Melzer, R.; Theissen, G. MIKC-type MADS-domain proteins: Structural modularity, protein interactions and network evolution in land plants. Gene 2005, 347, 183–198. [Google Scholar] [CrossRef]
- Freeling, M.; Lyons, E.; Pedersen, B.; Alam, M.; Ming, R.; Lisch, D. Many or most genes in Arabidopsis transposed after the origin of the order Brassicales. Genome Res. 2008, 18, 1924–1937. [Google Scholar] [CrossRef]
- Masiero, S.; Colombo, L.; Grini, P.E.; Schnittger, A.; Kater, M.M. The emerging importance of type I MADS box transcription factors for plant reproduction. Plant Cell 2011, 23, 865–872. [Google Scholar] [CrossRef]
- Walia, H.; Josefsson, C.; Dilkes, B.; Kirkbride, R.; Harada, J.; Comai, L. Dosage-dependent deregulation of an AGAMOUS-LIKE gene cluster contributes to interspecific incompatibility. Curr. Biol. 2009, 19, 1128–1132. [Google Scholar] [CrossRef]
- De Bodt, S.; Raes, J.; Van de Peer, Y.; Theissen, G. And then there were many: MADS goes genomic. Trends Plant Sci. 2003, 8, 475–483. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Tang, H.; Tan, X.; Ficklin, S.P.; Feltus, F.A.; Paterson, A.H. Modes of gene duplication contribute differently to genetic novelty and redundancy, but show parallels across divergent angiosperms. PLoS One 2011, 6, e28150. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gramzow, L.; Theißen, G. Phylogenomics of MADS-Box Genes in Plants — Two Opposing Life Styles in One Gene Family. Biology 2013, 2, 1150-1164. https://doi.org/10.3390/biology2031150
Gramzow L, Theißen G. Phylogenomics of MADS-Box Genes in Plants — Two Opposing Life Styles in One Gene Family. Biology. 2013; 2(3):1150-1164. https://doi.org/10.3390/biology2031150
Chicago/Turabian StyleGramzow, Lydia, and Günter Theißen. 2013. "Phylogenomics of MADS-Box Genes in Plants — Two Opposing Life Styles in One Gene Family" Biology 2, no. 3: 1150-1164. https://doi.org/10.3390/biology2031150