Glycerophosphate/Acylglycerophosphate Acyltransferases

Abstract
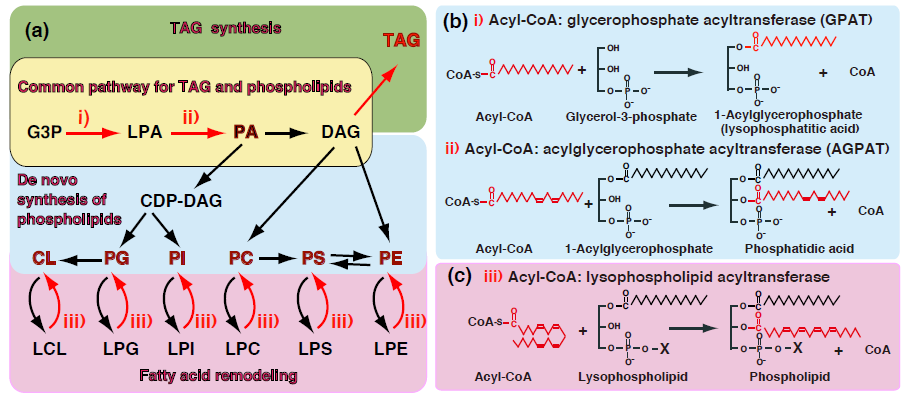
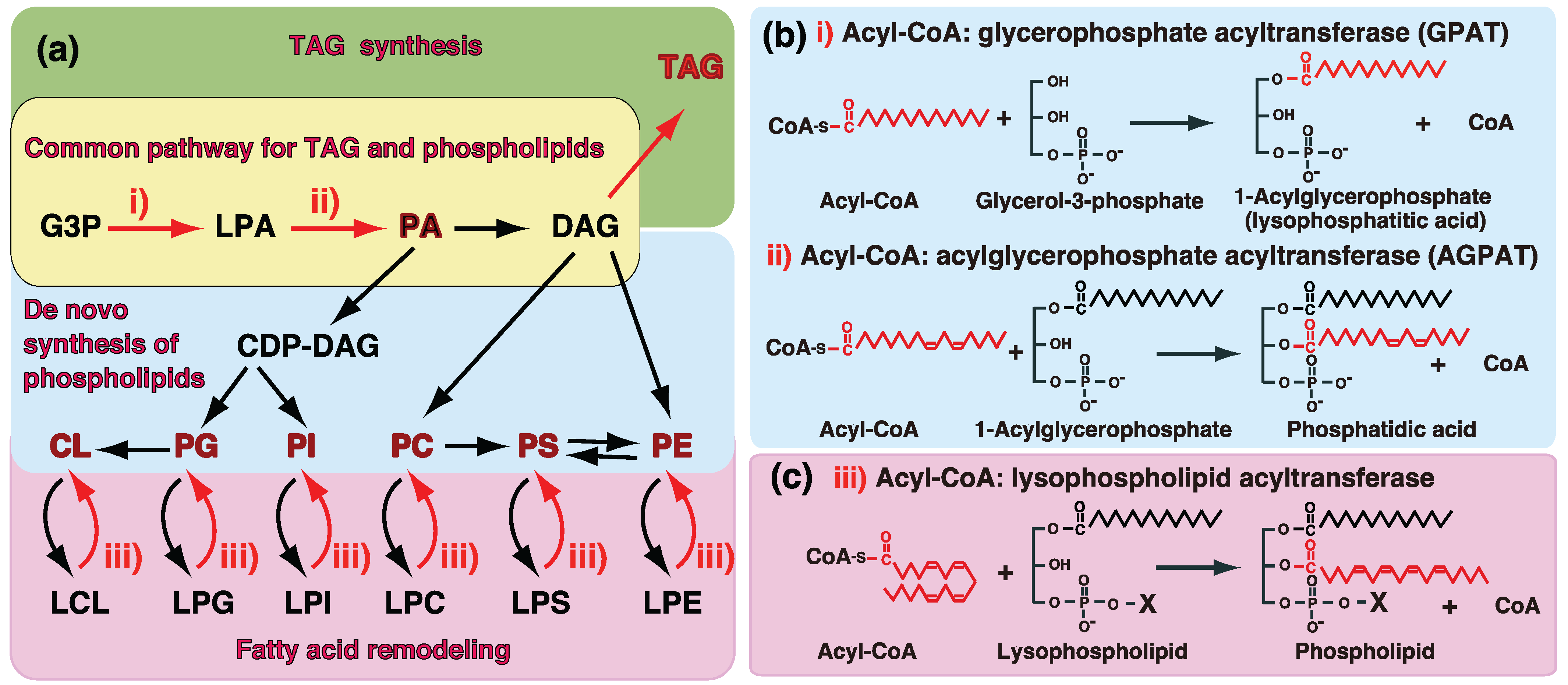
: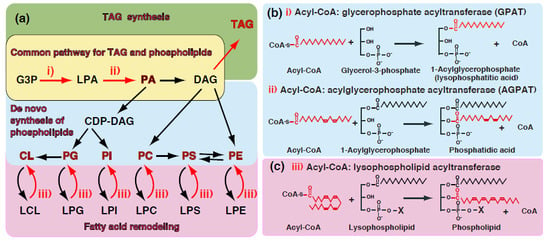
1. Introduction—GPAT and AGPAT in Glycerolipid Metabolism

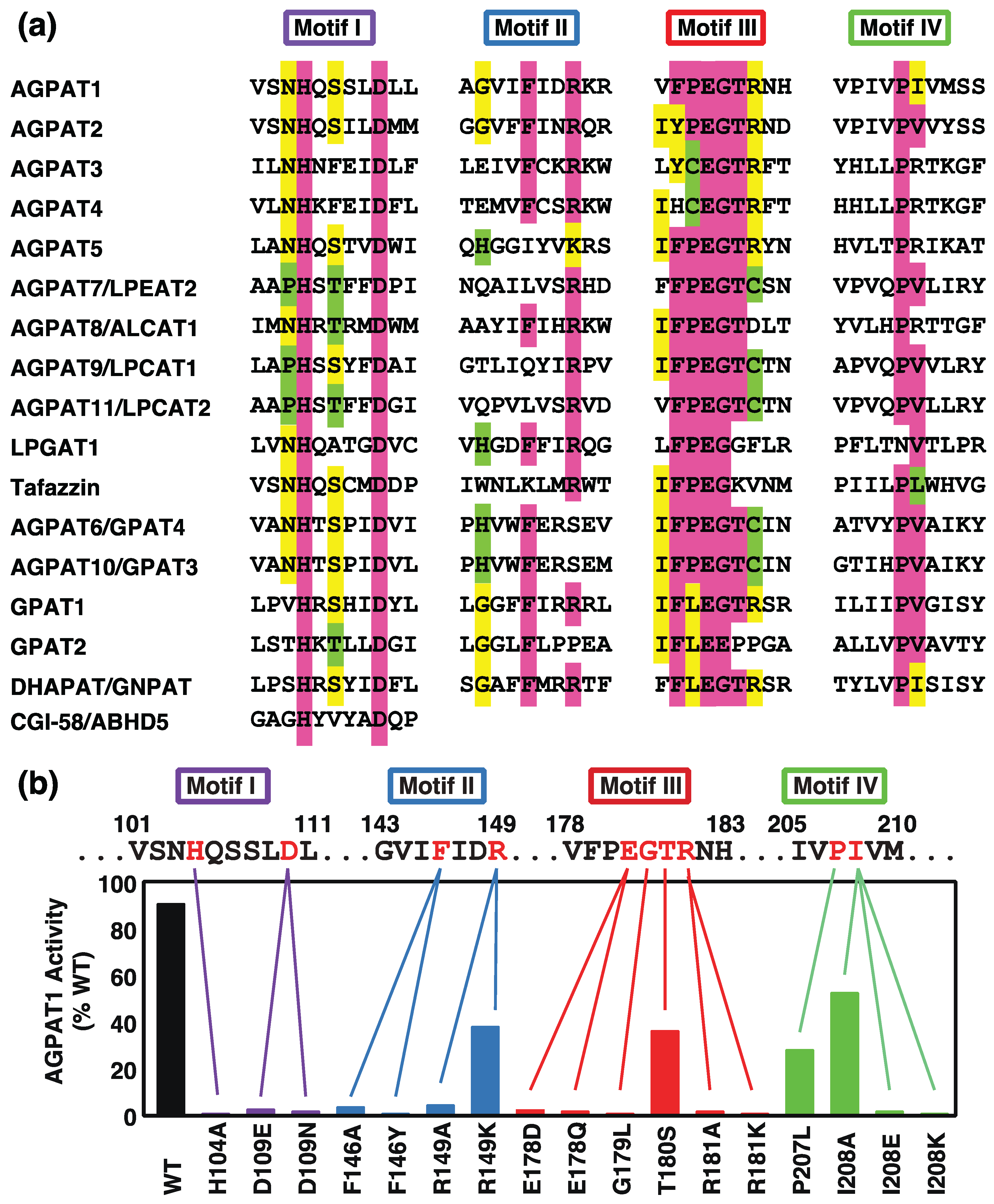
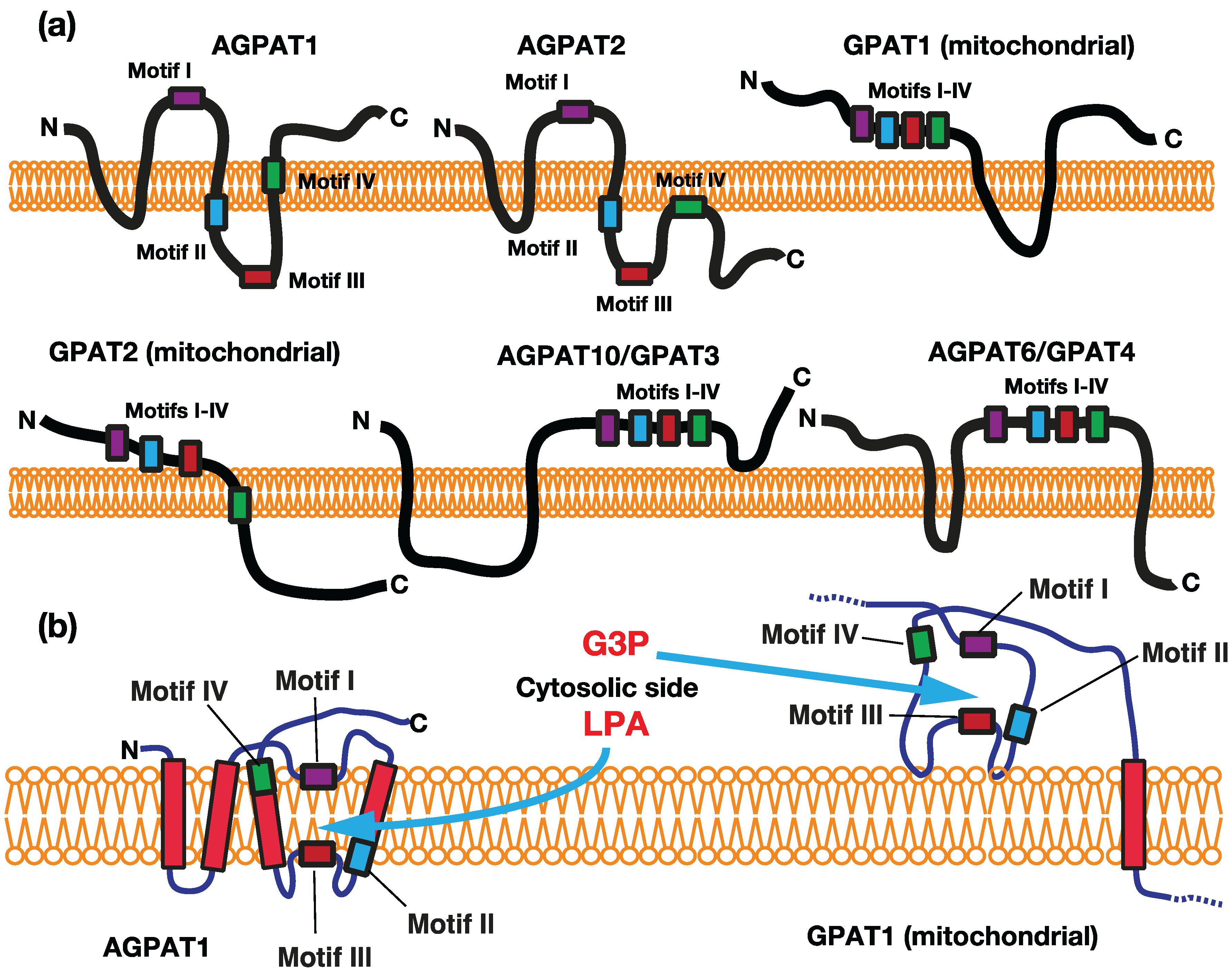
2. Acyltransferase Motifs Conserved in GPAT/AGPAT Family Enzymes
2.1. Acyltransferase Motif I

2.2. Acyltransferase Motif II
2.3. Acyltransferase Motif III
2.4. Acyltransferase Motif IV
2.5. Substrate Specificity and Membrane Topology of Acyltransferase Motifs

3. Glycerophosphate Acyltransferases (GPATs)
3.1. Mitochondrial GPATs (GPAT1 and GPAT2)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Other Symbol | Acyl Acceptor | Acyl donor (Acyl-CoA) | Notes | Diseases in Human, Phenotype of Gene-Manipulated Animal |
|---|---|---|---|---|---|
| GPAT1 | G3P | 16:0 > 18:1n-9 (Preference to saturated species) | Mitochondrial GPAT, NEM-resistant | ||
| GPAT2 | G3P | No preference to saturated species | Mitochondrial GPAT, NEM-sensitive | ||
| GPAT3 | AGPAT8, AGPAT10 | G3P | No preference to saturated species | Microsomal GPAT, NEM-sensitive | |
| GPAT4 | AGPAT6 | G3P | No preference to saturated species | Microsomal GPAT, NEM-sensitive | Causative gene of lipodystrophy |
| DHAPAT | GNPAT | DHAP | Biosynthesis of ether-linked phospholipids | Causative gene of TYPE 2 rhizomelic chondrodysplasia punctata | |
| AGPAT1 | LPAATα, LPAAT1 | LPA | |||
| AGPAT2 | LPAATβ, LPAAT2 | LPA | Causative gene of lipodystrophy | ||
| AGPAT3 | LPAATγ, LPAAT3 | LPA. LPI | Preference to PUFA species | ||
| AGPAT4 | LPAATδ, LPAAT4 | LPA | Location of mitochondria? | ||
| AGPAT5 | LPAATε, LPAAT5 | Location of mitochondria? | |||
| AGPAT7 | LPAATη, AYTL3, LPEAT2 | LPE | Preference to oleic acid | ||
| AGPAT8 | ALCAT1, LCLAT1, LYCAT1 | Lysocardiolipin, LPG, LPI (1-acyl or 2-acyl) | PUFA for LCL, 18:0 for 2-acyl LPI | Mitochondrial dysfunction associated with hypertrophic cardiomyopathy, blood lineages | |
| AGPAT9 | LPCAT1, AYTL2 | LPC, LPE, LysoPAF | Preference to saturated species, Acetyl-CoA | Surfactant biosynthesis, EF-hand motif | |
| AGPAT11 | LPCAT2, AYTL1, LysoPAFAT | LPC, LPE, LysoPAF | Acetyl-CoA, Acyl-CoA | Inducible enzyme for PAF biosynthesis, EF-hand motif | |
| LPGAT1 | LPG | ||||
| TFZ | Tafazzin | Lysocardiolipin | Causative gene of Barth syndrome |
| Symbol | Tissue Distribution |
|---|---|
| GPAT1 | BAT > WAT > liver > muscle > brain |
| GPAT2 | testis > liver > adipose tissue, skeletal muscle, brain, adrenal grand, kidney, lung, heart |
| GPAT3/AGPAT10 | adipose tissue > small intestine > heart > brain > liver |
| GPAT4/AGPAT6 | brown adipose tissue, testis > liver, kidney, brain, intestine, WAT > heart, skeletal muscle |
| AGPAT1/LPAATα | testis > spleen, thymus, prostate, ovary, small intestine, colon, PBL (nearly ubiquitous) |
| AGPAT2/LPAATβ | liver > pancreas >lung, heart, small intestine, skeletal muscle > colon, PBL > spleen, prostate >> brain |
| AGPAT3/LPAATγ | testis > kidney > liver > heart > brain |
| AGPAT4/LPAATδ | brain > skeletal muscle > spleen |
| AGPAT5/LPAATε | testis > prostate > placenta > brain |
| AGPAT7/LPEAT2 | brain > stomach > heart > liver |
| AGPAT8/ALCAT1 | heart, liver, kidney > small intestine, skin, brain, lung > spleen, thymus, testis > muscle, stomach |
| AGPAT9/LPCAT1 | lung (alveolar type II cells) >> spleen > brain, heart, skeletal muscle, ovary, pancreas |
| AGPAT11/LPCAT2 | macrophage >> neutrophil >> skin > brain, heart, stomach, colon, spleen > lung, liver, ovary, placenta |
| LPGAT1 | liver, placenta > peripheral blood, lung, kidney, brain >> colon |
| BAT; brown adipose tissue, WAT; white adipose tissue, PBL; peripheral blood leukocytes |
3.2. Microsomal GPATs—GPAT3/AGPAT10 and GPAT4/AGPAT6
3.3. Dihydroxyacetone Phosphate Acyltransferase (DHAPAT/GNPAT)
4. Acylglycerophosphate Acyltransferases (AGPATs) Involved in the de novo Synthesis of TAG and Phospholipids
4.1. AGPAT1/LPAATα
4.2. AGPAT2/LPAATβ
4.3. AGPAT3/LPAATγ
4.4. AGPAT4 (LPAATδ) and AGPAT5 (LPAATε)
5. Acylglycerophosphate Acyltransferases (AGPATs) Involved in the Remodeling of Phospholipids
5.1. LPCAT1/AGPAT9
5.2. LPCAT2/AGPAT11
5.3. AGPAT7/LPEAT2
5.4. AGPAT8/ALCAT1/2-Acyl LPIAT
5.5. Tafazzins and the Fatty Acid Remodeling of Cardiolipin
5.6. Lysophosphatidylglycerol Acyltransferase 1 (LPGAT1)
6. Conclusions
Conflicts of Interest
References
- Coleman, R.A.; Lee, D.P. Enzymes of triacylglycerol synthesis and their regulation. Prog. Lipid Res. 2004, 43, 134–176. [Google Scholar] [CrossRef] [PubMed]
- Holub, B.J.; Kuksis, A. Metabolism of molecular species of diacylglycerophospholipids. Adv. Lipid Res. 1978, 16, 1–125. [Google Scholar] [PubMed]
- Lands, W.E. Metabolism of glycerolipids: A comparison of lecithin and triglyceride synthesis. J. Biol. Chem. 1958, 231, 883–888. [Google Scholar] [PubMed]
- Leung, D.W. The structure and functions of human lysophosphatidic acid acyltransferases. Front. Biosci. 2001, 6, D944–D953. [Google Scholar] [CrossRef] [PubMed]
- Shindou, H.; Shimizu, T. Acyl-CoA:lysophospholipid acyltransferases. J. Biol. Chem. 2009, 284, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Sugiura, T.; Waku, K. Acyltransferases and transacylases involved in fatty acid remodeling of phospholipids and metabolism of bioactive lipids in mammalian cells. J. Biochem. 1997, 122, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Hayashi, Y.; Nemoto-Sasaki, Y.; Ito, M.; Oka, S.; Tanikawa, T.; Waku, K.; Sugiura, T. Acyltransferases and transacylases that determine the fatty acid composition of glycerolipids and the metabolism of bioactive lipid mediators in mammalian cells and model organisms. Prog. Lipid Res. 2014, 53, 18–81. [Google Scholar] [CrossRef] [PubMed]
- Kitson, A.P.; Stark, K.D.; Duncan, R.E. Enzymes in brain phospholipid docosahexaenoic acid accretion: A PL-ethora of potential PL-ayers. Prostaglandins Leukot. Essent. Fatty Acids 2012, 87, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Heath, R.J.; Rock, C.O. A conserved histidine is essential for glycerolipid acyltransferase catalysis. J. Bacteriol. 1998, 180, 1425–1430. [Google Scholar] [PubMed]
- Lewin, T.M.; Wang, P.; Coleman, R.A. Analysis of amino acid motifs diagnostic for the sn-glycerol-3-phosphate acyltransferase reaction. Biochemistry 1999, 38, 5764–5771. [Google Scholar] [CrossRef] [PubMed]
- Dircks, L.K.; Ke, J.; Sul, H.S. A conserved seven amino acid stretch important for murine mitochondrial glycerol-3-phosphate acyltransferase activity. Significance of arginine 318 in catalysis. J. Biol. Chem. 1999, 274, 34728–34734. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Nakanishi, H.; Suzuki, H.; Kamata, R.; Tanaka, K.; Waku, K.; Sugiura, T. Topology of acyltransferase motifs and substrate specificity and accessibility in 1-acyl-sn-glycero-3-phosphate acyltransferase 1. Biochim. Biophys. Acta 2007, 1771, 1202–1215. [Google Scholar] [CrossRef] [PubMed]
- Harayama, T.; Shindou, H.; Ogasawara, R.; Suwabe, A.; Shimizu, T. Identification of a novel noninflammatory biosynthetic pathway of platelet-activating factor. J. Biol. Chem. 2008, 283, 11097–11106. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, A.P.; Rafferty, J.B.; Sedelnikova, S.E.; Slabas, A.R.; Schierer, T.P.; Kroon, J.T.; Simon, J.W.; Fawcett, T.; Nishida, I.; Murata, N.; et al. Analysis of the structure, substrate specificity, and mechanism of squash glycerol-3-phosphate (1)-acyltransferase. Structure 2001, 9, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Ramakrishnan, G.; Chandramohan, C.; Rajasekharan, R. CGI-58, the causative gene for Chanarin-Dorfman syndrome, mediates acylation of lysophosphatidic acid. J. Biol. Chem. 2008, 283, 24525–24533. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Paulauskis, J.D.; Moustaid, N.; Sul, H.S. Transcriptional regulation of p90 with sequence homology to Escherichia coli glycerol-3-phosphate acyltransferase. J. Biol. Chem. 1991, 266, 23834–23839. [Google Scholar] [PubMed]
- Yet, S.F.; Lee, S.; Hahm, Y.T.; Sul, H.S. Expression and identification of p90 as the murine mitochondrial glycerol-3-phosphate acyltransferase. Biochemistry 1993, 32, 9486–9491. [Google Scholar] [CrossRef] [PubMed]
- Jerkins, A.A.; Liu, W.R.; Lee, S.; Sul, H.S. Characterization of the murine mitochondrial glycerol-3-phosphate acyltransferase promoter. J. Biol. Chem. 1995, 270, 1416–1421. [Google Scholar] [CrossRef] [PubMed]
- Vancura, A.; Haldar, D. Purification and characterization of glycerophosphate acyltransferase from rat liver mitochondria. J. Biol. Chem. 1994, 269, 27209–27215. [Google Scholar] [PubMed]
- Hammond, L.E.; Gallagher, P.A.; Wang, S.; Hiller, S.; Kluckman, K.D.; Posey-Marcos, E.L.; Maeda, N.; Coleman, R.A. Mitochondrial glycerol-3-phosphate acyltransferase-deficient mice have reduced weight and liver triacylglycerol content and altered glycerolipid fatty acid composition. Mol. Cell Biol. 2002, 22, 8204–8214. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wilcox, D.; Nguyen, P.; Voorbach, M.; Suhar, T.; Morgan, S.J.; An, W.F.; Ge, L.; Green, J.; Wu, Z.; et al. Hepatic knockdown of mitochondrial GPAT1 in ob/ob mice improves metabolic profile. Biochem. Biophys. Res. Commun. 2006, 349, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Lindén, D.; William-Olsson, L.; Ahnmark, A.; Ekroos, K.; Hallberg, C.; Sjögren, H.P.; Becker, B.; Svensson, L.; Clapham, J.C.; Oscarsson, J.; et al. Liver-directed overexpression of mitochondrial glycerol-3-phosphate acyltransferase results in hepatic steatosis, increased triacylglycerol secretion and reduced fatty acid oxidation. FASEB J. 2006, 20, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Nagle, C.A.; An, J.; Shiota, M.; Torres, T.P.; Cline, G.W.; Liu, Z.X.; Wang, S.; Catlin, R.L.; Shulman, G.I.; Newgard, C.B.; et al. Hepatic overexpression of glycerol-sn-3-phosphate acyltransferase 1 in rats causes insulin resistance. J. Biol. Chem. 2007, 282, 14807–14815. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Lee, D.P.; Gong, N.; Schwerbrock, N.M.; Mashek, D.G.; Gonzalez-Baró, M.R.; Stapleton, C.; Li, L.O.; Lewin, T.M.; Coleman, R.A. Cloning and functional characterization of a novel mitochondrial N-ethylmaleimide sensitive glycerol-3-phosphate acyltransferase (GPAT2). Arch. Biochem. Biophys. 2007, 465, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Baró, M.R.; Lewin, T.M.; Coleman, R.A. Regulation of triglyceride metabolism II. Function of mitochondrial GPAT1 in the regulation of triacylglycerol biosynthesis and insulin action. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1195–G1199. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.Y.; Repa, J.J. The liver X receptor (LXR) and hepatic lipogenesis. The carbohydrate-response element-binding protein is a target gene of LXR. J. Biol. Chem. 2007, 282, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Bronnikov, G.E.; Aboulaich, N.; Vener, A.V.; Stralfors, P. Acute effects of insulin on the activity of mitochondrial GPAT1 in primary adipocytes. Biochem. Biophys. Res. Commun. 2008, 367, 201–207. [Google Scholar] [CrossRef]
- Wendel, A.A.; Cooper, D.E.; Ilkayeva, O.R.; Muoio, D.M.; Coleman, R.A. Glycerol-3-phosphate acyltransferase (GPAT)-1, but not GPAT4, incorporates newly synthesized fatty acids into triacylglycerol and diminishes fatty acid oxidation. J. Biol. Chem. 2013, 288, 27299–27306. [Google Scholar] [CrossRef] [PubMed]
- Ohba, Y.; Sakuragi, T.; Kage-Nakadai, E.; Tomioka, N.H.; Kono, N.; Imae, R.; Inoue, A.; Aoki, J.; Ishihara, N.; Inoue, T.; et al. Mitochondria-type GPAT is required for mitochondrial fusion. EMBO J. 2013, 32, 1265–1279. [Google Scholar] [CrossRef] [PubMed]
- Lewin, T.M.; Schwerbrock, N.M.; Lee, D.P.; Coleman, R.A. Identification of a new glycerol-3-phosphate acyltransferase isoenzyme, mtGPAT2, in mitochondria. J. Biol. Chem. 2004, 279, 13488–13495. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Hara, S.; Yoshida, M.; Zenitani, T.; Mawatari, K.; Nakano, M.; Takahashi, A.; Hosaka, T.; Yoshimoto, K.; Nakaya, Y. Molecular cloning of a murine glycerol-3-phosphate acyltransferase-like protein 1 (xGPAT1). Mol. Cell Biochem. 2007, 297, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Yuan, J.; Chen, X.; Gu, X.; Luo, K.; Li, J.; Wan, B.; Wang, Y.; Yu, L. Identification of a novel human lysophosphatidic acid acyltransferase, LPAAT-theta, which activates mTOR pathway. J. Biochem. Mol. Biol. 2006, 39, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Li, J.L.; Li, D.; Tobin, J.F.; Gimeno, R.E. Molecular identification of microsomal acyl-CoA:glycerol-3-phosphate acyltransferase, a key enzyme in de novo triacylglycerol synthesis. Proc. Natl. Acad. Sci. USA 2006, 103, 19695–19700. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yu, L.; Wu, H.; Shan, Y.; Guo, J.; Dang, Y.; Wei, Y.; Zhao, S. Cloning and identification of the human LPAAT-zeta gene, a novel member of the lysophosphatidic acid acyltransferase family. J. Hum. Genet. 2003, 48, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Beigneux, A.P.; Vergnes, L.; Qiao, X.; Quatela, S.; Davis, R.; Watkins, S.M.; Coleman, R.A.; Walzem, R.L.; Philips, M.; Reue, K.; et al. Agpat6—A novel lipid biosynthetic gene required for triacylglycerol production in mammary epithelium. J. Lipid Res. 2006, 47, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Vergnes, L.; Beigneux, A.P.; Davis, R.; Watkins, S.M.; Young, S.G.; Reue, K. Agpat6 deficiency causes subdermal lipodystrophy and resistance to obesity. J. Lipid Res. 2006, 47, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Q.; Kuo, M.S.; Li, S.; Bui, H.H.; Peake, D.A.; Sanders, P.E.; Thibodeaux, S.J.; Chu, S.; Qian, Y.W.; Zhao, Y.; et al. AGPAT6 is a novel microsomal glycerol-3-phosphate acyltransferase. J. Biol. Chem. 2008, 283, 10048–10057. [Google Scholar] [CrossRef] [PubMed]
- Nagle, C.A.; Vergnes, L.; Dejong, H.; Wang, S.; Lewin, T.M.; Reue, K.; Coleman, R.A. Identification of a novel sn-glycerol-3-phosphate acyltransferase isoform, GPAT4, as the enzyme deficient in Agpat6−/− mice. J. Lipid Res. 2008, 49, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, S.; Barnes, R.; Garg, A.; Agarwal, A. Functional characterization of the human 1-acylglycerol-3-phosphate-O-acyltransferase isoform 10/glycerol-3-phosphate acyltransferase isoform 3 (AGPAT10/GPAT3). J. Mol. Endocrinol. 2009, 42, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Perez, S.; Goodwin, B.; Lin, Q.; Peng, H.; Qadri, A.; Zhou, Y.; Clark, R.W.; Perreault, M.; Tobin, J.F.; et al. Mice deleted for GPAT3 have reduced GPAT activity in white adipose tissue and altered energy and cholesterol homeostasis in diet-induced obesity. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1176–E1187. [Google Scholar] [CrossRef] [PubMed]
- Shan, D.; Li, J.L.; Wu, L.; Li, D.; Hurov, J.; Tobin, J.F.; Gimeno, R.E.; Cao, J. GPAT3 and GPAT4 are regulated by insulin-stimulated phosphorylation and play distinct roles in adipogenesis. J. Lipid Res. 2010, 51, 1971–1981. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Cooper, D.E.; Grevengoed, T.J.; Li, L.O.; Klett, E.L.; Eaton, J.M.; Harris, T.E.; Coleman, R.A. Glycerol-3-phosphate acyltransferase-4-deficient mice are protected from diet-induced insulin resistance by the enhanced association of mTOR and rictor. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E305–E315. [Google Scholar] [CrossRef] [PubMed]
- Thai, T.P.; Heid, H.; Rackwitz, H.R.; Hunziker, A.; Gorgas, K.; Just, W.W. Ether lipid biosynthesis: Isolation and molecular characterization of human dihydroxyacetonephosphate acyltransferase. FEBS Lett. 1997, 420, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Ofman, R.; Hettema, E.H.; Hogenhout, E.M.; Caruso, U.; Muijsers, A.O.; Wanders, R.J. Acyl-CoA: Dihydroxyacetonephosphate acyltransferase: Cloning of the human cDNA and resolution of the molecular basis in rhizomelic chondrodysplasia punctata type 2. Hum. Mol. Genet. 1998, 7, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Elias, E.R.; Mobassaleh, M.; Hajra, A.K.; Moser, A.B. Developmental delay and growth failure caused by a peroxisomal disorder, dihydroxyacetonephosphate acyltransferase (DHAP-AT) deficiency. Am. J. Med. Genet. 1998, 80, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Thai, T.P.; Rodemer, C.; Jauch, A.; Hunziker, A.; Moser, A.; Gorgas, K.; Just, W.W. Impaired membrane traffic in defective ether lipid biosynthesis. Hum. Mol. Genet. 2001, 10, 127–136. [Google Scholar] [CrossRef] [PubMed]
- West, J.; Tompkins, C.K.; Balantac, N.; Nudelman, E.; Meengs, B.; White, T.; Bursten, S.; Coleman, J.; Kumar, A.; Singer, J.W.; et al. Cloning and expression of two human lysophosphatidic acid acyltransferase cDNAs that enhance cytokine-induced signaling responses in cells. DNA Cell Biol. 1997, 16, 691–701. [Google Scholar] [PubMed]
- Stamps, A.C.; Elmore, M.A.; Hill, M.E.; Kelly, K.; Makda, A.A.; Finnen, M.J. A human cDNA sequence with homology to non-mammalian lysophosphatidic acid acyltransferases. Biochem. J. 1997, 326, 455–461. [Google Scholar] [PubMed]
- Eberhardt, C.; Gray, P.W.; Tjoelker, L.W. Human lysophosphatidic acid acyltransferase. cDNA cloning, expression, and localization to chromosome 9q34.3. J. Biol. Chem. 1997, 272, 20299–20305. [Google Scholar] [CrossRef] [PubMed]
- Aguado, B.; Campbell, R.D. Characterization of a human lysophosphatidic acid acyltransferase that is encoded by a gene located in the class III region of the human major histocompatibility complex. J. Biol. Chem. 1998, 273, 20299–20305. [Google Scholar] [CrossRef]
- Kume, K.; Shimizu, T. cDNA cloning and expression of murine 1-acyl-sn-glycerol-3-phosphate acyltransferase. Biochem. Biophys. Res. Commun. 1997, 237, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Kawagishi, N.; Miyashita, T.; Nagatsuka, T.; Sugiura, T.; Kume, K.; Shimizu, T.; Waku, K. ATP-independent fatty acyl-coenzyme A synthesis from phospholipid. Coenzyme A-dependent transacylation activity toward lysophosphatidic acid catalyzed by acyl-coenzyme A:lysophosphatidic acid acyltransferase. J. Biol. Chem. 2001, 276, 26745–26752. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Pownall, H.J. Overexpression of 1-acyl-glycerol-3-phosphate acyltransferase-alpha enhances lipid storage in cellular models of adipose tissue and skeletal muscle. Diabetes 2001, 50, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Subauste, A.R.; Elliott, B.; Das, A.K.; Burant, C.F. A role for 1-acylglycerol-3-phosphate-O-acyltransferase-1 in myoblast differentiation. Differentiation 2010, 80, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Hollenback, D.; Bonham, L.; Law, L.; Rossnagle, E.; Romero, L.; Carew, H.; Tompkins, C.K.; Leung, D.W.; Singer, J.W.; White, T. Substrate specificity of lysophosphatidic acid acyltransferase β-evidence from membrane and whole cell assays. J. Lipid Res. 2006, 47, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.K.; Arioglu, E.; de Almeida, S.; Akkoc, N.; Taylor, S.I.; Bowcock, A.M.; Barnes, R.I.; Garg, A. AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nat. Genet. 2002, 31, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Haque, W.; Garg, A.; Agarwal, A.K. Enzymatic activity of naturally occurring 1-acylglycerol-3-phosphate-O-acyltransferase 2 mutants associated with congenital generalized lipodystrophy. Biochem. Biophys. Res. Commun. 2005, 327, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Gomes, K.B.; Pardini, V.C.; Ferreira, A.C.; Fernandes, A.P. Phenotypic heterogeneity in biochemical parameters correlates with mutations in AGPAT2 or Seipin genes among Berardinelli-Seip congenital lipodystrophy patients. J. Inherit. Metab. Dis. 2005, 28, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Haque, W.A.; Shimomura, I.; Matsuzawa, Y.; Garg, A. Serum adiponectin and leptin levels in patients with lipodystrophies. J. Clin. Endocrinol. Metab. 2002, 87, 2395–2398. [Google Scholar] [CrossRef] [PubMed]
- Gale, S.E.; Frolov, A.; Han, X.; Bickel, P.E.; Cao, L.; Bowcock, A.; Schaffer, J.E.; Ory, D.S. A regulatory role for 1-acylglycerol-3-phosphate-O-acyltransferase 2 in adipocyte differentiation. J. Biol. Chem. 2006, 281, 11082–11089. [Google Scholar] [CrossRef] [PubMed]
- Cortes, V.A.; Curtis, D.E.; Sukumaran, S.; Shao, X.; Parameswara, V.; Rashid, S.; Smith, A.R.; Ren, J.; Esser, V.; Hammer, R.E.; et al. Molecular mechanisms of hepatic steatosis and insulin resistance in the AGPAT2-deficient mouse model of congenital generalized lipodystrophy. Cell Metab. 2009, 9, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Coon, M.; Ball, A.; Pound, J.; Ap, S.; Hollenback, D.; White, T.; Tulinsky, J.; Bonham, L.; Morrison, D.K.; Finney, R.; et al. Inhibition of lysophosphatidic acid acyltransferase β disrupts proliferative and survival signals in normal cells and induces apoptosis of tumor cells. Mol. Cancer Ther. 2003, 2, 1067–1078. [Google Scholar] [PubMed]
- Hideshima, T.; Chauhan, D.; Ishitsuka, K.; Yasui, H.; Raje, N.; Kumar, S.; Podar, K.; Mitsiades, C.; Hideshima, H.; Bonham, L.; et al. Molecular characterization of PS-341 (bortezomib) resistance: Implications for overcoming resistance using lysophosphatidic acid acyltransferase (LPAAT)-β inhibitors. Oncogene 2005, 24, 3121–3129. [Google Scholar] [CrossRef] [PubMed]
- Pagel, J.M.; Laugen, C.; Bonham, L.; Hackman, R.C.; Hockenbery, D.M.; Bhatt, R.; Hollenback, D.; Carew, H.; Singer, J.W.; Press, O.W. Induction of apoptosis using inhibitors of lysophosphatidic acid acyltransferase-β and anti-CD20 monoclonal antibodies for treatment of human non-Hodgkin’s lymphomas. Clin. Cancer Res. 2005, 11, 4857–4866. [Google Scholar] [CrossRef] [PubMed]
- Springett, G.M.; Bonham, L.; Hummer, A.; Linkov, I.; Misra, D.; Ma, C.; Pezzoni, G.; di Giovine, S.; Singer, J.; Kawasaki, H.; et al. Lysophosphatidic acid acyltransferase-β is a prognostic marker and therapeutic target in gynecologic malignancies. Cancer Res. 2005, 65, 9415–9425. [Google Scholar] [CrossRef] [PubMed]
- Douvas, M.G.; Hogan, K.N.; Ji, Y.; Hollenback, D.; Bonham, L.; Singer, J.W.; Mitchell, B.S. Effect of lysophosphatidic acid acyltransferase-β inhibition in acute leukemia. Leuk. Res. 2006, 30, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- La Rosée, P.; Jia, T.; Demehri, S.; Härtel, N.; de Vries, P.; Bonham, L.; Hollenback, D.; Singer, J.W.; Melo, J.V.; Druker, B.J.; et al. Antileukemic activity of lysophosphatidic acid acyltransferase-β inhibitor CT32228 in chronic myelogenous leukemia sensitive and resistant to imatinib. Clin. Cancer Res. 2006, 12, 6540–6546. [Google Scholar] [CrossRef] [PubMed]
- Niesporek, S.; Denkert, C.; Weichert, W.; Köbel, M.; Noske, A.; Sehouli, J.; Singer, J.W.; Dietel, M.; Hauptmann, S. Expression of lysophosphatidic acid acyltransferase β (LPAAT-β) in ovarian carcinoma: correlation with tumour grading and prognosis. Br. J. Cancer 2005, 92, 1729–1736. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, C.S.; Soslow, R.A.; Iasonos, A.; Linkov, I.; Hedvat, C.; Bonham, L.; Singer, J.; Barakat, R.R.; Aghajanian, C.; Dupont, J. Lysophosphatidic acid acyltransferase-β (LPAAT-β) is highly expressed in advanced ovarian cancer and is associated with aggressive histology and poor survival. Cancer 2006, 107, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Jiang, Y.J.; Zhou, Y.; Xu, F.Y.; Hatch, G.M.; Choy, P.C. Cloning and characterization of murine 1-acyl-sn-glycerol 3-phosphate acyltransferases and their regulation by PPARα in murine heart. Biochem. J. 2005, 385, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Jiang, Y.J.; Man, M.Q.; Brown, B.; Wlias, P.M.; Feingold, K.R. Expression and regulation of 1-acyl-sn-glycerol-3-phosphate acyltransferases in the epidermis. J. Lipid Res. 2005, 46, 2448–2457. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.S.; Garg, A.; Agarwal, A.K. Enzymatic activities of the human AGPAT isoform 3 and isoform 5: Localization of AGPAT5 to mitochondria. J. Lipid Res. 2011, 52, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Yuki, K.; Shindou, H.; Hishikawa, D.; Shimizu, T. Characterization of mouse lysophosphatidic acid acyltransferase 3: An enzyme with dual functions in the testis. J. Lipid Res. 2009, 50, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.A.; Brown, W.J. Lysophosphatidic acid acyltransferase 3 regulates Golgi complex structure and function. J. Cell Biol. 2009, 186, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Koeberle, A.; Shindou, H.; Harayama, T.; Shimizu, T. Role of lysophosphatidic acid acyltransferase 3 for the supply of highly polyunsaturated fatty acids in TM4 Sertoli cells. FASEB J. 2010, 24, 4929–4938. [Google Scholar] [CrossRef] [PubMed]
- Koeberle, A.; Shindou, H.; Harayama, T.; Yuki, K.; Shimizu, T. Polyunsaturated fatty acids are incorporated into maturating male mouse germ cells by lysophosphatidic acid acyltransferase 3. FASEB J. 2012, 26, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Eto, M.; Shindou, H.; Shimizu, T. A novel lysophosphatidic acid acyltransferase enzyme (LPAAT4) with a possible role for incorporating docosahexaenoic acid into brain glycerophospholipids. Biochem. Biophys. Res. Commun. 2014, 443, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, H.; Shindou, H.; Hishikawa, D.; Harayama, T.; Ogasawara, R.; Suwabe, A.; Taguchi, R.; Shimizu, T. Cloning and characterization of mouse lung-type acyl-CoA: Lysophosphatidylcholine acyltransferase 1 (LPCAT1). Expression in alveolar type II cells and possible involvement in surfactant production. J. Biol. Chem. 2006, 281, 20140–20147. [Google Scholar] [PubMed]
- Chen, X.; Hyatt, B.A.; Mucenski, M.L.; Mason, R.J.; Shannon, J.M. Identification and characterization of a lysophosphatidylcholine acyltransferase in alveolar type II cells. Proc. Natl. Acad. Sci. USA 2006, 103, 11724–11729. [Google Scholar] [CrossRef] [PubMed]
- Soupene, E.; Fyrst, H.; Kuypers, F.A. Mammalian acyl-CoA:lysophosphatidylcholine acyltransferase enzymes. Proc. Natl. Acad. Sci. USA 2008, 105, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Harayama, T.; Shindou, H.; Shimizu, T. Biosynthesis of phosphatidylcholine by human lysophosphatidylcholine acyltransferase 1. J. Lipid Res. 2009, 50, 1824–1831. [Google Scholar] [CrossRef] [PubMed]
- Bridges, J.P.; Ikegami, M.; Brilli, L.L.; Chen, X.; Mason, R.J.; Shannon, J.M. LPCAT1 regulates surfactant phospholipid synthesis and is required for transitioning to air breathing in mice. J. Clin. Invest. 2010, 120, 1736–1748. [Google Scholar] [CrossRef] [PubMed]
- Harayama, T.; Eto, M.; Shindou, H.; Kita, Y.; Otsubo, E.; Hishikawa, D.; Ishii, S.; Sakimura, K.; Mishina, M.; Shimizu, T. Lysophospholipid acyltransferases mediate phosphatidylcholine diversification to achieve the physical properties required in vivo. Cell. Metab. 2014, 20, 295–305. [Google Scholar] [PubMed]
- Friedman, J.S.; Chang, B.; Krauth, D.S.; Lopez, I.; Waseem, N.H.; Hurd, R.E.; Feathers, K.L.; Branham, K.E.; Shaw, M.; Thomas, G.E.; et al. Loss of lysophosphatidylcholine acyltransferase 1 leads to photoreceptor degeneration in rd11 mice. Proc. Natl. Acad. Sci. USA 2010, 107, 15523–15528. [Google Scholar] [PubMed]
- Butler, P.L.; Mallampalli, R.K. Cross-talk between remodeling and de novo pathways maintains phospholipid balance through ubiquitination. J. Biol. Chem. 2010, 285, 6246–6258. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Butler, P.L.; Coon, T.A.; Smith, R.M.; Hammen, G.; Zhao, Y.; Chen, B.B.; Mallampalli, R.K. LPS impairs phospholipid synthesis by triggering beta-transducin repeat-containing protein (beta-TrCP)-mediated polyubiquitination and degradation of the surfactant enzyme acyl-CoA:lysophosphatidylcholine acyltransferase I (LPCAT1). J. Biol. Chem. 2011, 286, 2719–2727. [Google Scholar] [CrossRef] [PubMed]
- Moessinger, C.; Kuerschner, L.; Spandl, J.; Shevchenko, A.; Thiele, C. Human lysophosphatidylcholine acyltransferases 1 and 2 are located in lipid droplets where they catalyze the formation of phosphatidylcholine. J. Biol. Chem. 2011, 286, 21330–21339. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Ellis, B.M.; Smith, R.M.; Chen, B.B.; Zhao, Y.; Mallampalli, R.K. Acyl-CoA:lysophosphatidylcholine acyltransferase I (Lpcat1) catalyzes histone protein O-palmitoylation to regulate mRNA synthesis. J. Biol. Chem. 2011, 286, 28019–28025. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Lawrence, T.J.; He, Z.; Pound, C.R.; Mao, J.; Bigler, S.A. The expression level of lysophosphatidylcholine acyltransferase 1 (LPCAT1) correlates to the progression of prostate cancer. Exp. Mol. Pathol. 2011, 92, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Mansilla, F.; da Costa, K.A.; Wang, S.; Kruhøffer, M.; Lewin, T.M.; Orntoft, T.F.; Coleman, R.A.; Birkenkamp-Demtröder, K. Lysophosphatidylcholine acyltransferase 1 (LPCAT1) overexpression in human colorectal cancer. J. Mol. Med. (Berl.) 2009, 87, 85–97. [Google Scholar] [CrossRef]
- Shindou, H.; Hishikawa, D.; Nakanishi, H.; Harayama, T.; Ishii, S.; Taguchi, R.; Shimizu, T. A single enzyme catalyzes both platelet-activating factor production and membrane biogenesis of inflammatory cells. Cloning and characterization of acetyl-CoA:Lyso-PAF acetyltransferase. J. Biol. Chem. 2007, 282, 6532–6539. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.K.; Garg, A. Enzymatic activity of the human 1-acylglycerol-3-phosphate-O-acyltransferase isoform 11: Upregulated in breast and cervical cancers. J. Lipid Res. 2010, 51, 2143–2152. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.; Shindou, H.; Oda, Y.; Shimizu, T. Phosphorylation of lysophosphatidylcholine acyltransferase 2 at Ser34 enhances platelet-activating factor production in endotoxin-stimulated macrophages. J. Biol. Chem. 2010, 285, 29857–29862. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.; Shindou, H.; Tarui, M.; Shimizu, T. Rapid production of platelet-activating factor is induced by protein kinase Cα-mediated phosphorylation of lysophosphatidylcholine acyltransferase 2 protein. J. Biol. Chem. 2014, 289, 15566–15576. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Yasuda, K.; Matsuyama, T.; Ishihara, T.; Higa, R.; Sawairi, T.; Yamaguchi, M.; Egi, M.; Akai, S.; Miyase, T.; Ikari, A.; Miwa, M.; Sugatani, J. A Penicillium. sp. F33 metabolite and its synthetic derivatives inhibit acetyl-CoA:1-O-alkyl-sn-glycero-3-phosphocholine acetyltransferase (a key enzyme in platelet-activating factor biosynthesis) and carrageenan-induced paw edema in mice. Biochem. Pharmacol. 2013, 86, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Tarui, M.; Shindou, H.; Kumagai, K.; Morimoto, R.; Harayama, T.; Hashidate, T.; Kojima, H.; Okabe, T.; Nagano, T.; Nagase, T.; et al. Selective inhibitors of a PAF biosynthetic enzyme lysophosphatidylcholine acyltransferase 2. J. Lipid Res. 2014, 55, 1386–1396. [Google Scholar] [CrossRef] [PubMed]
- Bouchoux, J.; Beilstein, F.; Pauquai, T.; Guerrera, I.C.; Chateau, D.; Ly, N.; Alqub, M.; Klein, C.; Chambaz, J.; Rousset, M.; et al. The proteome of cytosolic lipid droplets isolated from differentiated Caco-2/TC7 enterocytes reveals cell-specific characteristics. Biol. Cell 2011, 103, 499–517. [Google Scholar] [CrossRef] [PubMed]
- Athenstaedt, K.; Daum, G. Phosphatidic acid, a key intermediate in lipid metabolism. Eur. J. Biochem. 1999, 266, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, A.; Clem, B.; Makoni, S.; Clem, A.; Nelson, K.; Thornburg, J.; Siow, D.; Lane, A.N.; Brock, S.E.; Goswami, U.; et al. Selective inhibition of choline kinase simultaneously attenuates MAPK and PI3K/AKT signaling. Oncogene 2010, 29, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Du, G. Phosphatidic acid signaling regulation of Ras superfamily of small guanosine triphosphatases. Biochim. Biophys. Acta 2009, 1791, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.M.; Chen, C.; Huang, S.; Han, D.D.; Guo, J.H.; Wan, B.; Yu, L. Cloning and characterization a novel human 1-acyl-sn-glycerol-3-phosphate acyltransferase gene AGPAT7. DNA Seq. 2005, 16, 386–390. [Google Scholar] [PubMed]
- Cao, J.; Shan, D.; Revett, T.; Li, D.; Wu, L.; Liu, W.; Tobin, J.F.; Gimeno, R.E. Molecular identification of a novel mammalian brain isoform of acyl-CoA:lysophospholipid acyltransferase with prominent ethanolamine lysophospholipid acylating activity, LPEAT2. J. Biol. Chem. 2008, 283, 19049–19057. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Liu, Y.; Lockwood, J.; Burn, P.; Shi, Y. A novel cardiolipin-remodeling pathway revealed by a gene encoding an endoplasmic reticulum-associated acyl-CoA:lysocardiolipin acyltransferase (ALCAT1) in mouse. J. Biol. Chem. 2004, 279, 31727–31734. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Romestaing, C.; Han, X.; Li, Y.; Hao, X.; Wu, Y.; Sun, C.; Liu, X.; Jefferson, L.S.; Xiong, J.; et al. Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab. 2010, 12, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Shen, W.; Chang, Z.; Shi, Y. ALCAT1 is a polyglycerophospholipid acyltransferase potently regulated by adenine nucleotide and thyroid status. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E647–E653. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ye, B.; Miller, S.; Yuan, H.; Zhang, H.; Tian, L.; Nie, J.; Imae, R.; Arai, H.; Li, Y.; et al. Ablation of ALCAT1 mitigates hypertrophic cardiomyopathy through effects on oxidative stress and mitophagy. Mol. Cell Biol. 2012, 32, 4493–4504. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.K.; Barnes, R.I.; Garg, A. Functional characterization of human 1-acylglycerol-3-phosphate acyltransferase isoform 8: Cloning, tissue distribution, gene structure, and enzymatic activity. Arch. Biochem. Biophys. 2006, 449, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chen, Y.Q.; Li, S.; Konrad, R.J.; Cao, G. The microsomal cardiolipin remodeling enzyme acyl-CoA lysocardiolipin acyltransferase is an acyltransferase of multiple anionic lysophospholipids. J. Lipid Res. 2009, 50, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Le Guédard, M.; Bessoule, J.J.; Boyer, V.; Ayciriex, S.; Velours, G.; Kulik, W.; Ejsing, C.S.; Shevchenko, A.; Coulon, D.; Lessire, R.; et al. PSI1 is responsible for the stearic acid enrichment that is characteristic of phosphatidylinositol in yeast. FEBS J. 2009, 276, 6412–6424. [Google Scholar] [CrossRef] [PubMed]
- Imae, R.; Inoue, T.; Kimura, M.; Kanamori, T.; Tomioka, N.H.; Kage-Nakadai, E.; Mitani, S.; Arai, H. Intracellular phospholipase A1 and acyltransferase, which are involved in Caenorhabditis elegans stem cell divisions, determine the sn-1 fatty acyl chain of phosphatidylinositol. Mol. Biol. Cell 2010, 21, 3114–3124. [Google Scholar] [CrossRef] [PubMed]
- Imae, R.; Inoue, T.; Nakasaki, Y.; Uchida, Y.; Ohba, Y.; Kono, N.; Nakanishi, H.; Sasaki, T.; Mitani, S.; Arai, H. LYCAT, a homologue of C. elegans acl-8, acl-9, and acl-10, determines the fatty acid composition of phosphatidylinositol in mice. J. Lipid Res. 2012, 53, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Faloon, P.W.; Tan, Z.; Lv, Y.; Zhang, P.; Ge, Y.; Deng, H.; Xiong, J.W. Mouse lysocardiolipin acyltransferase controls the development of hematopoietic and endothelial lineages during in vitro embryonic stem-cell differentiation. Blood 2007, 110, 3601–3609. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.W.; Yu, Q.; Zhang, J.; Mably, J.D. An acyltransferase controls the generation of hematopoietic and endothelial lineages in zebrafish. Circ. Res. 2008, 102, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Barth, P.G.; Scholte, J.A.; Berden, J.A.; van der Klei-Van Moorsel, J.M.; Luyt-Houwen, I.E.M.; Van’t Veer-Korthof, E.T.; van der Harten, J.J.; Sobotka-Plojhar, M.A. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 1983, 62, 327–355. [Google Scholar] [CrossRef] [PubMed]
- Bione, S.; D’Adamo, P.; Maestrini, E.; Gedeon, A.K.; Bolhuis, P.A.; Toniolo, D. A novel X-linked gene, G4.5, is responsible for Barth syndrome. Nat. Genet. 1996, 12, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Schlame, M.; Towbin, J.A.; Heerdt, P.M.; Jehle, R.; DiMauro, S.; Blanck, T.J.J. Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann. Neurol. 2002, 51, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Kelley, R.I.; Blanck, T.J.; Schlame, M. Remodeling of cardiolipin by phospholipid transacylation. J. Biol. Chem. 2003, 278, 51380–51385. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Malhotra, A.; Ren, M.; Schlame, M. The enzymatic function of tafazzin. J. Biol. Chem. 2006, 281, 39217–39224. [Google Scholar] [PubMed]
- Malhotra, A.; Xu, Y.; Ren, M.; Schlame, M. Formation of molecular species of mitochondrial cardiolipin. 1. A novel transacylation mechanism to shuttle fatty acids between sn-1 and sn-2 positions of multiple phospholipid species. Biochim. Biophys. Acta 2009, 1791, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Soustek, M.S.; Falk, D.J.; Mah, C.S.; Toth, M.J.; Schlame, M.; Lewin, A.S.; Byrne, B.J. Characterization of a transgenic short hairpin RNA-induced murine model of Tafazzin deficiency. Hum. Gene Ther. 2011, 22, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Cao, J.; Shi, Y. Identification and characterization of a gene encoding human LPGAT1, an endoplasmic reticulum-associated lysophosphatidylglycerol acyltransferase. J. Biol. Chem. 2004, 279, 55866–55874. [Google Scholar] [CrossRef] [PubMed]
- Hiramine, Y.; Emoto, H.; Takasuga, S.; Hiramatsu, R. Novel acyl-coenzyme A: Monoacylglycerol acyltransferase plays an important role in hepatic triacylglycerol secretion. J. Lipid Res. 2010, 51, 1424–1431. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamashita, A.; Hayashi, Y.; Matsumoto, N.; Nemoto-Sasaki, Y.; Oka, S.; Tanikawa, T.; Sugiura, T. Glycerophosphate/Acylglycerophosphate Acyltransferases. Biology 2014, 3, 801-830. https://doi.org/10.3390/biology3040801
Yamashita A, Hayashi Y, Matsumoto N, Nemoto-Sasaki Y, Oka S, Tanikawa T, Sugiura T. Glycerophosphate/Acylglycerophosphate Acyltransferases. Biology. 2014; 3(4):801-830. https://doi.org/10.3390/biology3040801
Chicago/Turabian StyleYamashita, Atsushi, Yasuhiro Hayashi, Naoki Matsumoto, Yoko Nemoto-Sasaki, Saori Oka, Takashi Tanikawa, and Takayuki Sugiura. 2014. "Glycerophosphate/Acylglycerophosphate Acyltransferases" Biology 3, no. 4: 801-830. https://doi.org/10.3390/biology3040801