
Developmental Control of NRAMP1 (SLC11A1) Expression in Professional Phagocytes

Abstract
:1. Introduction
2. Results and Discussion
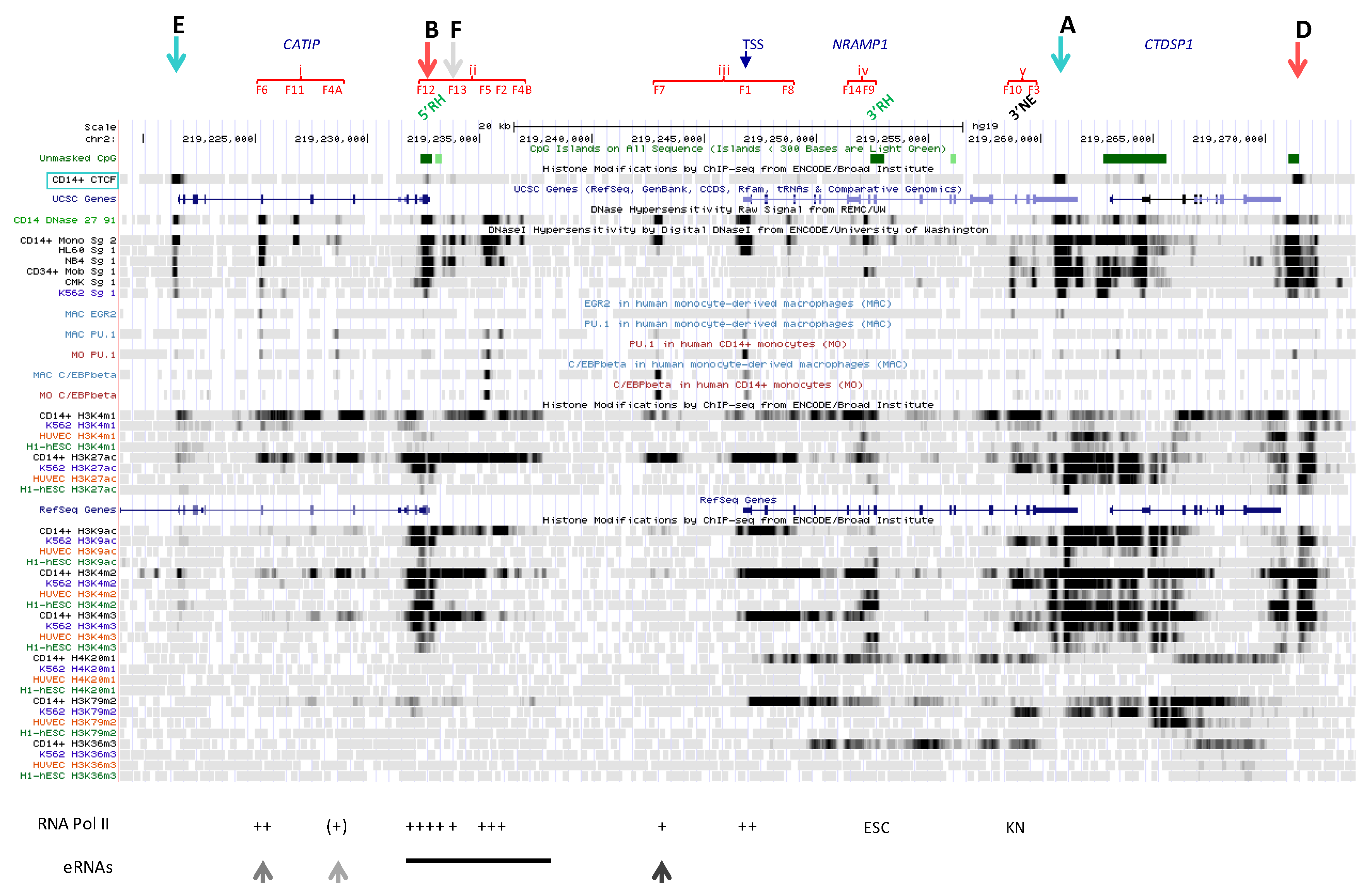
2.1. Functional Delineation of NRAMP1 Locus by CTCF Sites
2.2. Regulatory Determinants Identified by Cap Analysis of NRAMP1 Expression
2.2.1. BloodCAGE (TrackHub CAGE of Haematopoietic Cell Types)
2.2.1.1. NRAMP1 Expressing Blood Cells
2.2.1.2. Blood Cells Not Expressing NRAMP1
2.2.1.3. Blood Cells Expressing NRAMP1 at Intermediate Levels
2.2.2. NRAMP1 CAGE in AML Cells
2.2.2.1. An Alternative NRAMP1 TSS in AMLs?
2.2.2.2. Correlating CAGE Signals and Other Marks of NRAMP1 Expression in Myeloid Leukemias
2.2.3. Sites Showing Bidirectional CAGE Signals in NRAMP1 Expressing Cells
2.2.3.1. Upstream Ubiquitous Regulatory Hub (DHS F12)
2.2.3.2. Intermediate Cluster of Myeloid-Specific TF Binding Sites (DHS F13)
2.2.3.3. Complex Cluster of Myelomocytic-Specific Binding Sites (DHS F5-F2)
2.2.3.4. Candidate Super-Enhancer Domain
2.2.3.5. Simple Myelo-Monocytic Proximal Enhancer (DHS F7)
2.2.3.6. DNase Footprint in NRAMP1 Intron 3 (DHS F8)
2.3. Other Candidate NRAMP1 Regulatory Elements
2.3.1. Determinants Partially CAGE Positive
2.3.1.1. Upstream Footprint That Overlaps CATIP Exon V (DHS F6)
2.3.1.2. Pair of DNase1 Footprints in CATIP Intron 6 (DHS F4A)
2.3.1.3. NRAMP1 TSS Area (DHS F1)
2.3.1.4. Putative Element in NRAMP1 Intron 5 (DHS F14)
2.3.1.5. Candidate Regulatory Hub within NRAMP1 ORF (DHS F9)
2.3.1.6. Negative Regulatory Element in NRAMP1 Intron 12 (DHS F10)
2.3.1.7. Potential Negative Element Overlapping NRAMP1 Exon XV (DHS F3)
2.3.2. CAGE Negative Determinants
2.3.2.1. Seemingly Primed 5′ Element (DHS F11)
2.3.2.2. Potential Element at the 3′ Boundary of NRAMP1 Candidate S-E (DHS F4B)
2.3.3. CTCF Sites at NRAMP1 Locus
2.3.4. Area Downstream of NRAMP1 3′ CTCF
2.3.5. Autonomous Regulation of NRAMP1 Locus
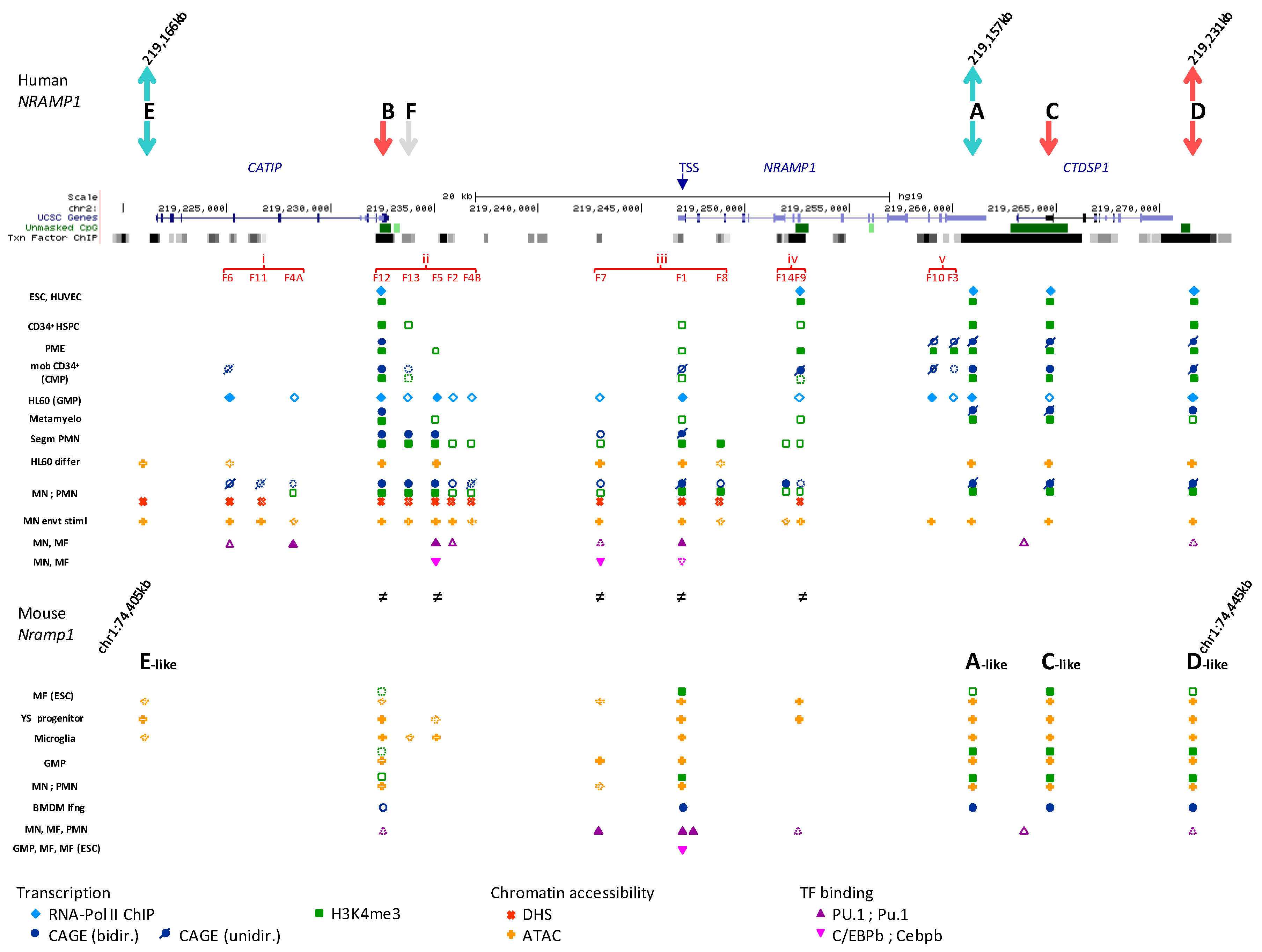
2.4. Recapitulating the Process of NRAMP1 Gene Activation during Development
2.4.1. Reappraisal of NRAMP1 Tissue-Specificity Using Large Scale Transcriptomic Datasets
2.4.2. Segmenting NRAMP1 Locus in Five Regulatory Regions
2.4.3. Regional Predictions of Potential TFBSs
2.4.4. NRAMP1 Locus Activity in Early Developmental Stages of Hematopoiesis
2.4.5. Activation of NRAMP1 Regulatory Regions i–v during Myelo-Monopoiesis
2.4.5.1. Region ii: 5′ Regulatory Hub and Candidate Super-Enhancer
2.4.5.2. Region i: Most Upstream Elements
2.4.5.3. Region iii: Around NRAMP1 TSS
2.4.5.4. Region iv: Intragenic 3′ Regulatory Hub
2.4.5.5. Region v: NRAMP1 3′ Negative Elements
2.4.6. Myelo-Monocytic Programming of NRAMP1 Gene Expression
2.5. Transcriptomic Analyses of NRAMP1 and Other Genes Contributing to MF Iron Trafficking
2.6. Expression Studies of the Mouse Ortholog Nramp1
2.6.1. Nramp1 Locus Organization
2.6.2. Regulation of Expression
2.6.3. Predicted Regulatory Elements
2.6.4. TF Binding at Nramp1 Locus
3. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Forbes, J.R.; Gros, P. Divalent-metal transport by NRAMP proteins at the interface of host-pathogen interactions. Trends Microbiol. 2001, 9, 397–403. [Google Scholar] [CrossRef]
- Cesar-Razquin, A.; Snijder, B.; Frappier-Brinton, T.; Isserlin, R.; Gyimesi, G.; Bai, X.; Reithmeier, R.A.; Hepworth, D.; Hediger, M.A.; Edwards, A.M.; et al. A Call for Systematic Research on Solute Carriers. Cell 2015, 162, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Cellier, M.F. Nutritional immunity: Homology modeling of Nramp metal import. Adv. Exp. Med. Biol. 2012, 946, 335–351. [Google Scholar] [PubMed]
- Soe-Lin, S.; Apte, S.S.; Mikhael, M.R.; Kayembe, L.K.; Nie, G.; Ponka, P. Both Nramp1 and DMT1 are necessary for efficient macrophage iron recycling. Exp. Hematol. 2010, 38, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Delaby, C.; Rondeau, C.; Pouzet, C.; Willemetz, A.; Pilard, N.; Desjardins, M.; Canonne-Hergaux, F. Subcellular localization of iron and heme metabolism related proteins at early stages of erythrophagocytosis. PLoS ONE 2012, 7, e42199. [Google Scholar] [CrossRef]
- Peracino, B.; Wagner, C.; Balest, A.; Balbo, A.; Pergolizzi, B.; Noegel, A.A.; Steinert, M.; Bozzaro, S. Function and mechanism of action of Dictyostelium Nramp1 (Slc11a1) in bacterial infection. Traffic 2006, 7, 22–38. [Google Scholar] [CrossRef] [PubMed]
- Peracino, B.; Buracco, S.; Bozzaro, S. The Nramp (Slc11) proteins regulate development, resistance to pathogenic bacteria and iron homeostasis in Dictyostelium discoideum. J. Cell Sci. 2013, 126 Pt 1, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Buracco, S.; Peracino, B.; Cinquetti, R.; Signoretto, E.; Vollero, A.; Imperiali, F.; Castagna, M.; Bossi, E.; Bozzaro, S. Dictyostelium Nramp1, which is structurally and functionally similar to mammalian DMT1 transporter, mediates phagosomal iron efflux. J. Cell Sci. 2015, 128, 3304–3316. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Thurman, R.E.; Rynes, E.; Humbert, R.; Vierstra, J.; Maurano, M.T.; Haugen, E.; Sheffield, N.C.; Stergachis, A.B.; Wang, H.; Vernot, B.; et al. The accessible chromatin landscape of the human genome. Nature 2012, 489, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Cellier, M.F. Cell-Type Specific Determinants of NRAMP1 Expression in Professional Phagocytes. Biology 2013, 2, 233–283. [Google Scholar] [CrossRef] [PubMed]
- Richer, E.; Campion, C.G.; Dabbas, B.; White, J.H.; Cellier, M.F. Transcription factors Sp1 and C/EBP regulate NRAMP1 gene expression. FEBS J. 2008, 275, 5074–5089. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.H.; Benner, C.; Lichtinger, M.; Schwarzfischer, L.; Hu, Y.; Andreesen, R.; Chen, W.; Rehli, M. Dynamic epigenetic enhancer signatures reveal key transcription factors associated with monocytic differentiation states. Blood 2012, 119, e161–e171. [Google Scholar] [CrossRef] [PubMed]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M. J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Saeed, S.; Quintin, J.; Kerstens, H.H.; Rao, N.A.; Aghajanirefah, A.; Matarese, F.; Cheng, S.C.; Ratter, J.; Berentsen, K.; van der Ent, M.A.; et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 2014, 345, 1251086. [Google Scholar] [CrossRef] [PubMed]
- Andersson, R.; Gebhard, C.; Miguel-Escalada, I.; Hoof, I.; Bornholdt, J.; Boyd, M.; Chen, Y.; Zhao, X.; Schmidl, C.; Suzuki, T.; et al. An atlas of active enhancers across human cell types and tissues. Nature 2014, 507, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Forrest, A.R.; Kawaji, H.; Rehli, M.; Baillie, J.K.; de Hoon, M.J.; Haberle, V.; Lassmann, T.; Kulakovskiy, I.V.; Lizio, M.; Itoh, M.; et al. A promoter-level mammalian expression atlas. Nature 2014, 507, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Vernimmen, D.; Bickmore, W.A. The Hierarchy of Transcriptional Activation: From Enhancer to Promoter. Trends Genet. 2015, 31, 696–708. [Google Scholar] [CrossRef] [PubMed]
- Pott, S.; Lieb, J.D. What are super-enhancers? Nat. Genet. 2015, 47, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Witte, S.; O’Shea, J.J.; Vahedi, G. Super-enhancers: Asset management in immune cell genomes. Trends Immunol. 2015, 36, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Soufi, A.; Garcia, M.F.; Jaroszewicz, A.; Osman, N.; Pellegrini, M.; Zaret, K.S. Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 2015, 161, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Romanoski, C.E.; Link, V.M.; Heinz, S.; Glass, C.K. Exploiting genomics and natural genetic variation to decode macrophage enhancers. Trends Immunol. 2015, 36, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Zaret, K.S.; Mango, S.E. Pioneer transcription factors, chromatin dynamics, and cell fate control. Curr. Opin. Genet. Dev. 2016, 37, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Schubeler, D. Function and information content of DNA methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Lara-Astiaso, D.; Weiner, A.; Lorenzo-Vivas, E.; Zaretsky, I.; Jaitin, D.A.; David, E.; Keren-Shaul, H.; Mildner, A.; Winter, D.; Jung, S.; et al. Immunogenetics. Chromatin state dynamics during blood formation. Science 2014, 345, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010, 465, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Garcia-Bassets, I.; Benner, C.; Li, W.; Su, X.; Zhou, Y.; Qiu, J.; Liu, W.; Kaikkonen, M.U.; Ohgi, K.A.; et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 2011, 474, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Melo, C.A.; Drost, J.; Wijchers, P.J.; van de Werken, H.; de Wit, E.; Oude Vrielink, J.A.; Elkon, R.; Melo, S.A.; Leveille, N.; Kalluri, R.; et al. eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol. Cell 2013, 49, 524–535. [Google Scholar] [CrossRef] [PubMed]
- Jiao, A.L.; Slack, F.J. RNA-mediated gene activation. Epigenetics 2014, 9, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.T.; Li, W.; Rosenfeld, M.G.; Glass, C.K. Enhancer RNAs and regulated transcriptional programs. Trends Biochem. Sci. 2014, 39, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Murakawa, Y.; Yoshihara, M.; Kawaji, H.; Nishikawa, M.; Zayed, H.; Suzuki, H.; Fantom Consortium; Hayashizaki, Y. Enhanced Identification of Transcriptional Enhancers Provides Mechanistic Insights into Diseases. Trends Genet. 2016, 32, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, K.; Zare, H.; Koulnis, M.; Sartorelli, V. The emerging roles of eRNAs in transcriptional regulatory networks. RNA Biol. 2014, 11, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Kaikkonen, M.U.; Spann, N.J.; Heinz, S.; Romanoski, C.E.; Allison, K.A.; Stender, J.D.; Chun, H.B.; Tough, D.F.; Prinjha, R.K.; Benner, C.; et al. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol. Cell 2013, 51, 310–325. [Google Scholar] [CrossRef] [PubMed]
- Heinz, S.; Romanoski, C.E.; Benner, C.; Glass, C.K. The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol. 2015, 16, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Sigova, A.A.; Abraham, B.J.; Ji, X.; Molinie, B.; Hannett, N.M.; Guo, Y.E.; Jangi, M.; Giallourakis, C.C.; Sharp, P.A.; Young, R.A. Transcription factor trapping by RNA in gene regulatory elements. Science 2015, 350, 978–981. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Nord, A.S.; Akiyama, J.A.; Shoukry, M.; Afzal, V.; Rubin, E.M.; Pennacchio, L.A.; Visel, A. Tissue-specific RNA expression marks distant-acting developmental enhancers. PLoS Genet. 2014, 10, e1004610. [Google Scholar] [CrossRef] [PubMed]
- IIott, N.E.; Heward, J.A.; Roux, B.; Tsitsiou, E.; Fenwick, P.S.; Lenzi, L.; Goodhead, I.; Hertz-Fowler, C.; Heger, A.; Hall, N.; et al. Long non-coding RNAs and enhancer RNAs regulate the lipopolysaccharide-induced inflammatory response in human monocytes. Nat. Commun. 2014, 5, 3979. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, D.; Glass, C.K. Epigenomics of macrophages. Immunol. Rev. 2014, 262, 96–112. [Google Scholar] [CrossRef] [PubMed]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Adam, R.C.; Yang, H.; Rockowitz, S.; Larsen, S.B.; Nikolova, M.; Oristian, D.S.; Polak, L.; Kadaja, M.; Asare, A.; Zheng, D.; et al. Pioneer factors govern super-enhancer dynamics in stem cell plasticity and lineage choice. Nature 2015, 521, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Schuijers, J.; Lin, C.Y.; Weintraub, A.S.; Abraham, B.J.; Lee, T.I.; Bradner, J.E.; Young, R.A. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol. Cell 2015, 58, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Hah, N.; Benner, C.; Chong, L.W.; Yu, R.T.; Downes, M.; Evans, R.M. Inflammation-sensitive super enhancers form domains of coordinately regulated enhancer RNAs. Proc. Natl. Acad. Sci. USA 2015, 112, E297–E302. [Google Scholar] [CrossRef] [PubMed]
- Stasevich, T.J.; Hayashi-Takanaka, Y.; Sato, Y.; Maehara, K.; Ohkawa, Y.; Sakata-Sogawa, K.; Tokunaga, M.; Nagase, T.; Nozaki, N.; McNally, J.G.; et al. Regulation of RNA polymerase II activation by histone acetylation in single living cells. Nature 2014, 516, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Yu, L.R.; Wang, L.; Zhang, Z.; Kasper, L.H.; Lee, J.E.; Wang, C.; Brindle, P.K.; Dent, S.Y.; Ge, K. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 2011, 30, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Sun, L.; Chen, Z.; Whitaker, J.W.; Wang, T.; Wang, W. Predicting enhancer transcription and activity from chromatin modifications. Nucleic Acids Res. 2013, 41, 10032–10043. [Google Scholar] [CrossRef] [PubMed]
- Schwalb, B.; Michel, M.; Zacher, B.; Fruhauf, K.; Demel, C.; Tresch, A.; Gagneur, J.; Cramer, P. TT-seq maps the human transient transcriptome. Science 2016, 352, 1225–1228. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, K.R.; Armstrong, J.; Barber, G.P.; Casper, J.; Clawson, H.; Diekhans, M.; Dreszer, T.R.; Fujita, P.A.; Guruvadoo, L.; Haeussler, M.; et al. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res. 2015, 43, D670–D681. [Google Scholar] [CrossRef] [PubMed]
- Danko, C.G.; Hyland, S.L.; Core, L.J.; Martins, A.L.; Waters, C.T.; Lee, H.W.; Cheung, V.G.; Kraus, W.L.; Lis, J.T.; Siepel, A. Identification of active transcriptional regulatory elements from GRO-seq data. Nat. Methods 2015, 12, 433–438. [Google Scholar] [CrossRef] [PubMed]
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar]
- Fullwood, M.J.; Han, Y.; Wei, C.L.; Ruan, X.; Ruan, Y. Chromatin interaction analysis using paired-end tag sequencing. Curr. Protoc. Mol. Biol. 2010. [Google Scholar] [CrossRef]
- Li, G.; Fullwood, M.J.; Xu, H.; Mulawadi, F.H.; Velkov, S.; Vega, V.; Ariyaratne, P.N.; Mohamed, Y.B.; Ooi, H.S.; Tennakoon, C.; et al. ChIA-PET tool for comprehensive chromatin interaction analysis with paired-end tag sequencing. Genome Biol. 2010, 11, R22. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Ruan, X.; Auerbach, R.K.; Sandhu, K.S.; Zheng, M.; Wang, P.; Poh, H.M.; Goh, Y.; Lim, J.; Zhang, J.; et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 2012, 148, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Nedelec, Y.; Sanz, J.; Baharian, G.; Szpiech, Z.A.; Pacis, A.; Dumaine, A.; Grenier, J.C.; Freiman, A.; Sams, A.J.; Hebert, S.; et al. Genetic Ancestry and Natural Selection Drive Population Differences in Immune Responses to Pathogens. Cell 2016, 167, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Rudan, M.V.; Hadjur, S.; Sexton, T. Detecting Spatial Chromatin Organization by Chromosome Conformation Capture II: Genome-Wide Profiling by Hi-C. Methods Mol. Biol. 2017, 1589, 47–74. [Google Scholar]
- Ji, X.; Dadon, D.B.; Powell, B.E.; Fan, Z.P.; Borges-Rivera, D.; Shachar, S.; Weintraub, A.S.; Hnisz, D.; Pegoraro, G.; Lee, T.I.; et al. 3D Chromosome Regulatory Landscape of Human Pluripotent Cells. Cell Stem Cell 2016, 18, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Nichols, M.H.; Corces, V.G. A CTCF Code for 3D Genome Architecture. Cell 2015, 162, 703–705. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Xu, Q.; Canzio, D.; Shou, J.; Li, J.; Gorkin, D.U.; Jung, I.; Wu, H.; Zhai, Y.; Tang, Y.; et al. CRISPR Inversion of CTCF Sites Alters Genome Topology and Enhancer/Promoter Function. Cell 2015, 162, 900–910. [Google Scholar] [CrossRef] [PubMed]
- De Wit, E.; Vos, E.S.; Holwerda, S.J.; Valdes-Quezada, C.; Verstegen, M.J.; Teunissen, H.; Splinter, E.; Wijchers, P.J.; Krijger, P.H.; de Laat, W. CTCF Binding Polarity Determines Chromatin Looping. Mol. Cell 2015, 60, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Fudenberg, G.; Imakaev, M.; Lu, C.; Goloborodko, A.; Abdennur, N.; Mirny, L.A. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 2016, 15, 2038–2049. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhuang, J.; Iyer, S.; Lin, X.Y.; Greven, M.C.; Kim, B.H.; Moore, J.; Pierce, B.G.; Dong, X.; Virgil, D.; et al. Factorbook.org: A Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. Nucleic Acids Res. 2013, 41, D171–D176. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Monahan, K.; Wu, H.; Gertz, J.; Varley, K.E.; Li, W.; Myers, R.M.; Maniatis, T.; Wu, Q. CTCF/cohesin-mediated DNA looping is required for protocadherin alpha promoter choice. Proc. Natl. Acad. Sci. USA 2012, 109, 21081–21086. [Google Scholar] [CrossRef] [PubMed]
- Mathelier, A.; Fornes, O.; Arenillas, D.J.; Chen, C.Y.; Denay, G.; Lee, J.; Shi, W.; Shyr, C.; Tan, G.; Worsley-Hunt, R.; et al. JASPAR 2016: A major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2016, 44, D110–D115. [Google Scholar] [CrossRef] [PubMed]
- Shiraki, T.; Kondo, S.; Katayama, S.; Waki, K.; Kasukawa, T.; Kawaji, H.; Kodzius, R.; Watahiki, A.; Nakamura, M.; Arakawa, T.; et al. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc. Natl. Acad. Sci. USA 2003, 100, 15776–15781. [Google Scholar] [CrossRef] [PubMed]
- Duttke, S.H.; Lacadie, S.A.; Ibrahim, M.M.; Glass, C.K.; Corcoran, D.L.; Benner, C.; Heinz, S.; Kadonaga, J.T.; Ohler, U. Perspectives on Unidirectional versus Divergent Transcription. Mol. Cell 2015, 60, 348–349. [Google Scholar] [CrossRef] [PubMed]
- Andersson, R.; Chen, Y.; Core, L.; Lis, J.T.; Sandelin, A.; Jensen, T.H. Human Gene Promoters Are Intrinsically Bidirectional. Mol. Cell 2015, 60, 346–347. [Google Scholar] [CrossRef] [PubMed]
- Duttke, S.H.; Lacadie, S.A.; Ibrahim, M.M.; Glass, C.K.; Corcoran, D.L.; Benner, C.; Heinz, S.; Kadonaga, J.T.; Ohler, U. Human promoters are intrinsically directional. Mol. Cell 2015, 57, 674–684. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Pooley, C.; Freeman, T.C.; Lennartsson, A.; Babina, M.; Schmidl, C.; Geijtenbeek, T.; Michoel, T.; Severin, J.; Itoh, M.; et al. Transcription factor, promoter, and enhancer utilization in human myeloid cells. J. Leukoc. Biol. 2015, 97, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kim, Y.; Genoud, N.; Gao, J.; Kelly, J.W.; Pfaff, S.L.; Gill, G.N.; Dixon, J.E.; Noel, J.P. Determinants for dephosphorylation of the RNA polymerase II C-terminal domain by Scp1. Mol. Cell 2006, 24, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Yeo, M.; Lin, P.S.; Dahmus, M.E.; Gill, G.N. A novel RNA polymerase II C-terminal domain phosphatase that preferentially dephosphorylates serine 5. J. Biol. Chem. 2003, 278, 26078–26085. [Google Scholar] [CrossRef] [PubMed]
- Marquet, S.; Lepage, P.; Hudson, T.J.; Musser, J.M.; Schurr, E. Complete nucleotide sequence and genomic structure of the human NRAMP1 gene region on chromosome region 2q35. Mamm. Genome 2000, 11, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.J.; Setty, M.; Leslie, C.S. Early enhancer establishment and regulatory locus complexity shape transcriptional programs in hematopoietic differentiation. Nat. Genet. 2015, 47, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Bontems, F.; Fish, R.J.; Borlat, I.; Lembo, F.; Chocu, S.; Chalmel, F.; Borg, J.P.; Pineau, C.; Neerman-Arbez, M.; Bairoch, A.; et al. C2orf62 and TTC17 are involved in actin organization and ciliogenesis in zebrafish and human. PLoS ONE 2014, 9, e86476. [Google Scholar] [CrossRef] [PubMed]
- Cellier, M.; Shustik, C.; Dalton, W.; Rich, E.; Hu, J.; Malo, D.; Schurr, E.; Gros, P. Expression of the human NRAMP1 gene in professional primary phagocytes: Studies in blood cells and in HL-60 promyelocytic leukemia. J. Leukoc. Biol. 1997, 61, 96–105. [Google Scholar] [PubMed]
- Kowalczyk, M.S.; Hughes, J.R.; Garrick, D.; Lynch, M.D.; Sharpe, J.A.; Sloane-Stanley, J.A.; McGowan, S.J.; De, G.M.; Hosseini, M.; Vernimmen, D.; et al. Intragenic enhancers act as alternative promoters. Mol. Cell 2012, 45, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Roig, E.A.; Richer, E.; Canonne-Hergaux, F.; Gros, P.; Cellier, M.F. Regulation of NRAMP1 gene expression by 1alpha,25-dihydroxy-vitamin D(3) in HL-60 phagocytes. J. Leukoc. Biol. 2002, 71, 890–904. [Google Scholar] [PubMed]
- Canonne-Hergaux, F.; Calafat, J.; Richer, E.; Cellier, M.; Grinstein, S.; Borregaard, N.; Gros, P. Expression and subcellular localization of NRAMP1 in human neutrophil granules. Blood 2002, 100, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Neph, S.; Vierstra, J.; Stergachis, A.B.; Reynolds, A.P.; Haugen, E.; Vernot, B.; Thurman, R.E.; John, S.; Sandstrom, R.; Johnson, A.K.; et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 2012, 489, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Sugi, Y.; Takahashi, K.; Nakano, K.; Hosono, A.; Kaminogawa, S. Transcription of the Tollip gene is elevated in intestinal epithelial cells through impaired O-GlcNAcylation-dependent nuclear translocation of the negative regulator Elf-1. Biochem. Biophys. Res. Commun. 2011, 412, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Nobori, H.; Shimizu, M.; Watanabe, M.; Yonekura, M.; Nakai, T.; Kamikawa, Y.; Wakimura, A.; Funahashi, N.; Naruse, H.; et al. Multiple ETS family proteins regulate PF4 gene expression by binding to the same ETS binding site. PLoS ONE 2011, 6, e24837. [Google Scholar] [CrossRef] [PubMed]
- Xiang, P.; Lo, C.; Argiropoulos, B.; Lai, C.B.; Rouhi, A.; Imren, S.; Jiang, X.; Mager, D.; Humphries, R.K. Identification of E74-like factor 1 (ELF1) as a transcriptional regulator of the Hox cofactor MEIS1. Exp. Hematol. 2010, 38. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.H.; Nishiyama, C.; Nakano, N.; Shimokawa, N.; Hara, M.; Kanada, S.; Ogawa, H.; Okumura, K. Suppressive effect of Elf-1 on FcepsilonRI alpha-chain expression in primary mast cells. Immunogenetics 2008, 60, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Jalagadugula, G.; Dhanasekaran, D.N.; Kim, S.; Kunapuli, S.P.; Rao, A.K. Early growth response transcription factor EGR-1 regulates Galphaq gene in megakaryocytic cells. J. Thromb. Haemost. 2006, 4, 2678–2686. [Google Scholar] [CrossRef] [PubMed]
- Bjerregaard, M.D.; Jurlander, J.; Klausen, P.; Borregaard, N.; Cowland, J.B. The in vivo profile of transcription factors during neutrophil differentiation in human bone marrow. Blood 2003, 101, 4322–4332. [Google Scholar] [CrossRef] [PubMed]
- Rolli-Derkinderen, M.; Machavoine, F.; Baraban, J.M.; Grolleau, A.; Beretta, L.; Dy, M. ERK and p38 inhibit the expression of 4E-BP1 repressor of translation through induction of Egr-1. J. Biol. Chem. 2003, 278, 18859–18867. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, D.; Quinn, C.M.; El-Shanawany, T.; Gordon, S.; Greaves, D.R. Multiple Ets factors and interferon regulatory factor-4 modulate CD68 expression in a cell type-specific manner. J. Biol. Chem. 2003, 278, 21909–21919. [Google Scholar] [CrossRef] [PubMed]
- Guha, M.; Mackman, N. The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J. Biol. Chem. 2002, 277, 32124–32132. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Dziarski, R.; Wang, Q.; Swartz, K.; Sakamoto, K.M.; Gupta, D. Bacterial peptidoglycan-induced tnf-alpha transcription is mediated through the transcription factors Egr-1, Elk-1, and NF-kappaB. J. Immunol. 2001, 167, 6975–6982. [Google Scholar] [CrossRef] [PubMed]
- Voo, K.S.; Skalnik, D.G. Elf-1 and PU.1 induce expression of gp91(phox) via a promoter element mutated in a subset of chronic granulomatous disease patients. Blood 1999, 93, 3512–3520. [Google Scholar] [PubMed]
- Di Battista, J.A.; Martel-Pelletier, J.; Pelletier, J. Suppression of tumor necrosis factor (TNF-alpha) gene expression by prostaglandin E(2). Role Of early growth response protein-1 (Egr-1). Osteoarthr. Cartil. 1999, 7, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Nimer, S.; Zhang, J.; Avraham, H.; Miyazaki, Y. Transcriptional regulation of interleukin-3 expression in megakaryocytes. Blood 1996, 88, 66–74. [Google Scholar] [PubMed]
- Affymetrix ENCODE Transcriptome Project; Cold Spring Harbor Laboratory ENCODE Transcriptome Project. Post-transcriptional processing generates a diversity of 5′-modified long and short RNAs. Nature 2009, 457, 1028–1032. [Google Scholar]
- Kawaji, H.; Severin, J.; Lizio, M.; Waterhouse, A.; Katayama, S.; Irvine, K.M.; Kamikawa, Y.; Wakimura, A.; Funahashi, N.; Naruse, H.; et al. The FANTOM web resource: From mammalian transcriptional landscape to its dynamic regulation. Genome Biol. 2009, 10, R40. [Google Scholar] [CrossRef] [PubMed]
- Kawaji, H.; Kasukawa, T.; Fukuda, S.; Katayama, S.; Kai, C.; Kawai, J.; Carninci, P.; Hayashizaki, Y. CAGE Basic/Analysis Databases: The CAGE resource for comprehensive promoter analysis. Nucleic Acids Res. 2006, 34, D632–D636. [Google Scholar] [CrossRef] [PubMed]
- Hodges, E.; Molaro, A.; Dos Santos, C.O.; Thekkat, P.; Song, Q.; Uren, P.J.; Park, J.; Butler, J.; Rafii, S.; McCombie, W.R.; et al. Directional DNA methylation changes and complex intermediate states accompany lineage specificity in the adult hematopoietic compartment. Mol. Cell 2011, 44, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Heyn, H.; Li, N.; Ferreira, H.J.; Moran, S.; Pisano, D.G.; Gomez, A.; Diez, J.; Sanchez-Mut, J.V.; Setien, F.; Carmona, F.J.; et al. Distinct DNA methylomes of newborns and centenarians. Proc. Natl. Acad. Sci. USA 2012, 109, 10522–10527. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhu, J.; Tian, G.; Li, N.; Li, Q.; Ye, M.; Zheng, H.; Yu, J.; Wu, H.; Sun, J.; et al. The DNA methylome of human peripheral blood mononuclear cells. PLoS Biol. 2010, 8, e1000533. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.T.; Corces, V.G. CTCF: An architectural protein bridging genome topology and function. Nat. Rev. Genet. 2014, 15, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Giannopoulou, E.G.; Chan, C.H.; Park, S.H.; Gong, S.; Chen, J.; Hu, X.; Elemento, O.; Ivashkiv, L.B. Synergistic activation of inflammatory cytokine genes by interferon-gamma-induced chromatin remodeling and toll-like receptor signaling. Immunity 2013, 39, 454–469. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Errico, D.; Vento-Tormo, R.; Sieweke, M.; Ballestar, E. Epigenetic control of myeloid cell differentiation, identity and function. Nat. Rev. Immunol. 2015, 15, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Zhang, X. dbSUPER: A database of super-enhancers in mouse and human genome. Nucleic Acids Res. 2016, 44, D164–D171. [Google Scholar] [CrossRef] [PubMed]
- Pelish, H.E.; Liau, B.B.; Nitulescu, I.I.; Tangpeerachaikul, A.; Poss, Z.C.; Da Silva, D.H.; Caruso, B.T.; Arefolov, A.; Fadeyi, O.; Christie, A.L.; et al. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature 2015, 526, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Tausendschon, M.; Rehli, M.; Dehne, N.; Schmidl, C.; Doring, C.; Hansmann, M.L.; Brune, B. Genome-wide identification of hypoxia-inducible factor-1 and -2 binding sites in hypoxic human macrophages alternatively activated by IL-10. Biochim. Biophys. Acta 2015, 1849, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Searle, S.; Blackwell, J.M. Evidence for a functional repeat polymorphism in the promoter of the human NRAMP1 gene that correlates with autoimmune versus infectious disease susceptibility. J. Med. Genet. 1999, 36, 295–299. [Google Scholar] [PubMed]
- Bayele, H.K.; Peyssonnaux, C.; Giatromanolaki, A.; Arrais-Silva, W.W.; Mohamed, H.S.; Collins, H.; Giorgio, S.; Koukourakis, M.; Johnson, R.S.; Blackwell, J.M.; et al. HIF-1 regulates heritable variation and allele expression phenotypes of the macrophage immune response gene SLC11A1 from a Z-DNA forming microsatellite. Blood 2007, 110, 3039–3048. [Google Scholar] [CrossRef] [PubMed]
- Galeev, R.; Baudet, A.; Kumar, P.; Rundberg, N.A.; Nilsson, B.; Soneji, S.; Torngren, T.; Borg, A.; Kvist, A.; Larsson, J. Genome-wide RNAi Screen Identifies Cohesin Genes as Modifiers of Renewal and Differentiation in Human HSCs. Cell Rep. 2016, 14, 2988–3000. [Google Scholar] [CrossRef] [PubMed]
- Quach, H.; Rotival, M.; Pothlichet, J.; Loh, Y.E.; Dannemann, M.; Zidane, N.; Laval, G.; Patin, E.; Harmant, C.; Lopez, M.; et al. Genetic Adaptation and Neandertal Admixture Shaped the Immune System of Human Populations. Cell 2016, 167, 643–656. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Gorkin, D.U.; Ren, B. Chromatin Domains: The Unit of Chromosome Organization. Mol. Cell 2016, 62, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Yip, S.P.; Leung, K.H.; Lin, C.K. Extent and distribution of linkage disequilibrium around the SLC11A1 locus. Genes Immun. 2003, 4, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; Abel, L.; Tooker, H.; Poon, A.; Simkin, L.; Girard, M.; Adams, G.J.; Starke, J.R.; Smith, K.C.; Graviss, E.A.; et al. Alleles of the NRAMP1 gene are risk factors for pediatric tuberculosis disease. Proc. Natl. Acad. Sci. USA 2005, 102, 12183–12188. [Google Scholar] [CrossRef] [PubMed]
- Archer, N.S.; Nassif, N.T.; O’Brien, B.A. Genetic variants of SLC11A1 are associated with both autoimmune and infectious diseases: Systematic review and meta-analysis. Genes Immun. 2015, 16, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Cellier, M.; Govoni, G.; Vidal, S.; Kwan, T.; Groulx, N.; Liu, J.; Sanchez, F.; Skamene, E.; Schurr, E.; Gros, P. Human natural resistance-associated macrophage protein: cDNA cloning, chromosomal mapping, genomic organization, and tissue-specific expression. J. Exp. Med. 1994, 180, 1741–1752. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.Z.; Di, M.S.; Gallouzi, I.; Rola-Pleszczynski, M.; Radzioch, D. RNA-binding protein HuR is required for stabilization of SLC11A1 mRNA and SLC11A1 protein expression. Mol. Cell. Biol. 2005, 25, 8139–8149. [Google Scholar] [CrossRef] [PubMed]
- Moisan, J.; Thuraisingam, T.; Henault, J.; De Sanctis, J.; Radzioch, D. Role of SLC11A1 (formerly NRAMP1) in regulation of signal transduction induced by Toll-like receptor 7 ligands. FEMS Immunol. Med. Microbiol. 2006, 47, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.Z.; Thuraisingam, T.; Marino, R.; Radzioch, D. Recruitment of SWI/SNF complex is required for transcriptional activation of the SLC11A1 gene during macrophage differentiation of HL-60 cells. J. Biol. Chem. 2011, 286, 12839–12849. [Google Scholar] [CrossRef] [PubMed]
- Zaahl, M.G.; Robson, K.J.; Warnich, L.; Kotze, M.J. Expression of the SLC11A1 (NRAMP1) 5′-(GT)n repeat: Opposite effect in the presence of -237C-->T. Blood Cells Mol. Dis. 2004, 33, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Taka, S.; Gazouli, M.; Politis, P.K.; Pappa, K.I.; Anagnou, N.P. Transcription factor ATF-3 regulates allele variation phenotypes of the human SLC11A1 gene. Mol. Biol. Rep. 2012, 40, 2263–2271. [Google Scholar] [CrossRef] [PubMed]
- Mass, E.; Ballesteros, I.; Farlik, M.; Halbritter, F.; Gunther, P.; Crozet, L.; Jacome-Galarza, C.E.; Handler, K.; Klughammer, J.; Kobayashi, Y.; et al. Specification of tissue-resident macrophages during organogenesis. Science 2016, 353, aaf4238. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, D.; Link, V.M.; Romanoski, C.E.; Fonseca, G.J.; Eichenfield, D.Z.; Spann, N.J.; Stender, J.D.; Chun, H.B.; Garner, H.; Geissmann, F.; et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 2014, 159, 1327–1340. [Google Scholar] [CrossRef] [PubMed]
- Lavin, Y.; Winter, D.; Blecher-Gonen, R.; David, E.; Keren-Shaul, H.; Merad, M.; Jung, S.; Amit, I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 2014, 159, 1312–1326. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Cellier, M.F.; Courville, P.; Campion, C. Nramp1 phagocyte intracellular metal withdrawal defense. Microbes Infect. 2007, 9, 1662–1670. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.D.; Lin, C.Y.; Duan, Q.; Griffin, G.; Federation, A.J.; Paranal, R.M.; Bair, S.; Newton, G.; Lichtman, A.H.; Kung, A.L.; et al. NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol. Cell 2014, 56, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Peeters, J.G.; Vervoort, S.J.; Tan, S.C.; Mijnheer, G.; de Roock, S.; Vastert, S.J.; Nieuwenhuis, E.E.; van Wijk, F.; Prakken, B.J.; Creyghton, M.P.; et al. Inhibition of Super-Enhancer Activity in Autoinflammatory Site-Derived T Cells Reduces Disease-Associated Gene Expression. Cell Rep. 2015, 12, 1986–1996. [Google Scholar] [CrossRef] [PubMed]
- Roe, J.S.; Mercan, F.; Rivera, K.; Pappin, D.J.; Vakoc, C.R. BET Bromodomain Inhibition Suppresses the Function of Hematopoietic Transcription Factors in Acute Myeloid Leukemia. Mol. Cell 2015, 58, 1028–1039. [Google Scholar] [CrossRef] [PubMed]
- Quang, D.X.; Erdos, M.R.; Parker, S.C.; Collins, F.S. Motif signatures in stretch enhancers are enriched for disease-associated genetic variants. Epigenet. Chromatin 2015, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Novakovic, B.; Habibi, E.; Wang, S.Y.; Arts, R.J.; Davar, R.; Megchelenbrink, W.; Kim, B.; Kuznetsova, T.; Kox, M.; Zwaag, J.; et al. beta-Glucan Reverses the Epigenetic State of LPS-Induced Immunological Tolerance. Cell 2016, 167, 1354–1368. [Google Scholar] [CrossRef] [PubMed]
- Marinescu, V.D.; Kohane, I.S.; Riva, A. The MAPPER database: A multi-genome catalog of putative transcription factor binding sites. Nucleic Acids Res. 2005, 33, D91–D97. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Schmeier, S.; Arner, E.; Alam, T.; Parihar, S.P.; Ozturk, M.; Tamgue, O.; Kawaji, H.; de Hoon, M.J.; Itoh, M.; et al. Redefining the transcriptional regulatory dynamics of classically and alternatively activated macrophages by deepCAGE transcriptomics. Nucleic Acids Res. 2015, 43, 6969–6982. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Forrest, A.R.; van Nimwegen, E.; Daub, C.O.; Balwierz, P.J.; Irvine, K.M.; Lassmann, T.; Ravasi, T.; Hasegawa, Y.; de Hoon, M.J.; et al. The transcriptional network that controls growth arrest and differentiation in a human myeloid leukemia cell line. Nat. Genet. 2009, 41, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Drouin, J. Minireview: Pioneer transcription factors in cell fate specification. Mol. Endocrinol. 2014, 28, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Iwafuchi-Doi, M.; Zaret, K.S. Pioneer transcription factors in cell reprogramming. Genes Dev. 2014, 28, 2679–2692. [Google Scholar] [CrossRef] [PubMed]
- Salci, K.R.; McIntyre, B.A.; Bhatia, M. Foundational concepts of cell fate conversion to the hematopoietic lineage. Curr. Opin. Genet. Dev. 2013, 23, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Ebina, W.; Rossi, D.J. Transcription factor-mediated reprogramming toward hematopoietic stem cells. EMBO J. 2015, 34, 694–709. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef] [PubMed]
- McGrath, K.E.; Frame, J.M.; Fegan, K.H.; Bowen, J.R.; Conway, S.J.; Catherman, S.C.; Kingsley, P.D.; Koniski, A.D.; Palis, J. Distinct Sources of Hematopoietic Progenitors Emerge before HSCs and Provide Functional Blood Cells in the Mammalian Embryo. Cell Rep. 2015, 11, 1892–1904. [Google Scholar] [CrossRef] [PubMed]
- Cauchy, P.; James, S.R.; Zacarias-Cabeza, J.; Ptasinska, A.; Imperato, M.R.; Assi, S.A.; Piper, J.; Canestraro, M.; Hoogenkamp, M.; Raghavan, M.; et al. Chronic FLT3-ITD Signaling in Acute Myeloid Leukemia Is Connected to a Specific Chromatin Signature. Cell Rep. 2015, 12, 821–836. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, A.; Lennartsson, A.; Lehmann, S. Epigenetic aberrations in acute myeloid leukemia: Early key events during leukemogenesis. Exp. Hematol. 2015, 43, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Sotoca, A.M.; Prange, K.H.; Reijnders, B.; Mandoli, A.; Nguyen, L.N.; Stunnenberg, H.G.; Martens, J.H. The oncofusion protein FUS-ERG targets key hematopoietic regulators and modulates the all-trans retinoic acid signaling pathway in t(16;21) acute myeloid leukemia. Oncogene 2016, 35, 1965–1976. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, H.; Wang, X.; Jin, W.; Tan, Y.; Fang, H.; Chen, S.; Chen, Z.; Wang, K. Genome-wide studies identify a novel interplay between AML1 and AML1/ETO in t(8;21) acute myeloid leukemia. Blood 2016, 127, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Trombly, D.J.; Whitfield, T.W.; Padmanabhan, S.; Gordon, J.A.; Lian, J.B.; van Wijnen, A.J.; Zaidi, S.K.; Stein, J.L.; Stein, G.S. Genome-wide co-occupancy of AML1-ETO and N-CoR defines the t(8;21) AML signature in leukemic cells. BMC Genom. 2015, 16, 309. [Google Scholar] [CrossRef] [PubMed]
- Mandoli, A.; Singh, A.A.; Jansen, P.W.; Wierenga, A.T.; Riahi, H.; Franci, G.; Prange, K.; Saeed, S.; Vellenga, E.; Vermeulen, M.; et al. CBFB-MYH11/RUNX1 together with a compendium of hematopoietic regulators, chromatin modifiers and basal transcription factors occupies self-renewal genes in inv(16) acute myeloid leukemia. Leukemia 2014, 28, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Tober, J.; Yzaguirre, A.D.; Piwarzyk, E.; Speck, N.A. Distinct temporal requirements for Runx1 in hematopoietic progenitors and stem cells. Development 2013, 140, 3765–3776. [Google Scholar] [CrossRef] [PubMed]
- Lanotte, M.; Martin-Thouvenin, V.; Najman, S.; Balerini, P.; Valensi, F.; Berger, R. NB4, a maturation inducible cell line with t(15;17) marker isolated from a human acute promyelocytic leukemia (M3). Blood 1991, 77, 1080–1086. [Google Scholar] [PubMed]
- Brent, M.R. Past Roadblocks and New Opportunities in Transcription Factor Network Mapping. Trends Genet. 2016, 32, 736–750. [Google Scholar] [CrossRef] [PubMed]
- Frank, C.L.; Manandhar, D.; Gordan, R.; Crawford, G.E. HDAC inhibitors cause site-specific chromatin remodeling at PU.1-bound enhancers in K562 cells. Epigenet. Chromatin 2016, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Carlin, L.M.; Stamatiades, E.G.; Auffray, C.; Hanna, R.N.; Glover, L.; Vizcay-Barrena, G.; Hedrick, C.C.; Cook, H.T.; Diebold, S.; Geissmann, F. Nr4a1-dependent Ly6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell 2013, 153, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.; Tacke, R.; Hedrick, C.C.; Hanna, R.N. Nonclassical patrolling monocyte function in the vasculature. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.D.; Hanna, R.N.; Vasudevan, N.T.; Hamers, A.A.; Romanoski, C.E.; McArdle, S.; Ross, K.D.; Blatchley, A.; Yoakum, D.; Hamilton, B.A.; et al. Deleting an Nr4a1 Super-Enhancer Subdomain Ablates Ly6Clow Monocytes while Preserving Macrophage Gene Function. Immunity 2016, 45, 975–987. [Google Scholar] [CrossRef] [PubMed]
- Schmidl, C.; Renner, K.; Peter, K.; Eder, R.; Lassmann, T.; Balwierz, P.J.; Itoh, M.; Nagao-Sato, S.; Kawaji, H.; Carninci, P.; et al. Transcription and enhancer profiling in human monocyte subsets. Blood 2014, 123, e90–e99. [Google Scholar] [CrossRef] [PubMed]
- Gammella, E.; Buratti, P.; Cairo, G.; Recalcati, S. Macrophages: Central regulators of iron balance. Metallomics 2014, 6, 1336–1345. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Macrophages and Iron Metabolism. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Rawat, P.; Kumar, S.; Sheokand, N.; Raje, C.I.; Raje, M. The multifunctional glycolytic protein glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is a novel macrophage lactoferrin receptor. Biochem. Cell Biol. 2012, 90, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Tao, Y.; Zhang, Z.; Guo, X.; An, P.; Shen, Y.; Wu, Q.; Yu, Y.; Wang, F. Metalloreductase Steap3 coordinates the regulation of iron homeostasis and inflammatory responses. Haematologica 2012, 97, 1826–1835. [Google Scholar] [CrossRef] [PubMed]
- Jacolot, S.; Yang, Y.; Paitry, P.; Ferec, C.; Mura, C. Iron metabolism in macrophages from HFE hemochromatosis patients. Mol. Genet. Metab. 2010, 101, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, K.; Wong, C.O.; Zhu, M.X. The role of TRPMLs in endolysosomal trafficking and function. Cell Calcium 2015, 58, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Dayam, R.M.; Saric, A.; Shilliday, R.E.; Botelho, R.J. The Phosphoinositide-Gated Lysosomal Ca(2+) Channel, TRPML1, Is Required for Phagosome Maturation. Traffic 2015, 16, 1010–1026. [Google Scholar] [CrossRef] [PubMed]
- White, C.; Yuan, X.; Schmidt, P.J.; Bresciani, E.; Samuel, T.K.; Campagna, D.; Hall, C.; Bishop, K.; Calicchio, M.L.; Lapierre, A.; et al. HRG1 is essential for heme transport from the phagolysosome of macrophages during erythrophagocytosis. Cell Metab. 2013, 17, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Schaer, C.A.; Vallelian, F.; Imhof, A.; Schoedon, G.; Schaer, D.J. Heme carrier protein (HCP-1) spatially interacts with the CD163 hemoglobin uptake pathway and is a target of inflammatory macrophage activation. J. Leukoc. Biol. 2008, 83, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.P.; Shing, J.; Saraon, P.; Berger, L.C.; Eiden, M.V.; Wilde, A.; Tailor, C.S. The Fowler syndrome-associated protein FLVCR2 is an importer of heme. Mol. Cell. Biol. 2010, 30, 5318–5324. [Google Scholar] [CrossRef] [PubMed]
- Frey, A.G.; Nandal, A.; Park, J.H.; Smith, P.M.; Yabe, T.; Ryu, M.S.; Ghosh, M.C.; Lee, J.; Rouault, T.A.; Park, M.H.; et al. Iron chaperones PCBP1 and PCBP2 mediate the metallation of the dinuclear iron enzyme deoxyhypusine hydroxylase. Proc. Natl. Acad. Sci. USA 2014, 111, 8031–8036. [Google Scholar] [CrossRef] [PubMed]
- Dowdle, W.E.; Nyfeler, B.; Nagel, J.; Elling, R.A.; Liu, S.; Triantafellow, E.; Menon, S.; Wang, Z.; Honda, A.; Pardee, G.; et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol. 2014, 16, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Keel, S.B.; Doty, R.T.; Yang, Z.; Quigley, J.G.; Chen, J.; Knoblaugh, S.; Kingsley, P.D.; De Domenico, I.; Vaughn, M.B.; Kaplan, J.; et al. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science 2008, 319, 825–828. [Google Scholar] [CrossRef] [PubMed]
- Nakashige, T.G.; Zhang, B.; Krebs, C.; Nolan, E.M. Human calprotectin is an iron-sequestering host-defense protein. Nat. Chem. Biol. 2015, 11, 765–771. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Rozowsky, J.; Euskirchen, G.; Auerbach, R.K.; Zhang, Z.D.; Gibson, T.; Bjornson, R.; Carriero, N.; Snyder, M.; Gerstein, M.B. PeakSeq enables systematic scoring of ChIP-seq experiments relative to controls. Nat. Biotechnol. 2009, 27, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, R.K.; Euskirchen, G.; Rozowsky, J.; Lamarre-Vincent, N.; Moqtaderi, Z.; Lefrancois, P.; Struhl, K.; Gerstein, M.; Snyder, M. Mapping accessible chromatin regions using Sono-Seq. Proc. Natl. Acad. Sci. USA 2009, 106, 14926–14931. [Google Scholar] [CrossRef] [PubMed]
- Krijger, P.H.; Di Stefano, B.; de Wit, E.; Limone, F.; van Oevelen, C.; de Laat, W.; Graf, T. Cell-of-Origin-Specific 3D Genome Structure Acquired during Somatic Cell Reprogramming. Cell Stem Cell 2016, 18, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Goode, D.K.; Obier, N.; Vijayabaskar, M.S.; Lie, A.L.; Lilly, A.J.; Hannah, R.; Lichtinger, M.; Batta, K.; Florkowska, M.; Patel, R.; et al. Dynamic Gene Regulatory Networks Drive Hematopoietic Specification and Differentiation. Dev. Cell 2016, 36, 572–587. [Google Scholar] [CrossRef] [PubMed]
- Yue, F.; Cheng, Y.; Breschi, A.; Vierstra, J.; Wu, W.; Ryba, T.; Sandstrom, R.; Ma, Z.; Davis, C.; Pope, B.; et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature 2014, 515, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Luyten, A.; Zang, C.; Liu, X.S.; Shivdasani, R.A. Active enhancers are delineated de novo during hematopoiesis, with limited lineage fidelity among specified primary blood cells. Genes Dev. 2014, 28, 1827–1839. [Google Scholar] [CrossRef] [PubMed]
- Paul, F.; Arkin, Y.; Giladi, A.; Jaitin, D.A.; Kenigsberg, E.; Keren-Shaul, H.; Winter, D.; Lara-Astiaso, D.; Gury, M.; Weiner, A.; et al. Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors. Cell 2015, 163, 1663–1677. [Google Scholar] [CrossRef] [PubMed]
- Ferri, F.; Parcelier, A.; Petit, V.; Gallouet, A.S.; Lewandowski, D.; Dalloz, M.; van den Heuvel, A.; Kolovos, P.; Soler, E.; Squadrito, M.L.; et al. TRIM33 switches off Ifnb1 gene transcription during the late phase of macrophage activation. Nat. Commun. 2015, 6, 8900. [Google Scholar] [CrossRef] [PubMed]
- Langlais, D.; Barreiro, L.B.; Gros, P. The macrophage IRF8/IRF1 regulome is required for protection against infections and is associated with chronic inflammation. J. Exp. Med. 2016, 213, 585–603. [Google Scholar] [CrossRef] [PubMed]
- Matcovitch-Natan, O.; Winter, D.R.; Giladi, A.; Vargas, A.S.; Spinrad, A.; Sarrazin, S.; Ben-Yehuda, H.; David, E.; Zelada, G.F.; Perrin, P.; et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science 2016, 353, aad8670. [Google Scholar] [CrossRef] [PubMed]
- Govoni, G.; Gauthier, S.; Billia, F.; Iscove, N.N.; Gros, P. Cell-specific and inducible Nramp1 gene expression in mouse macrophages in vitro and in vivo. J. Leukoc. Biol. 1997, 62, 277–286. [Google Scholar] [PubMed]
- Weiner, A.; Lara-Astiaso, D.; Krupalnik, V.; Gafni, O.; David, E.; Winter, D.R.; Hanna, J.H.; Amit, I. Co-ChIP enables genome-wide mapping of histone mark co-occurrence at single-molecule resolution. Nat. Biotechnol. 2016, 34, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Rayan, N.A.; Muratani, M.; Lim, S.; Elanggovan, B.; Xin, L.; Lu, T.; Makhija, H.; Poschmann, J.; Lufkin, T.; et al. Comprehensive benchmarking reveals H2BK20 acetylation as a distinctive signature of cell-state-specific enhancers and promoters. Genome Res. 2016, 26, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Tong, A.J.; Liu, X.; Thomas, B.J.; Lissner, M.M.; Baker, M.R.; Senagolage, M.D.; Allred, A.L.; Barish, G.D.; Smale, S.T. A Stringent Systems Approach Uncovers Gene-Specific Mechanisms Regulating Inflammation. Cell 2016, 165, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Bhagwat, A.S.; Roe, J.S.; Mok, B.Y.; Hohmann, A.F.; Shi, J.; Vakoc, C.R. BET Bromodomain Inhibition Releases the Mediator Complex from Select cis-Regulatory Elements. Cell Rep. 2016, 15, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Hasemann, M.S.; Lauridsen, F.K.; Waage, J.; Jakobsen, J.S.; Frank, A.K.; Schuster, M.B.; Rapin, N.; Bagger, F.O.; Hoppe, P.S.; Schroeder, T.; et al. C/EBPalpha is required for long-term self-renewal and lineage priming of hematopoietic stem cells and for the maintenance of epigenetic configurations in multipotent progenitors. PLoS Genet. 2014, 10, e1004079. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Alberich-Jorda, M.; Amabile, G.; Yang, H.; Staber, P.B.; Di Ruscio, A.; Welner, R.S.; Ebralidze, A.; Zhang, J.; Levantini, E.; et al. Sox4 is a key oncogenic target in C/EBPalpha mutant acute myeloid leukemia. Cancer Cell 2013, 24, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Fossati, V.; Israel, M.; Snoeck, H.W. Lin-Sca1+kit-bone marrow cells contain early lymphoid-committed precursors that are distinct from common lymphoid progenitors. J. Immunol. 2008, 181, 7507–7513. [Google Scholar] [CrossRef] [PubMed]
- Kurotaki, D.; Osato, N.; Nishiyama, A.; Yamamoto, M.; Ban, T.; Sato, H.; Nakabayashi, J.; Umehara, M.; Miyake, N.; Matsumoto, N.; et al. Essential role of the IRF8-KLF4 transcription factor cascade in murine monocyte differentiation. Blood 2013, 121, 1839–1849. [Google Scholar] [CrossRef] [PubMed]
- KC, W.; Satpathy, A.T.; Rapaport, A.S.; Briseno, C.G.; Wu, X.; Albring, J.C.; Russler-Germain, E.V.; Kretzer, N.M.; Durai, V.; Persaud, S.P.; et al. L-Myc expression by dendritic cells is required for optimal T-cell priming. Nature 2014, 507, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Nagamura-Inoue, T.; Shmeltzer, Z.; Kuwata, T.; Ozato, K. ICSBP directs bipotential myeloid progenitor cells to differentiate into mature macrophages. Immunity 2000, 13, 155–165. [Google Scholar] [CrossRef]
- Yanez, A.; Goodridge, H.S. Interferon regulatory factor 8 and the regulation of neutrophil, monocyte, and dendritic cell production. Curr. Opin. Hematol. 2016, 23, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Oishi, Y.; Spann, N.J.; Link, V.M.; Muse, E.D.; Strid, T.; Edillor, C.; Kolar, M.J.; Matsuzaka, T.; Hayakawa, S.; Tao, J.; et al. SREBP1 Contributes to Resolution of Pro-inflammatory TLR4 Signaling by Reprogramming Fatty Acid Metabolism. Cell Metab. 2017, 25, 412–427. [Google Scholar] [CrossRef] [PubMed]
- Villar, D.; Berthelot, C.; Aldridge, S.; Rayner, T.F.; Lukk, M.; Pignatelli, M.; Park, T.J.; Deaville, R.; Erichsen, J.T.; Jasinska, A.J.; et al. Enhancer evolution across 20 mammalian species. Cell 2015, 160, 554–566. [Google Scholar] [CrossRef] [PubMed]
- Van den Bosch, T.; Boichenko, A.; Leus, N.G.; Ourailidou, M.E.; Wapenaar, H.; Rotili, D.; Mai, A.; Imhof, A.; Bischoff, R.; Haisma, H.J.; et al. The histone acetyltransferase p300 inhibitor C646 reduces pro-inflammatory gene expression and inhibits histone deacetylases. Biochem. Pharmacol. 2016, 102, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Joosten, L.A.; Latz, E.; Mills, K.H.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef] [PubMed]
- Tacchini, L.; Gammella, E.; De Ponti, C.; Recalcati, S.; Cairo, G. Role of HIF-1 and NF-kappaB transcription factors in the modulation of transferrin receptor by inflammatory and anti-inflammatory signals. J. Biol. Chem. 2008, 283, 20674–20686. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K. Genetic and genomic approaches to understanding macrophage identity and function. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Fattahi-Dolatabadi, M.; Mousavi, T.; Mohammadi-Barzelighi, H.; Irian, S.; Bakhshi, B.; Nilforoushzadeh, M.A.; Shirani-Bidabadi, L.; Hariri, M.M.; Ansari, N.; Akbari, N. NRAMP1 gene polymorphisms and cutaneous leishmaniasis: An evaluation on host susceptibility and treatment outcome. J. Vector Borne Dis. 2016, 53, 257–263. [Google Scholar] [PubMed]
- Kadkhodazadeh, M.; Ebadian, A.R.; Amid, R.; Zarnegarnia, P.; Mollaverdi, F.; Aghamohammadi, N. Natural Resistance Associated Macrophage Protein 1 Gene Polymorphism is Associated with Chronic Periodontitis Not Peri-Implantitis in an Iranian Population: A Cross Sectional Study. Acta Med. Iran. 2016, 54, 323–329. [Google Scholar] [PubMed]
- Brochado, M.J.; Gatti, M.F.; Zago, M.A.; Roselino, A.M. Association of the solute carrier family 11 member 1 gene polymorphisms with susceptibility to leprosy in a Brazilian sample. Mem. Inst. Oswaldo Cruz. 2016, 111, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Niño-Moreno, P.; Turrubiartes-Martínez, E.; Oceguera-Maldonado, B.; Baltazar-Benítez, N.; Negrete-González, C.; Oliva-Ramírez, B.; Baranda, L.; González-Amaro, R. The Role of NRAMP1/SLC11A1 Gene Variant D543N (1730G/A) in the Genetic Susceptibility to Develop Rheumatoid Arthritis in the Mexican Mestizo population. Rev. Investig. Clin. 2017, 69, 5–10. [Google Scholar]
- Gorenshteyn, D.; Zaslavsky, E.; Fribourg, M.; Park, C.Y.; Wong, A.K.; Tadych, A.; Hartmann, B.M.; Albrecht, R.A.; Garcia-Sastre, A.; Kleinstein, S.H.; et al. Interactive Big Data Resource to Elucidate Human Immune Pathways and Diseases. Immunity 2015, 43, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Dey, G.; Jaimovich, A.; Collins, S.R.; Seki, A.; Meyer, T. Systematic Discovery of Human Gene Function and Principles of Modular Organization through Phylogenetic Profiling. Cell Rep. 2015, 10, 993–1006. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CHROMOSOMAL REGIONS (Presented in Section) | i (2.4.5.2) | ii (2.4.5.1) | iii (2.4.5.3) | iv (2.4.5.4) | v (2.4.5.5) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DNAse1 FOOTPRINTS (described in section) | 5′ CTCF | F6 | F11 | F4A | F12 | F13 | F5 | F2 | F4B | F7 | F1 | F8 | F14 | F9 | F10 | F3 | 3′ CTCF stretch | |
| (2.3.3) | (2.3.1.1) | (2.3.2.1) | (2.3.1.2) | (2.2.3.1) | (2.2.3.2) | (2.2.3.3) | (2.2.3.3) | (2.3.2.2) | (2.2.3.5) | (2.3.1.3) | (2.2.3.6) | (2.3.1.4 | (2.3.1.5) | (2.3.1.6) | (2.3.1.7) | (2.3.3) | (2.3.4) | |
| DIFFERENTIATION STAGE | ||||||||||||||||||
| ESC HUVEC | + | − | − | − | (+) | +/− | − | − | − | − | − | − | − | + | − | − | + | + |
| +/− | − | − | − | (+) | − | − | − | − | − | − | − | − | +/− | − | − | + | + | |
| HSPC CD34+ cells EP (K562 cells) | + | (+) | − | − | ++ | (+) | (+) | − | − | +/− | +/− | − | − | + | (+) | (+) | ++ | ++ |
| + | +/− | − | − | + | − | − | − | − | − | − | − | − | − | ++ | +/− | ++ | ++ | |
| ITD FLT3 AML blasts | +/− | + | − | − | + | +/− | +/− | − | − | − | − | − | − | +/− | (+) | (+) | ++ | + |
| +/− | (+) | − | − | + | +/− | +/− | − | − | +/− | (+) | − | − | − | +/− | +/− | ++ | (+) | |
| CMP (mCD34 cells) | + | (+) | − | − | ++ | (+) | + | − | − | (+) | (+) | − | − | + | (+) | (+) | ++ | ++ |
| GMP (NB4 cells) GMP (HL-60 cells) | (+) | (+) | − | − | ++ | + | (+) | (+) | − | − | − | − | − | − | + | +/− | ++ | ++ |
| (+) | + | − | +/− | + | +/− | ++ | +/− | − | +/− | ++ | +/− | − | +/− | +/− | +/− | ++ | ++ | |
| MNs PMNs | + | + | (+) | +/− | ++ | (+) | ++ | (+) | (+) | + | ++ | (+) | +/− | (+) | +/− | +/− | ++ | ++ |
| + | + | (+) | +/− | ++ | (+) | ++ | (+) | (+) | + | ++ | (+) | +/− | (+) | +/− | +/− | ++ | ++ | |
| BOUND FACTORS | ||||||||||||||||||
| MN/Mac | CTCF | (CTCF) | (CTCF) | CTCF | CTCF | |||||||||||||
| C/EBPb | C/EBPb | (C/EBPb) | ||||||||||||||||
| (PU.1) | PU.1 | PU.1 | (PU.1) | (PU.1) | PU.1 | (PU.1) | (PU.1) | |||||||||||
| EGR2 | (EGR2) | (EGR2) | EGR2 | EGR2 | EGR2 | |||||||||||||
| Mac (hypox., +/− IL-10) | (HIF1/2a) | HIF1/2a | (HIF1/2a) | |||||||||||||||
| Mac (+/− LPS, IFNg) | (STAT1) | (STAT1) | STAT1 | STAT1 | STAT1 | STAT1 | STAT1 | (STAT1) | ||||||||||
| IRF1 | (IRF1) | (IRF1) | ||||||||||||||||
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cellier, M.F.M. Developmental Control of NRAMP1 (SLC11A1) Expression in Professional Phagocytes. Biology 2017, 6, 28. https://doi.org/10.3390/biology6020028
Cellier MFM. Developmental Control of NRAMP1 (SLC11A1) Expression in Professional Phagocytes. Biology. 2017; 6(2):28. https://doi.org/10.3390/biology6020028
Chicago/Turabian StyleCellier, Mathieu F. M. 2017. "Developmental Control of NRAMP1 (SLC11A1) Expression in Professional Phagocytes" Biology 6, no. 2: 28. https://doi.org/10.3390/biology6020028
APA StyleCellier, M. F. M. (2017). Developmental Control of NRAMP1 (SLC11A1) Expression in Professional Phagocytes. Biology, 6(2), 28. https://doi.org/10.3390/biology6020028