Pseudotime Dynamics in Melanoma Single-Cell Transcriptomes Reveals Different Mechanisms of Tumor Progression



Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Approval and Consent to Participate
2.2. Short-Term Cultures
2.3. Single-Cell RNA-seq and Data Preprocessing
2.4. Data Availability
2.5. Pseudotime Analysis
2.6. Functional PT-Profiles
3. Results
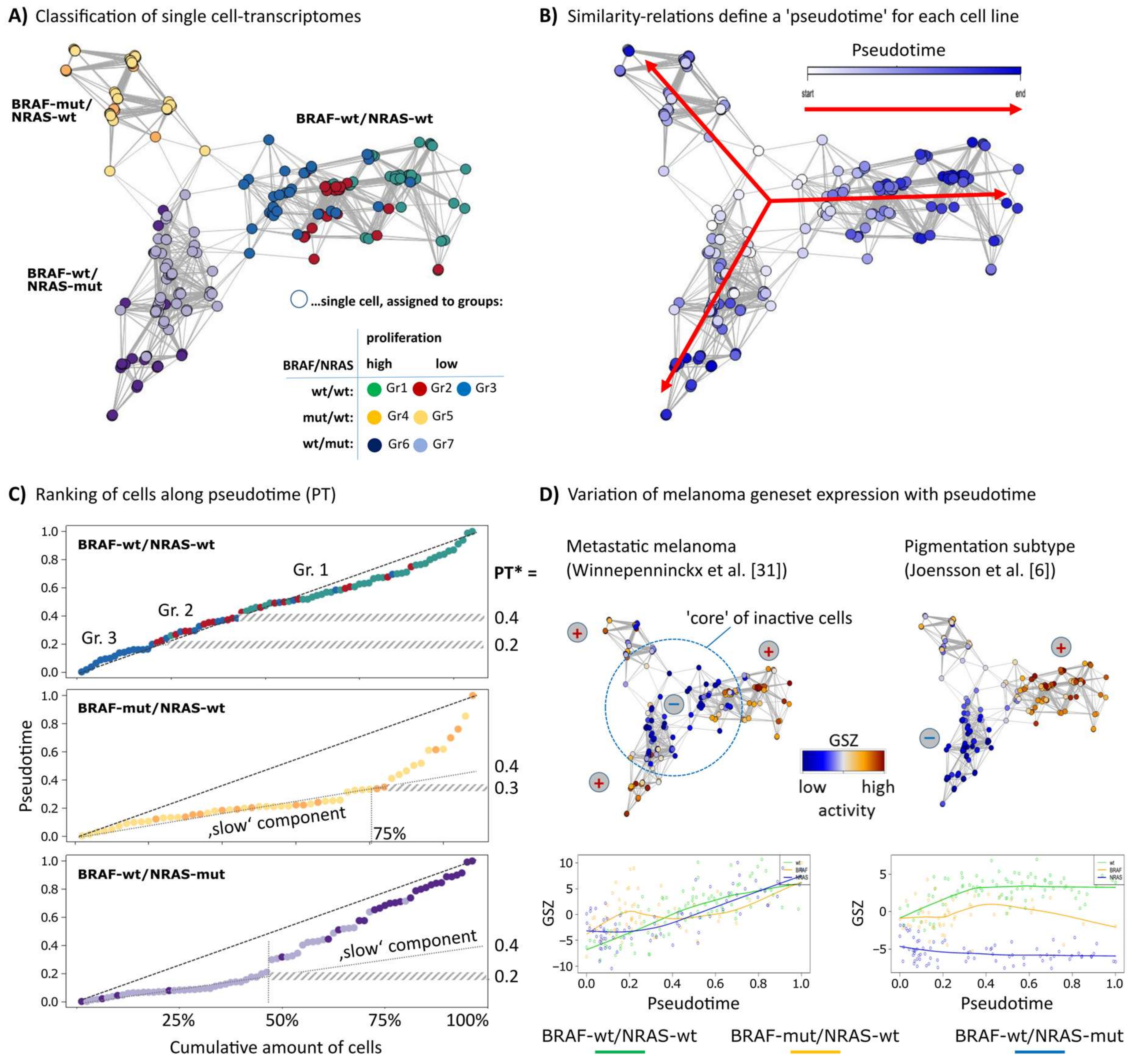
3.1. RNA-seq Based Pseudotime Dynamics in Patient-Derived Melanoma Cell Cultures
3.2. Cell-Culture Specific Dynamics of Functional Signatures
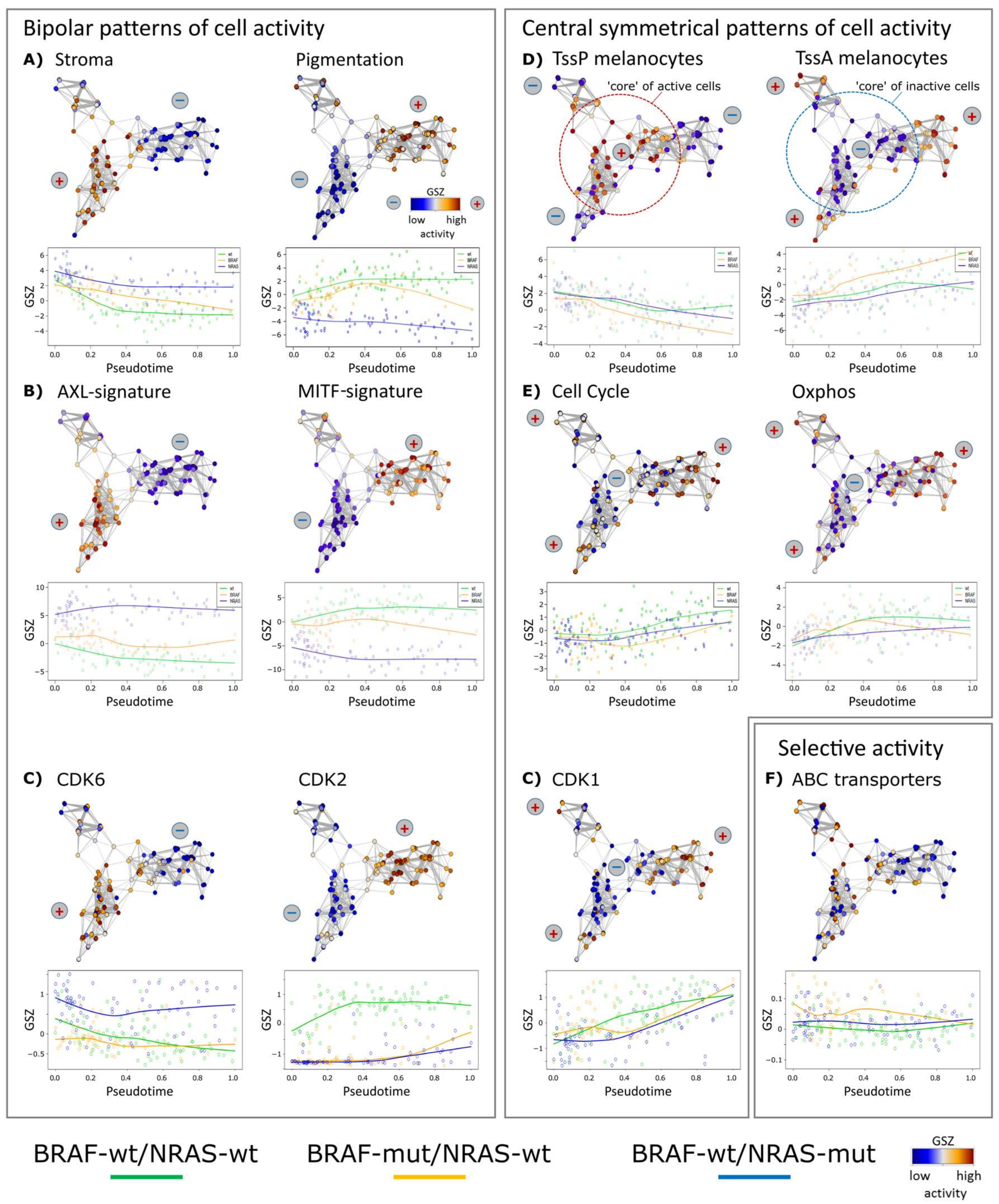
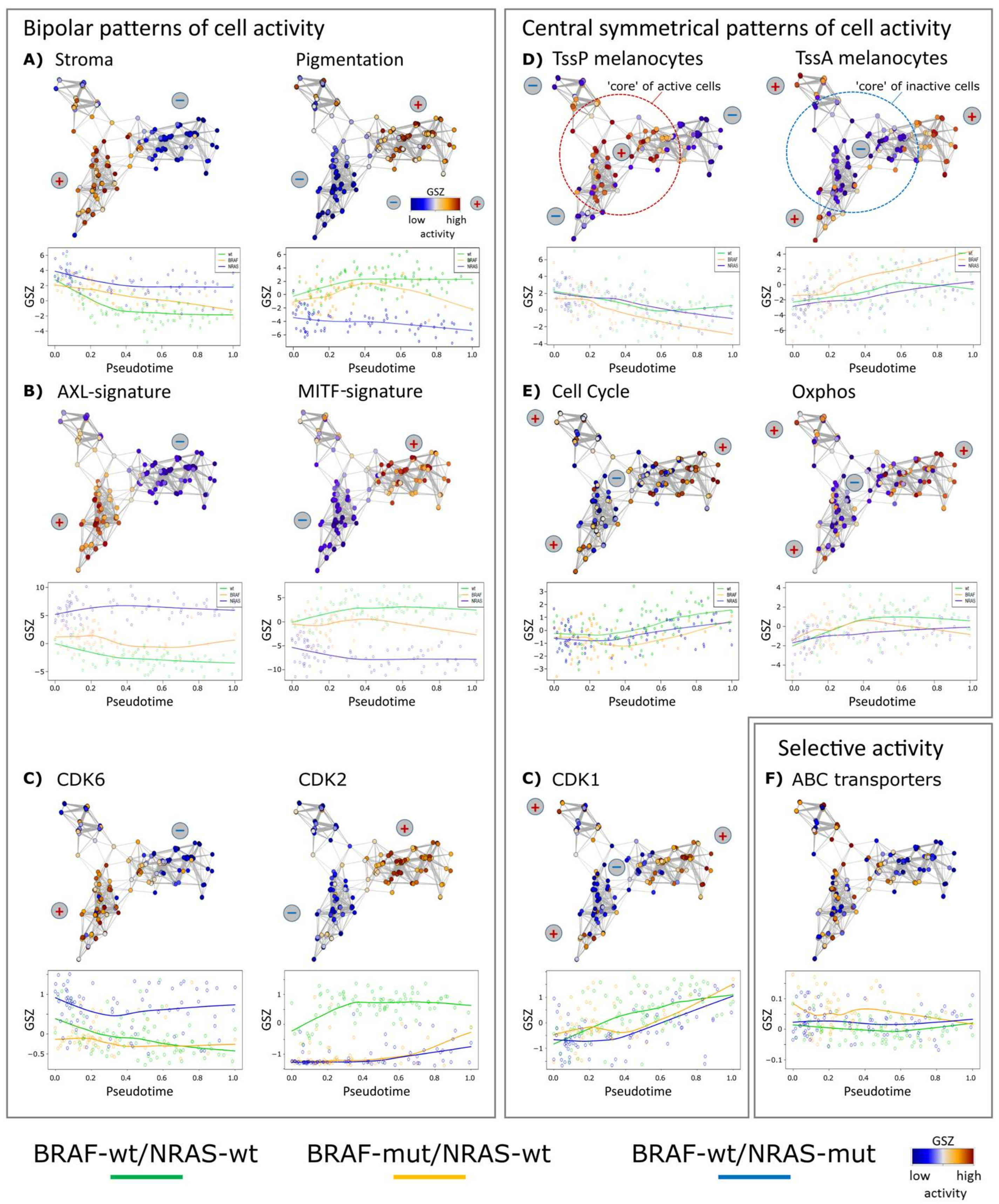
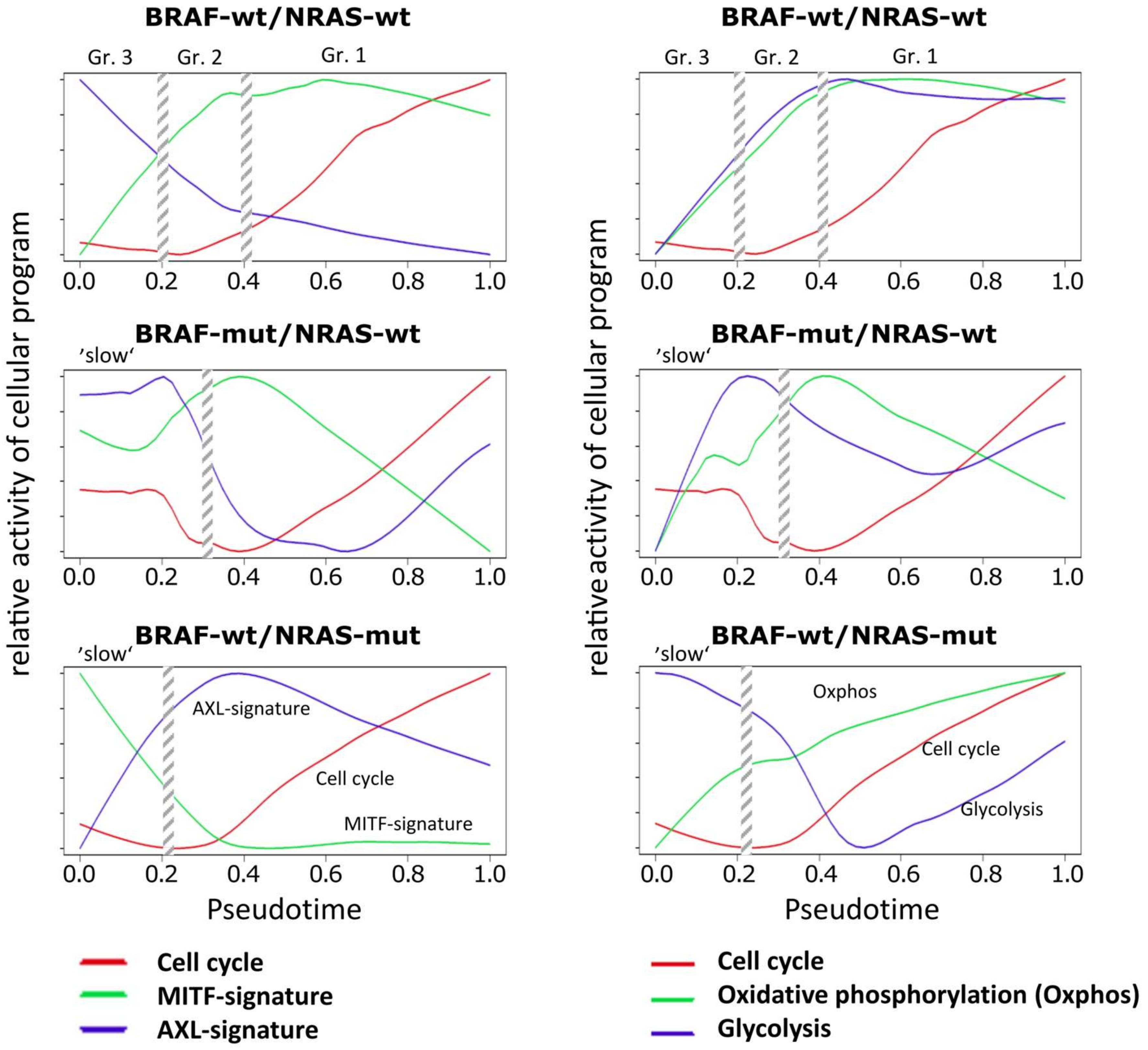
3.3. High Resolution PT-Dependent Activity of Cellular Programs
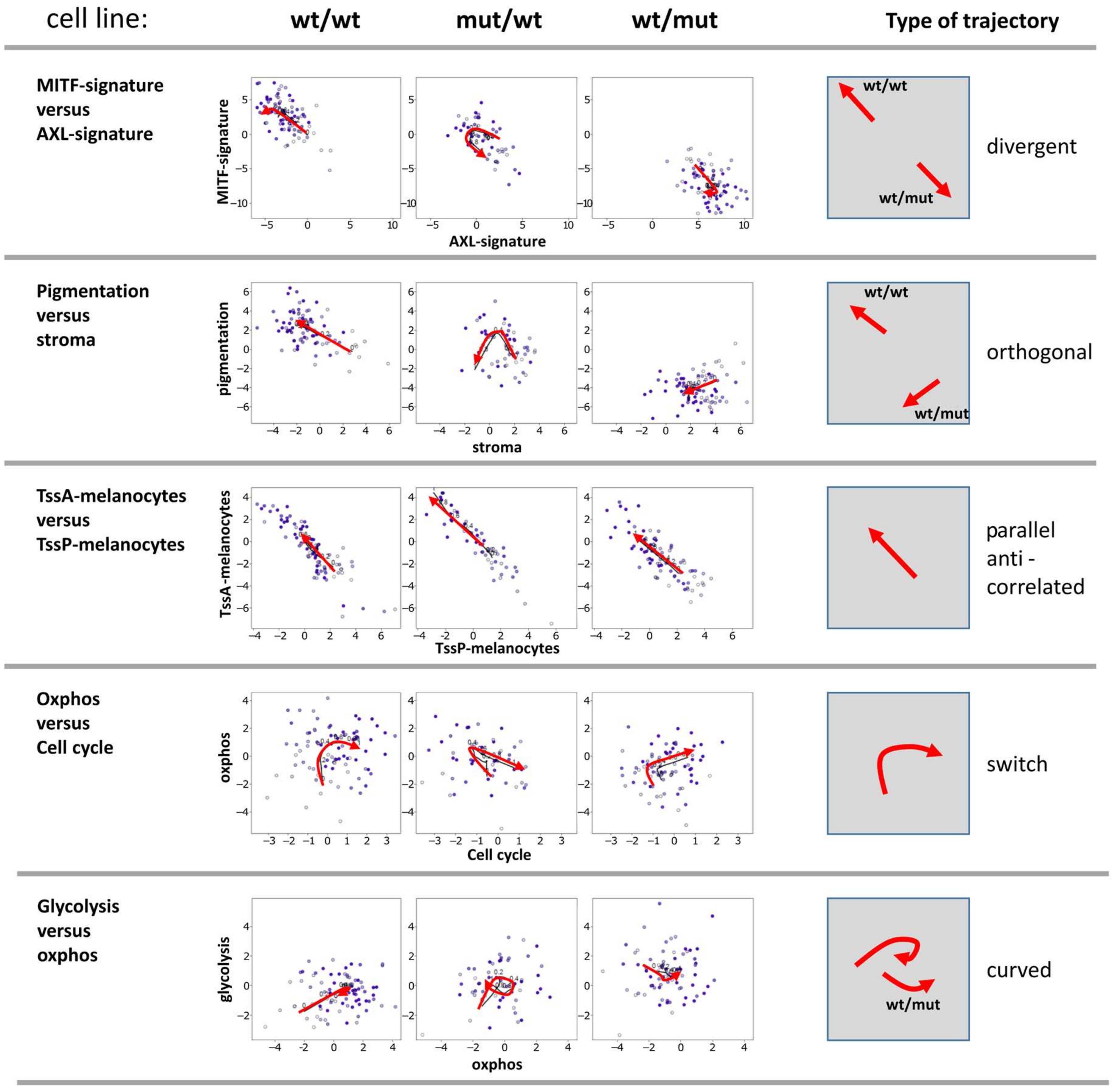
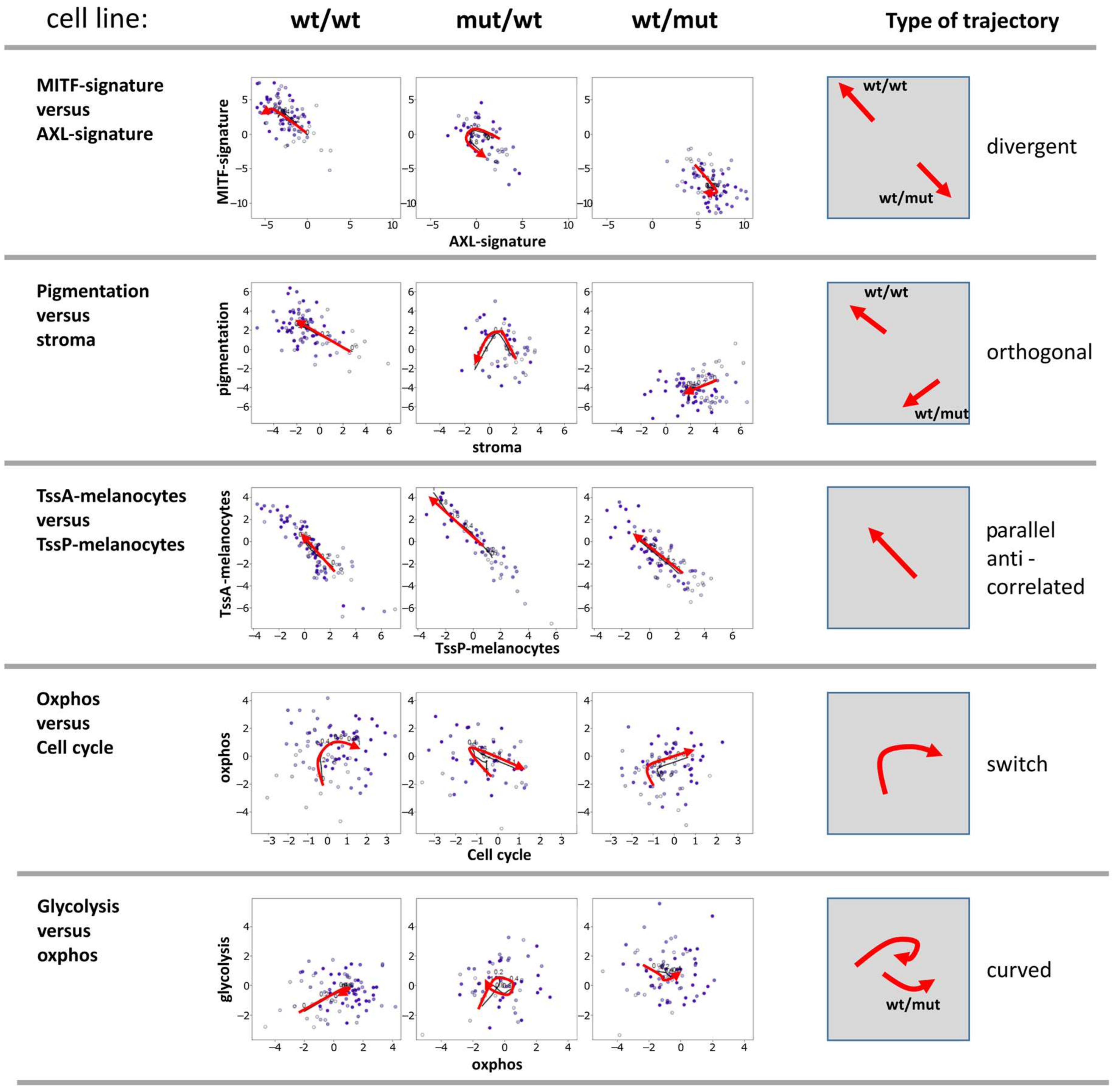
3.4. Two-Dimensional Trajectories Reveal Different Modes of Mutual Activities of Cellular Programs
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wong, D.J.L.; Ribas, A. Targeted therapy for melanoma. Cancer Treat. Res. 2016, 167, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Callahan, M.K.; Flaherty, C.R.; Postow, M.A. Checkpoint blockade for the treatment of advanced melanoma. Cancer Treat. Res. 2016, 167, 231–250. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, Z.; Ribas, A. Combination therapy with BRAF and MEK inhibitors for melanoma: Latest evidence and place in therapy. Ther. Adv. Med. Oncol. 2016, 8, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Welsh, S.J.; Rizos, H.; Scolyer, R.A.; Long, G.V. Resistance to combination BRAF and MEK inhibition in metastatic melanoma: Where to next? Eur. J. Cancer 2016, 62, 76–85. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Clonal heterogeneity and tumor evolution: Past, present, and the future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Jönsson, G.; Busch, C.; Knappskog, S.; Geisler, J.; Miletic, H.; Ringnér, M.; Lillehaug, J.R.; Borg, A.; Lønning, P.E. Gene expression profiling-based identification of molecular subtypes in stage IV melanomas with different clinical outcome. Clin. Cancer Res. 2010, 16, 3356–3367. [Google Scholar] [CrossRef] [PubMed]
- Harbst, K.; Staaf, J.; Lauss, M.; Karlsson, A.; Masback, A.; Johansson, I.; Bendahl, P.O.; Vallon-Christersson, J.; Törngren, T.; Ekedahl, H. Molecular profiling reveals low- and high-grade forms of primary melanoma. Clin. Cancer Res. 2012, 18, 4026–4036. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Harbst, K.; Lauss, M.; Cirenajwis, H.; Isaksson, K.; Rosengren, F.; Törngren, T.; Kvist, A.; Johansson, M.C.; Vallon-Christersson, J.; Baldetorp, B. Multiregion whole-exome sequencing uncovers the genetic evolution and mutational heterogeneity of early-stage metastatic melanoma. Cancer Res. 2016, 76, 4765–4774. [Google Scholar] [CrossRef] [PubMed]
- Gawad, C.; Koh, W.; Quake, S.R. Single-cell genome sequencing: Current state of the science. Nat. Rev. Genet. 2016, 17, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Tsoucas, D.; Yuan, G.C. Recent progress in single-cell cancer genomics. Curr. Opin. Genet. Dev. 2017, 42, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Gerber, T.; Willscher, E.; Loeffler-Wirth, H.; Hopp, L.; Schadendorf, D.; Schartl, M.; Anderegg, U.; Camp, G.; Treutlein, B.; Binder, H.; et al. Mapping heterogeneity in patient-derived melanoma cultures by single-cell RNA-seq. Oncotarget 2017, 8, 846–862. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Moris, N.; Pina, C.; Arias, A.M. Transition states and cell fate decisions in epigenetic landscapes. Nat. Rev. Genet. 2016, 17, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell–like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Suva, M.L.; Riggi, N.; Bernstein, B.E. Epigenetic Reprogramming in Cancer. Science 2013, 339, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Ugurel, S.; Thirumaran, R.K.; Bloethner, S.; Gast, A.; Sucker, A.; Mueller-Berghaus, J.; Rittgen, W.; Hemminki, K.; Becker, J.C.; Kumar, R.; et al. B-RAF and N-RAS mutations are preserved during short time in vitro propagation and differentially impact prognosis. PLoS ONE 2007, 2, e236. [Google Scholar] [CrossRef] [PubMed]
- Camp, J.G.; Badsha, F.; Florio, M.; Kanton, S.; Gerber, T.; Wilsch-Bräuninger, M.; Lewitus, E.; Sykes, A.; Hevers, W.; Lancaster, M.; et al. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc. Natl. Acad. Sci. USA 2015, 112, 15672–15677. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Scialdone, A.; Natarajan, K.N.; Saraiva, L.R.; Proserpio, V.; Teichmann, S.A.; Stegle, O.; Marioni, J.C.; Buettner, F. Computational assignment of cell-cycle stage from single-cell transcriptome data. Methods 2015, 85, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.R.; Yau, C. Order Under Uncertainty: Robust Differential Expression Analysis Using Probabilistic Models for Pseudotime Inference. PLoS Comput. Biol. 2016, 12, e1005212. [Google Scholar] [CrossRef] [PubMed]
- Bendall, S.C.; Davis, K.L.; Amir, E.-A.D.; Tadmor, M.D.; Simonds, E.F.; Chen, T.J.; Shenfeld, D.K.; Nolan, G.P.; Peer, D. Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell 2014, 157, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G.; Wright, G.; Dave, S.S.; Xiao, W.; Powell, J.; Zhao, H.; Xu, W.; Tan, B.; Goldschmidt, N.; Iqbal, J.; et al. Stromal gene signatures in large-B-cell lymphomas. N. Engl. J. Med. 2008, 359, 2313–2323. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, M.L.; Sherlock, G.; Saldanha, A.J.; Murray, J.I.; Ball, C.A.; Alexander, K.E.; Matese, J.C.; Perou, C.M.; Hurt, M.M.; Brown, P.O.; et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol. Biol. Cell 2002, 13, 1977–2000. [Google Scholar] [CrossRef] [PubMed]
- Törönen, P.; Ojala, P.J.; Marttinen, P.; Holm, L. Robust extraction of functional signals from gene set analysis using a generalized threshold free scoring function. BMC Bioinform. 2009, 10, 307. [Google Scholar] [CrossRef] [PubMed]
- Wirth, H.; von Bergen, M.; Binder, H. Mining SOM expression portraits: Feature selection and integrating concepts of molecular function. BioData Min. 2012, 5, 18–63. [Google Scholar] [CrossRef] [PubMed]
- Winnepenninckx, V.; Lazar, V.; Michiels, S.; Dessen, P.; Stas, M.; Alonso, S.R.; Avril, M.F.; Ortiz Romero, P.L.; Robert, T.; Balacescu, O.; et al. Gene expression profiling of primary cutaneous melanoma and clinical outcome. J. Natl. Cancer Inst. 2006, 98, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Lauss, M.; Nsengimana, J.; Staaf, J.; Newton-Bishop, J.; Jönsson, G. Consensus of Melanoma Gene Expression Subtypes Converges on Biological Entities. J. Investig. Dermatol. 2016, 136, 2502–2505. [Google Scholar] [CrossRef] [PubMed]
- Kasowski, M.; Kyriazopoulou-Panagiotopoulou, S.; Grubert, F.; Zaugg, J.B.; Kundaje, A.; Liu, Y.; Boyle, A.P.; Zhang, Q.C.; Zakharia, F.; Spacek, D.V.; et al. Extensive Variation in Chromatin States across Humans. Science 2013, 342, 750–752. [Google Scholar] [CrossRef] [PubMed]
- Roadmap Epigenomics Consortium; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Ratnikov, B.I.; Scott, D.A.; Osterman, A.L.; Smith, J.W.; Ronai, Z.A. Metabolic rewiring in melanoma. Oncogene 2017, 36, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Vincent, K.M.; Postovit, L.-M. Investigating the utility of human melanoma cell lines as tumour models. Oncotarget 2017, 8, 10498–10509. [Google Scholar] [CrossRef] [PubMed]
- Brandner, J.M.; Haass, N.K. Melanoma’s connections to the tumour microenvironment. Pathology 2013, 45, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.; Foppen, M.H.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef] [PubMed]
- Konieczkowski, D.J.; Johannessen, C.M.; Abudayyeh, O.; Kim, J.W.; Cooper, Z.A.; Piris, A.; Frederick, D.T.; Barzily-Rokni, M.; Straussman, R.; Haq, R.; et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014, 4, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Falletta, P.; Sanchez-Del-Campo, L.; Chauhan, J.; Effern, M.; Kenyon, A.; Kershaw, C.J.; Siddaway, R.; Lisle, R.; Freter, R.; Daniels, M.J.; et al. Translation reprogramming is an evolutionarily conserved driver of phenotypic plasticity and therapeutic resistance in melanoma. Genes Dev. 2017, 31, 18–33. [Google Scholar] [CrossRef] [PubMed]
- Ramanujan, V.K. Metabolic Plasticity in Cancer Cells: Reconnecting Mitochondrial Function to Cancer Control. J. Cell Sci. Ther. 2015, 6, 1000211. [Google Scholar] [CrossRef] [PubMed]
- Haq, R. Metabolic Dysregulation in Melanoma: Cause or Consequence? Cancer Discov. 2014, 4, 390–391. [Google Scholar] [CrossRef] [PubMed]
- Haq, R.; Fisher, D.E.; Widlund, H.R. Molecular pathways: BRAF induces bioenergetic adaptation by attenuating oxidative phosphorylation. Clin. Cancer Res. 2014, 20, 2257–2263. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Yang, G.; Hou, Y.; Tang, X.; Wu, C.; Wu, X.-A.; Guo, L.; Zhu, Q.; Luo, H.; Du, Y.E.; et al. Cytoplasmic GPER translocation in cancer-associated fibroblasts mediates cAMP/PKA/CREB/glycolytic axis to confer tumor cells with multidrug resistance. Oncogene 2017, 36, 2131–2145. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Top Gene Sets Changing with PT | Top-10 Genes | ||
|---|---|---|---|---|
| BRAF/NRAS | Correlated | Anti-Correlated | Correlated | Anti-Correlated |
| wt/wt | Sister chromatid cohesion (10−11) 1 | Cell adhesion (10−10) | H2AFZ CHCHD6 CD63 TRPM1 MLANA GSTP1 TUBA1B SLC25A5 HSPD1 PMEL (<10−8) | ANXA1 CALD1 (<10−5) LIMA1 CRYAB FN1 ANXA2 ABL2 FKBP9 A2M ARID5B (<0.002) |
| Chromosome, centromeric region (10−11) | Ras guanyl-nucleotide exchange factor activity (10−10) | |||
| DNA replication (10−11) | Extracellular matrix organization (10−9) | |||
| Kinetochore (10−11) | Proteinaceous extracellular matrix (10−9) | |||
| Cell division (10−10) | Skeletal muscle tissue development (10−9) | |||
| mut/wt | Mitochondrial inner membrane (0.008) | Extracellular matrix organization (0.008) | UQCRHL UQCRH MRPL24 (<0.07) FIBP CCDC167 RAB18 CYC1 KPNA2 RBMX RAB32 (<0.14) | EIF4G3 TUBG1 MITD1 TRIB2 (<0.2) SESN1 TSN EXO1 BIRC5 ITGAE SKA2 (<0.3) |
| Mitochondrial respiratory chain complex I assembly (0.01) | Collagen trimer (0.01) | |||
| Mitochondrion (0.01) | Negative regulation of cell adhesion (0.01) | |||
| Chromatin (0.01) | Ras guanyl-nucleotide exchange factor activity (0.02) | |||
| DNA metabolic process (0.01) | Cell adhesion (0.02) | |||
| Cell division (0.01) | ||||
| wt/mut | DNA replication (10−10) | Calcium ion binding (10−6) | RAD51AP1 PRIM1 (<10−5) KNTC1 NUP107 BARD1 RRM2 CDK1 HAT1 DEK HELLS (<0.001) | GIGYF2 SORBS2 TANC1 (0.02) SLITRK6 STAT2 (0.03) PTPRS FUNDC2 TMX2 LOXL3 TM9SF2 (0.05) |
| Cell division (10−9) | Carbohydrate binding (10−5) | |||
| Mitotic nuclear division (10−9) | Endoplasmic reticulum lumen (10−5) | |||
| Sister chromatid cohesion (10−9) | Extracellular region (10−5) | |||
| Mitotic sister chromatid segregation (10−9) | Golgi membrane (10−5) | |||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loeffler-Wirth, H.; Binder, H.; Willscher, E.; Gerber, T.; Kunz, M. Pseudotime Dynamics in Melanoma Single-Cell Transcriptomes Reveals Different Mechanisms of Tumor Progression. Biology 2018, 7, 23. https://doi.org/10.3390/biology7020023
Loeffler-Wirth H, Binder H, Willscher E, Gerber T, Kunz M. Pseudotime Dynamics in Melanoma Single-Cell Transcriptomes Reveals Different Mechanisms of Tumor Progression. Biology. 2018; 7(2):23. https://doi.org/10.3390/biology7020023
Chicago/Turabian StyleLoeffler-Wirth, Henry, Hans Binder, Edith Willscher, Tobias Gerber, and Manfred Kunz. 2018. "Pseudotime Dynamics in Melanoma Single-Cell Transcriptomes Reveals Different Mechanisms of Tumor Progression" Biology 7, no. 2: 23. https://doi.org/10.3390/biology7020023
APA StyleLoeffler-Wirth, H., Binder, H., Willscher, E., Gerber, T., & Kunz, M. (2018). Pseudotime Dynamics in Melanoma Single-Cell Transcriptomes Reveals Different Mechanisms of Tumor Progression. Biology, 7(2), 23. https://doi.org/10.3390/biology7020023