Deep Learning Neural Network Prediction Method Improves Proteome Profiling of Vascular Sap of Grapevines during Pierce’s Disease Development


 ,
,  and
and
Abstract
:1. Introduction
2. Material and Methods
2.1. Plant Material and X. fastidiosa Inoculation
2.2. Vascular Sap Extraction and X. fastidiosa Quantification
2.3. Protein Digestion of Vascular Leaf Sap
2.4. Liquid Chromatography Tandem Mass Spectrometry
2.5. Chromatogram Library Creation
2.6. Analytic Samples, Data Analysis and Raw Data Processing
2.7. Spectral Library Search
2.8. Quantification and Criteria for Protein Identification
2.9. Functional Enrichment Analysis
3. Results
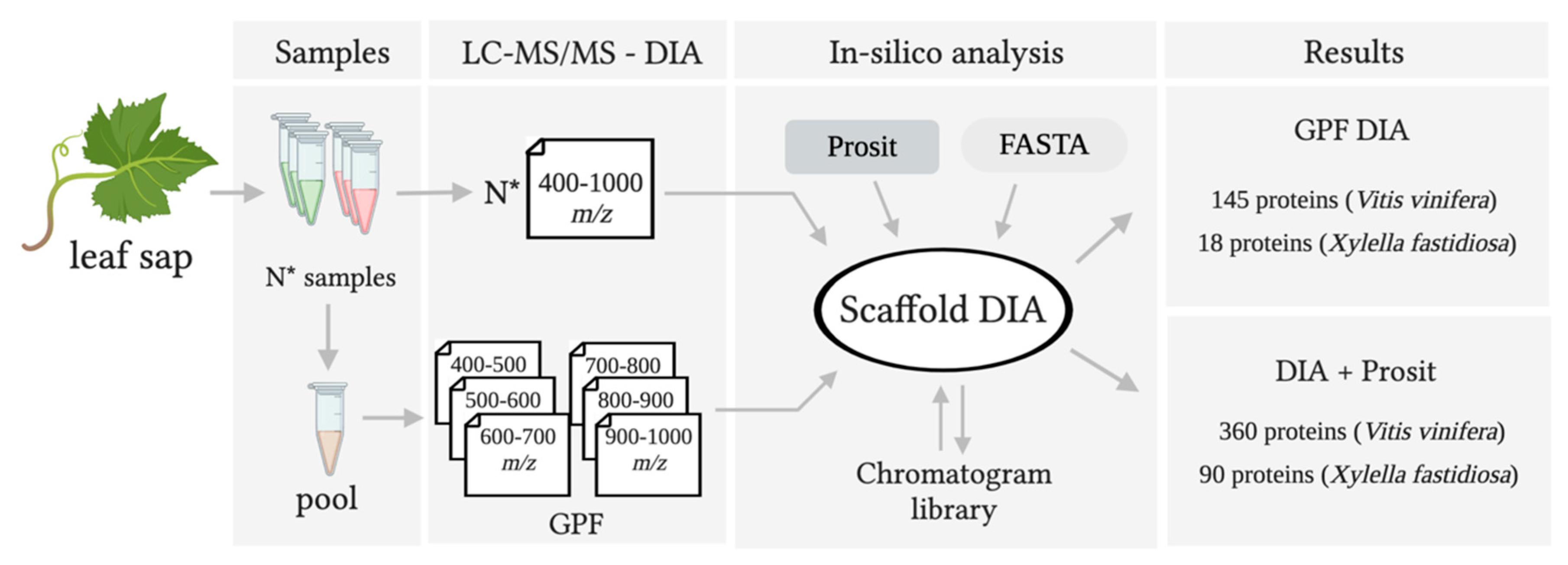
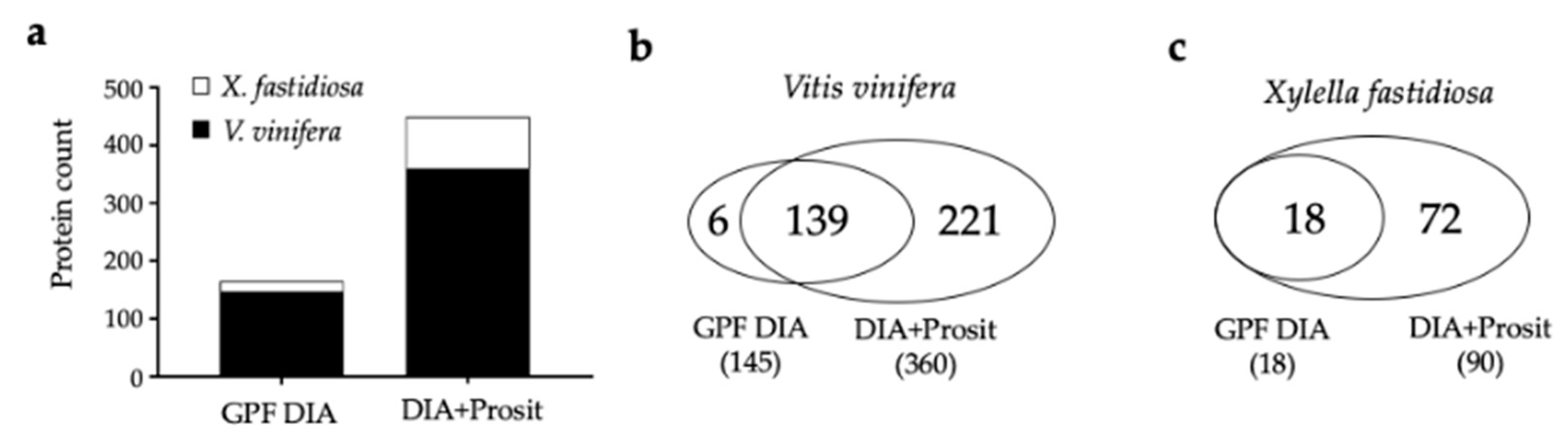
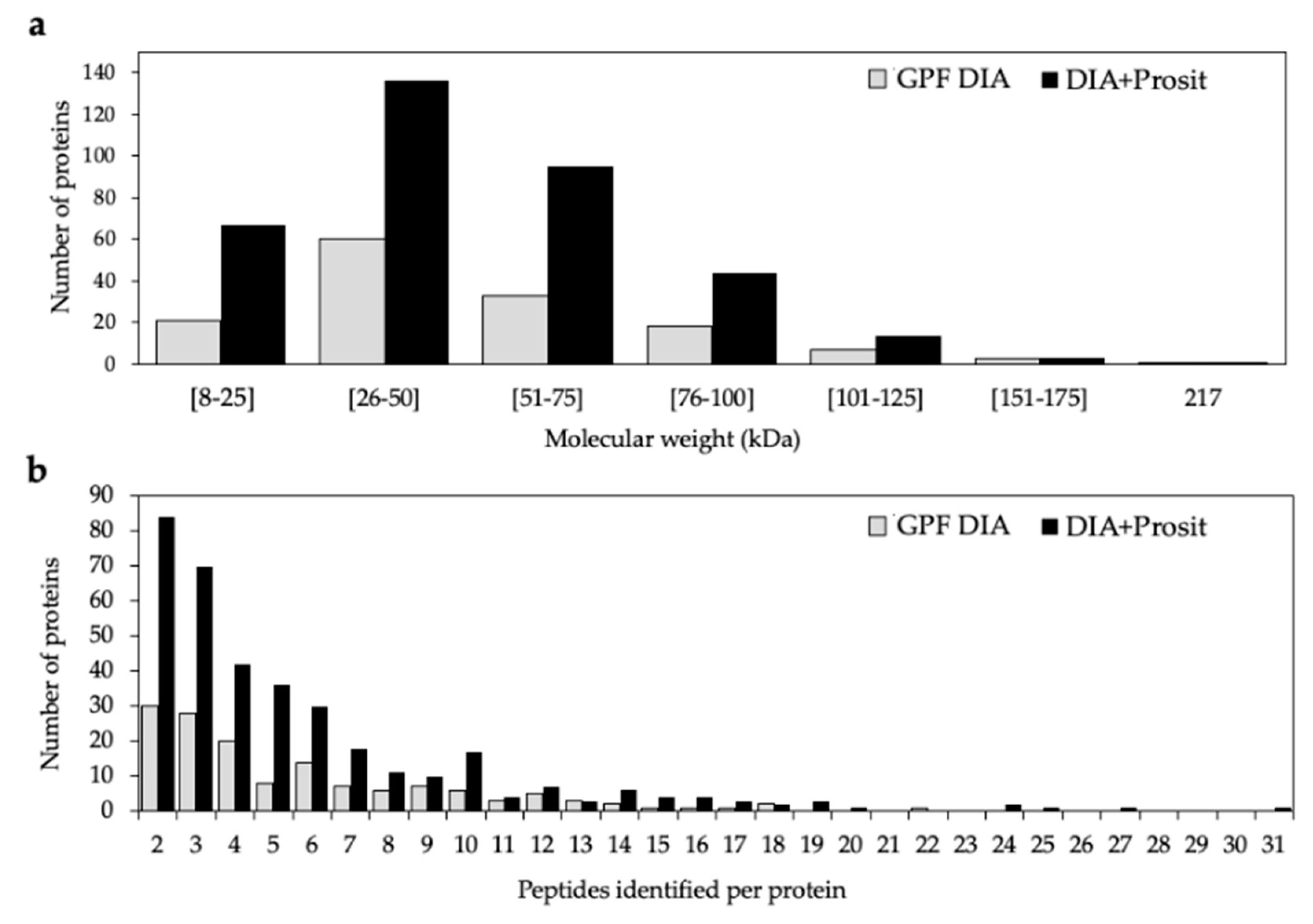
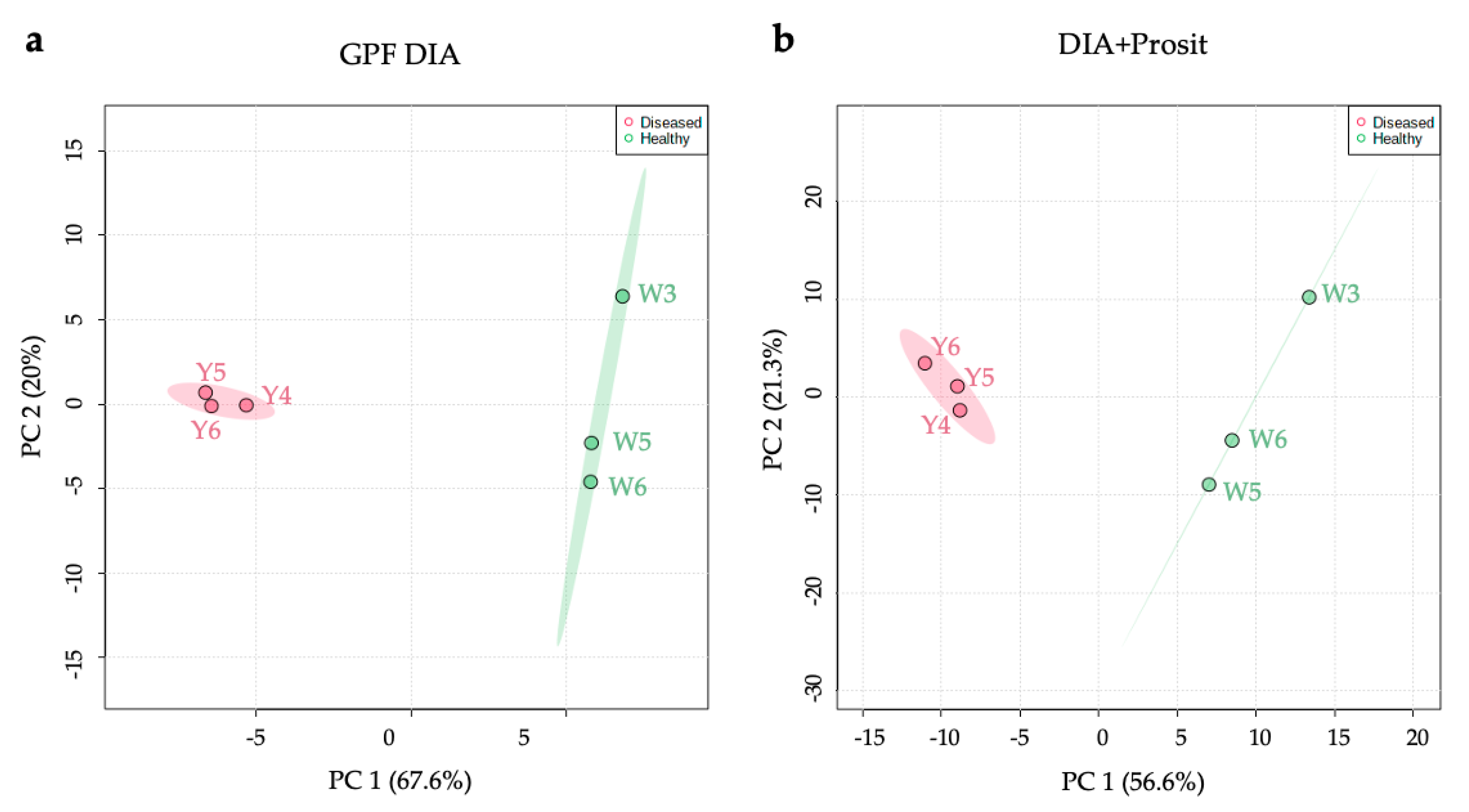
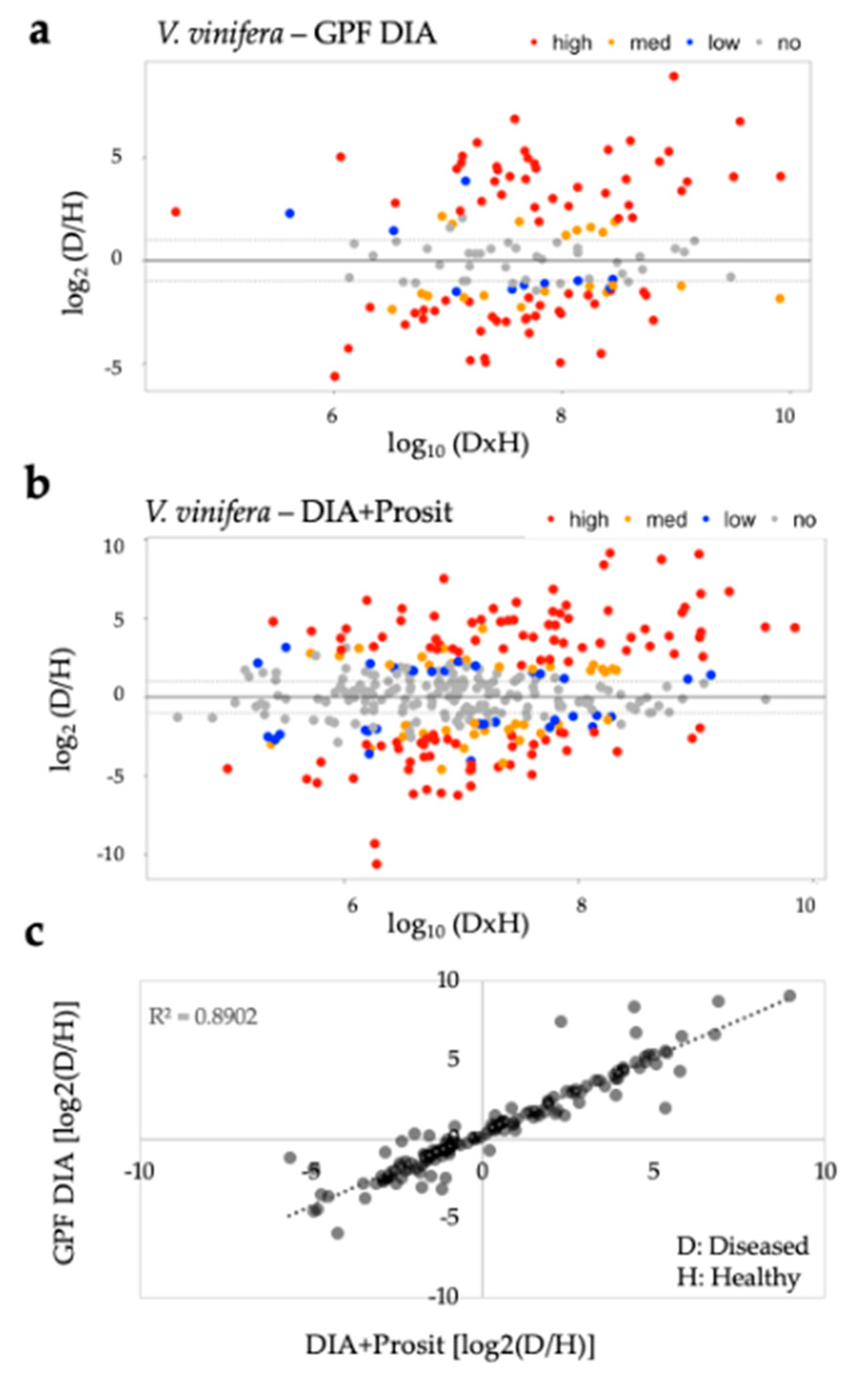
3.1. Creating a DIA Library and Improving the Data Mining of Vascular Sap Proteome Data
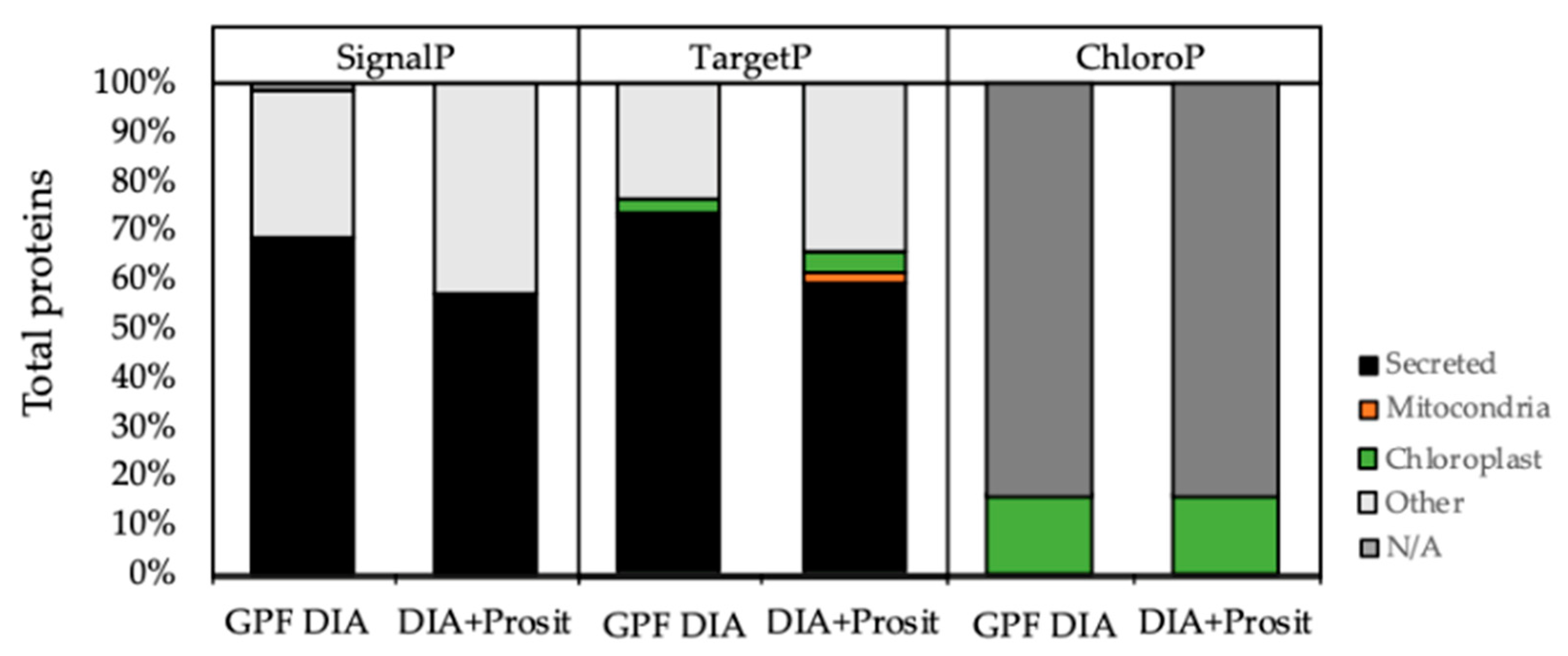
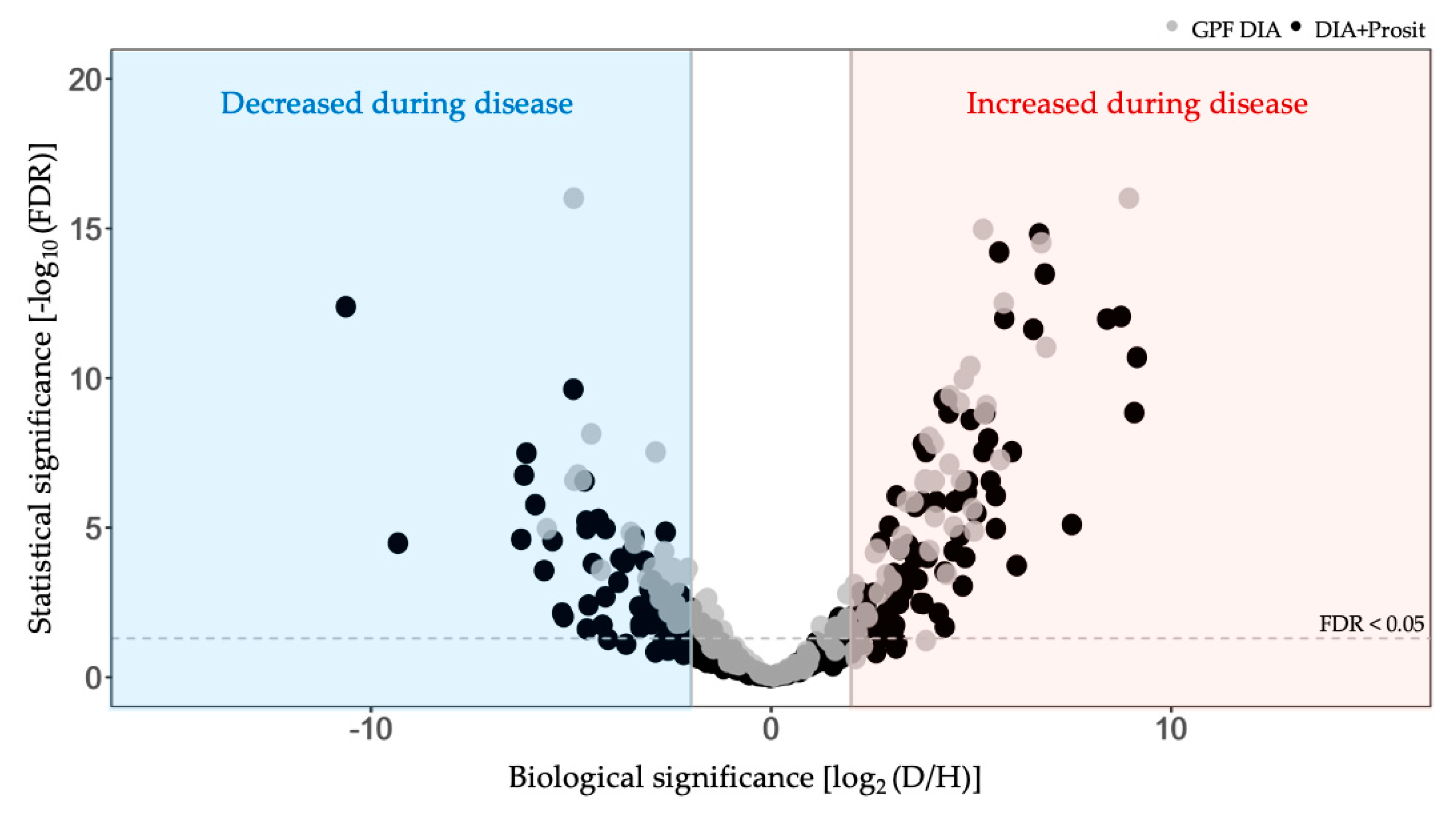
3.2. Plant Secreted Proteins in Response to Pierce’s Disease
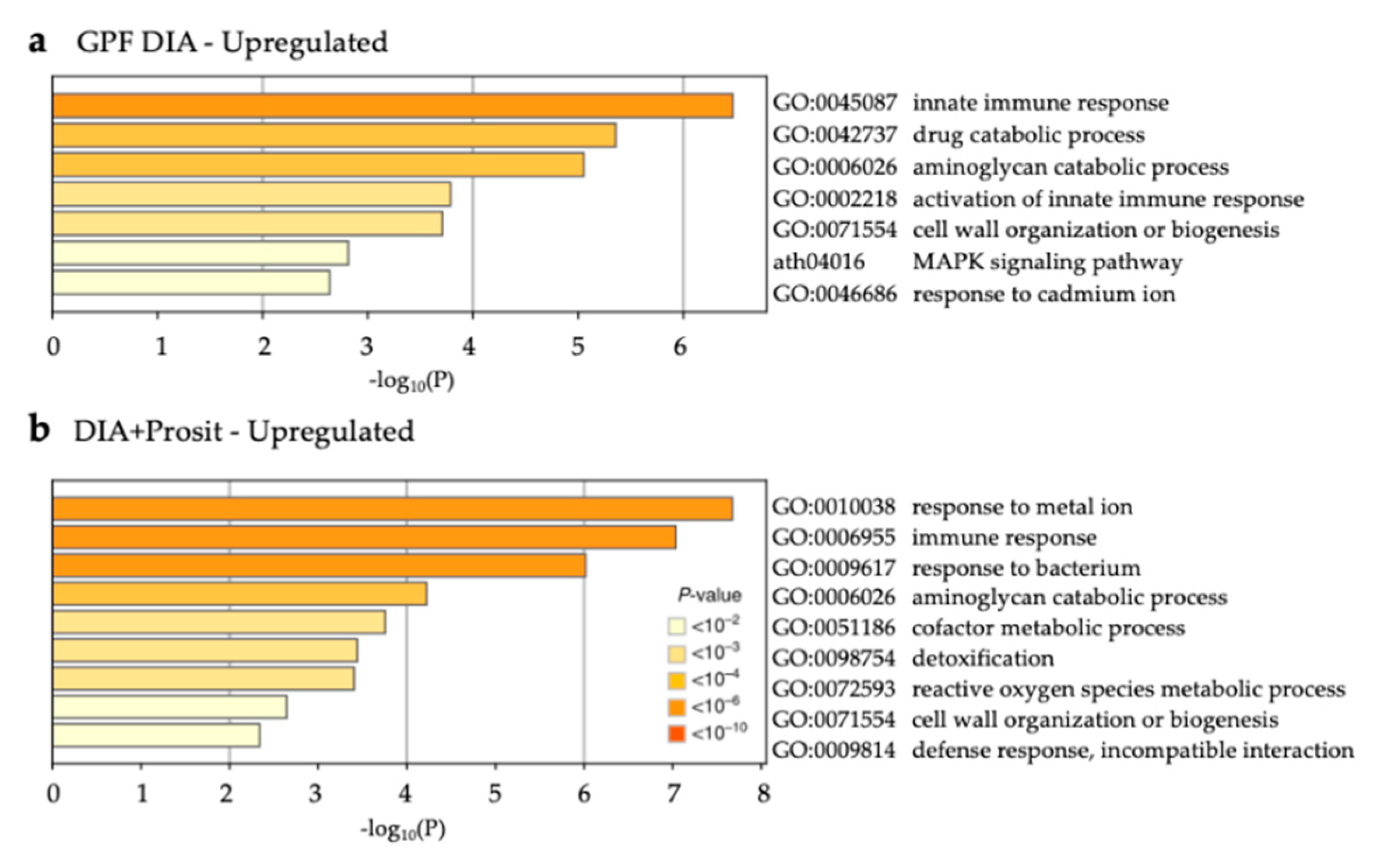
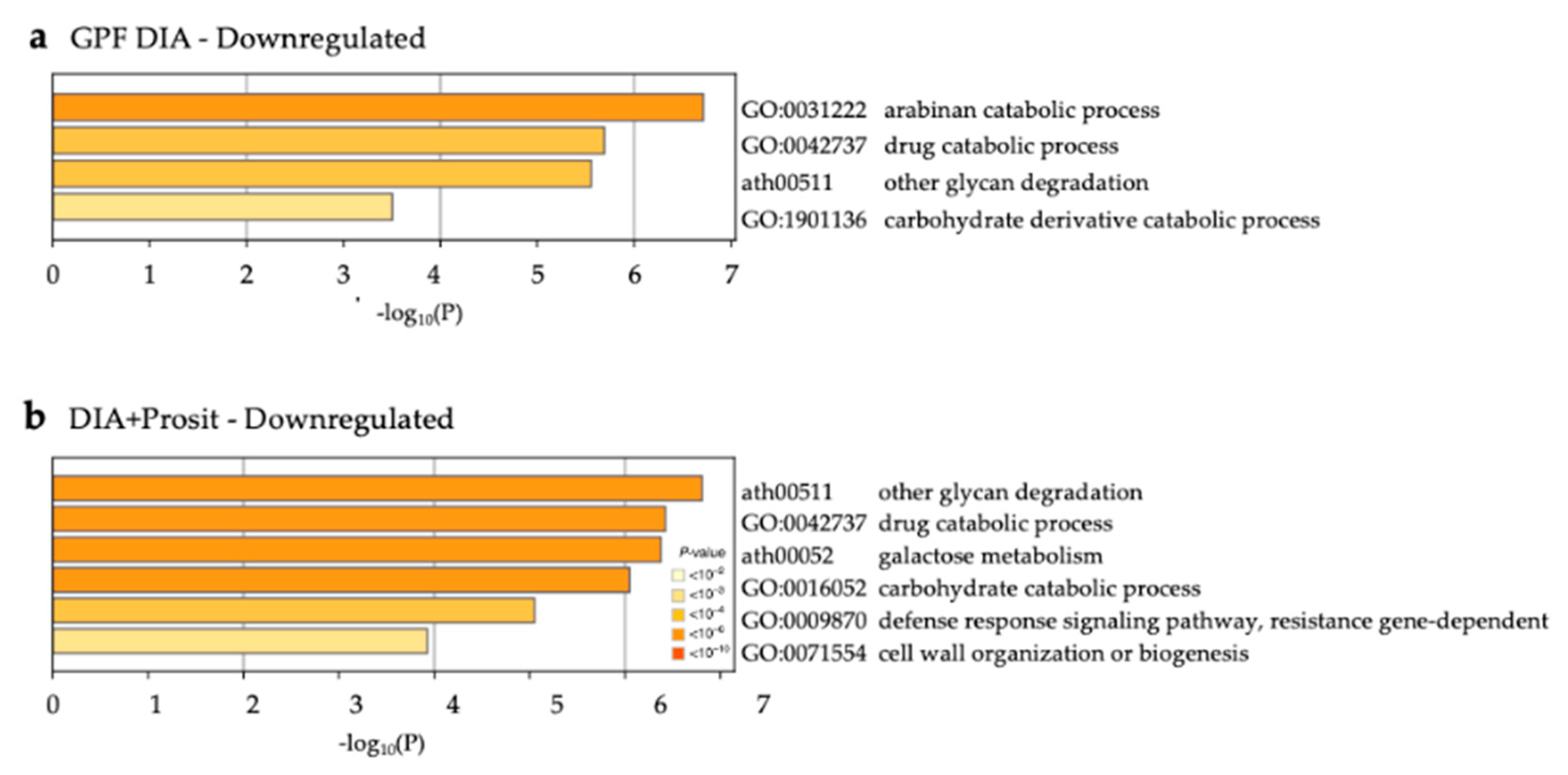
3.3. Enriched Biological Processes in Grapevine Vascular Leaf Sap
4. Discussion
4.1. A New Proteomic Approach for Vascular Sap Studies
4.2. Plant Response to X. fastidiosa Infection as Assessed Using the Vascular Sap
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Schepper, V.; De Swaef, T.; Bauweraerts, I.; Steppe, K. Phloem transport: A review of mechanisms and controls. J. Exp. Bot. 2013, 64, 4839–4850. [Google Scholar] [CrossRef] [Green Version]
- Tanner, W.; Beevers, H. Transpiration, a prerequisite for long-distance transport of minerals in plants? Proc. Natl. Acad. Sci. USA 2001, 98, 9443–9447. [Google Scholar] [CrossRef] [Green Version]
- Carella, P.; Wilson, D.C.; Kempthorne, C.J.; Cameron, R.K. Vascular sap proteomics: Providing insight into long-distance signaling during stress. Front. Plant Sci. 2016, 7, 651. [Google Scholar] [CrossRef] [Green Version]
- Drakakaki, G.; Dandekar, A. Protein secretion: How many secretory routes does a plant cell have? Plant Sci. 2013, 203–204, 74–78. [Google Scholar] [CrossRef]
- Agrawal, G.K.; Jwa, N.S.; Lebrun, M.H.; Job, D.; Rakwal, R. Plant secretome: Unlocking secrets of the secreted proteins. Proteomics 2010, 10, 799–827. [Google Scholar] [CrossRef]
- Yadeta, K.A.; Thomma, B.P.H.J. The xylem as battleground for plant hosts and vascular wilt pathogens. Front. Plant Sci. 2013, 4, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.J.; Purcell, A.H.; Thomson, S.V. Pierce’s disease of grapevines: Isolation of the causal bacterium. Science 1978, 199, 75–77. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, V.; Garnier, M.; Bove, J.M.; Beretta, M.J.G.; Reixeira, A.R.R.; Quaggio, J.A.; De Negri, J.D. Occurence of xylem-restricted bacteria in sweet orange trees affected by chlorotic variegation, a new citrus disease in Brazil. C. R. Acad. Sci. Ser. III 1990, 310, 345–349. [Google Scholar]
- Martelli, G.P. The current status of the quick decline syndrome of olive in southern Italy. Phytoparasitica 2016, 44, 1–10. [Google Scholar] [CrossRef]
- Simpson, A.J.G.; Reinach, F.C.; Arruda, P.; Abreu, F.A.; Acencio, M.; Alvarenga, R.; Alves, L.M.C.; Araya, J.E.; Baia, G.S.; Baptista, C.S.; et al. The genome sequence of the plant pathogen Xylella fastidiosa. Nature 2000, 406, 151–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorne, E.T.; Stevenson, J.F.; Rost, T.L.; Labavitch, J.M.; Matthews, M.A. Pierce’s disease symptoms: Comparison with symptoms of water deficit and the impact of water deficits. Am. J. Enol. Vitic. 2006, 57, 1–11. [Google Scholar]
- Chatterjee, S.; Wistrom, C.; Lindow, S.E. A cell-cell signaling sensor is required for virulence and insect transmission of Xylella fastidiosa. Proc. Natl. Acad. Sci. USA 2008, 105, 2670–2675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.; Almeida, R.P.; Lindow, S. Living in two worlds: The plant and insect lifestyles of Xylella fastidiosa. Annu. Rev. Phytopathol. 2008, 46, 243–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ionescu, M.; Baccari, C.; Da Silva, A.M.; Garcia, A.; Yokota, K.; Lindow, S.E. Diffusible signal factor (DSF) synthase RpfF of Xylella fastidiosa is a multifunction protein also required for response to DSF. J. Bacteriol. 2013, 195, 5273–5284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Sun, Y.; Andrew Walker, M.; Labavitch, J.M. Vascular occlusions in grapevines with Pierce’s disease make disease symptom development worse. Plant Physiol. 2013, 161, 1529–1541. [Google Scholar] [CrossRef] [Green Version]
- Nascimento, R.; Gouran, H.; Chakraborty, S.; Gillespie, H.W.; Almeida-Souza, H.O.; Tu, A.; Rao, B.J.; Feldstein, P.A.; Bruening, G.; Goulart, L.R.; et al. The type II secreted lipase/esterase LesA is a key virulence factor required for Xylella fastidiosa pathogenesis in grapevines. Sci. Rep. 2016, 6, 18598. [Google Scholar] [CrossRef] [Green Version]
- Gouran, H.; Gillespie, H.; Nascimento, R.; Chakraborty, S.; Zaini, P.A.; Jacobson, A.; Phinney, B.S.; Dolan, D.; Durbin-Johnson, B.P.; Antonova, E.S.; et al. The secreted protease PrtA controls cell growth, biofilm formation and pathogenicity in Xylella fastidiosa. Sci. Rep. 2016, 6, 31098. [Google Scholar] [CrossRef] [Green Version]
- da Santiago, A.S.; Mendes, J.S.; dos Santos, C.A.; de Toledo, M.A.S.; Beloti, L.L.; Crucello, A.; Horta, M.A.C.; Favaro, M.T.D.P.; Munar, D.M.M.; de Souza, A.A.; et al. The antitoxin protein of a toxin-antitoxin system from Xylella fastidiosa is secreted via outer membrane vesicles. Front. Microbiol. 2016, 7, 1–14. [Google Scholar] [CrossRef]
- Cianciotto, N.P.; White, R.C. Expanding role of type II secretion in bacterial pathogenesis and beyond. Infect. Immun. 2017, 85, e00014–e00017. [Google Scholar] [CrossRef] [Green Version]
- Feitosa-Junior, O.R.; Stefanello, E.; Zaini, P.A.; Nascimento, R.; Pierry, P.M.; Dandekar, A.M.; Lindow, S.E.; Da Silva, A.M. Proteomic and metabolomic analyses of Xylella fastidiosa OMV-enriched fractions reveal association with virulence factors and signaling molecules of the DSF family. Phytopathology 2019, 109, 1344–1353. [Google Scholar] [CrossRef]
- Agüero, C.B.; Thorne, E.T.; Ibáñez, A.M.; Gubler, W.D.; Dandekar, A.M. Xylem sap proteins from Vitis vinifera L. Chardonnay. Am. J. Enol. Vitic. 2008, 59, 306–311. [Google Scholar]
- Basha, S.M.; Mazhar, H.; Vasanthaiah, H.K.N. Proteomics approach to identify unique xylem sap proteins in Pierce’s disease-tolerant Vitis species. Appl. Biochem. Biotechnol. 2010, 160, 932–944. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lin, H.; Takahashi, Y.; Chen, F.; Walker, M.A.; Civerolo, E.L. Proteomic analysis of grapevine stem in response to Xylella fastidiosa inoculation. Physiol. Mol. Plant Pathol. 2011, 75, 90–99. [Google Scholar] [CrossRef]
- Delaunois, B.; Colby, T.; Belloy, N.; Conreux, A.; Harzen, A.; Baillieul, F.; Clément, C.; Schmidt, J.; Jeandet, P.; Cordelier, S. Large-scale proteomic analysis of the grapevine leaf apoplastic fluid reveals mainly stress-related proteins and cell wall modifying enzymes. BMC Plant Biol. 2013, 13, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kundai, R.K.; Lekan, M.C. Proteome biomarkers in xylem reveal Pierce’s disease tolerance in grape. J. Proteom. Bioinform. 2015, 8, 217–224. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.; Nascimento, R.; Zaini, P.A.; Gouran, H.; Rao, B.J.; Goulart, L.R.; Dandekar, A.M. Sequence/structural analysis of xylem proteome emphasizes pathogenesis-related proteins, chitinases an β-1, 3-glucanases as key players in grapevine defense against Xylella fastidiosa. PeerJ 2016, 4, e2007. [Google Scholar] [CrossRef] [Green Version]
- Venable, J.D.; Dong, M.-Q.; Wohlschlegel, J.; Dillin, A.; Yates, J.R. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat. Methods 2004, 1, 1–7. [Google Scholar] [CrossRef]
- Searle, B.C.; Swearingen, K.E.; Barnes, C.A.; Schmidt, T.; Gessulat, S.; Küster, B.; Wilhelm, M. Generating high quality libraries for DIA MS with empirically corrected peptide predictions. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Searle, B.C.; Pino, L.K.; Egertson, J.D.; Ting, Y.S.; Lawrence, R.T.; MacLean, B.X.; Villén, J.; MacCoss, M.J. Chromatogram libraries improve peptide detection and quantification by data independent acquisition mass spectrometry. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Liu, X.; Shen, C.; Lin, Y.; Yang, P.; Qiao, L. In silico spectral libraries by deep learning facilitate data-independent acquisition proteomics. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Gessulat, S.; Schmidt, T.; Zolg, D.P.; Samaras, P.; Schnatbaum, K.; Zerweck, J.; Knaute, T.; Rechenberger, J.; Delanghe, B.; Huhmer, A.; et al. Prosit: Proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat. Methods 2019, 16, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 2020, 17, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Ren, Y.; Yang, J.; Ren, Z.; Yang, H.; Liu, S. Improved peptide retention time prediction in liquid chromatography through deep learning. Anal. Chem. 2018, 90, 10881–10888. [Google Scholar] [CrossRef]
- Zhou, X.X.; Zeng, W.F.; Chi, H.; Luo, C.; Liu, C.; Zhan, J.; He, S.-M.; Zhang, Z. pDeep: Predicting MS/MS spectra of peptides with deep learning. Anal. Chem. 2017, 89, 12690–12697. [Google Scholar] [CrossRef] [PubMed]
- Tiwary, S.; Levy, R.; Gutenbrunner, P.; Salinas Soto, F.; Palaniappan, K.K.; Deming, L.; Berndl, M.; Brant, A.; Cimermancic, P.; Cox, J. High-quality MS/MS spectrum prediction for data-dependent and data-independent acquisition data analysis. Nat. Methods 2019, 16, 519–525. [Google Scholar] [CrossRef]
- Zolg, D.P.; Wilhelm, M.; Schmidt, T.; Médard, G.; Zerweck, J.; Knaute, T.; Wenschuh, H.; Reimer, U.; Schnatbaum, K.; Kuster, B. ProteomeTools: Systematic characterization of 21 post-translational protein modifications by liquid chromatography tandem mass spectrometry (LC-MS/MS) using synthetic peptides. Mol. Cell. Proteom. 2018, 17, 1850–1863. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Ge, W.; Ruan, G.; Cai, X.; Guo, T. Data-Independent Acquisition mass spectrometry-based proteomics and software tools: A glimpse in 2020. Proteomics 2020, 1900276, 1–12. [Google Scholar] [CrossRef]
- Francis, M.; Lin, H.; Rosa, J.C.-L.; Doddapaneni, H.; Civerolo, E.L. Genome-based PCR primers for specific and sensitive detection and quantification of Xylella fastidiosa. Eur. J. Plant Pathol. 2006, 115, 203–213. [Google Scholar] [CrossRef]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- Käll, L.; Canterbury, J.D.; Weston, J.; Noble, W.S.; MacCoss, M.J. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 2007, 4, 923–925. [Google Scholar] [CrossRef]
- Käll, L.; Storey, J.D.; MacCoss, M.J.; Noble, W.S. Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J. Proteome Res. 2008, 7, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Käll, L.; Storey, J.D.; Noble, W.S. Non-parametric estimation of posterior error probabilities associated with peptides identified by tandem mass spectrometry. Bioinformatics 2008, 24, 42–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef]
- Kalunke, R.M.; Tundo, S.; Benedetti, M.; Cervone, F.; De Lorenzo, G.; D’Ovidio, R. An update on polygalacturonase-inhibiting protein (PGIP), aleucine-rich repeat protein that protects crop plants against pathogens. Front. Plant Sci. 2015, 6, 146. [Google Scholar] [CrossRef] [Green Version]
- Héloir, M.-C.; Adrian, M.; Brulé, D.; Claverie, J.; Cordelier, S.; Daire, X.; Dorey, S.; Gauthier, A.; Lemaître-Guillier, C.; Negrel, J.; et al. Recognition of elicitors in grapevine: From MAMP and DAMP perception to induced resistance. Front. Plant Sci. 2019, 10, 1117. [Google Scholar] [CrossRef]
- Figueiredo, A.; Monteiro, F.; Sebastiana, M. Subtilisin-like proteases in plant-pathogen recognition and immune priming: A perspective. Front. Plant Sci. 2014, 5, 739. [Google Scholar] [CrossRef] [Green Version]
- Zaini, P.A.; Nascimento, R.; Gouran, H.; Cantu, D.; Chakraborty, S.; Phu, M.; Goulart, L.R.; Dandekar, A.M. Molecular profiling of Pierce’s disease outlines the response circuitry of Vitis vinifera to Xylella fastidiosa infection. Front. Plant Sci. 2018, 9, 771. [Google Scholar] [CrossRef]
- Giannakis, C.; Bucheli, C.S.; Skene, K.G.M.; Robinson, S.P.; Steele Scott, N. Chitinase and β-1,3-glucanase in grapevine leaves: A possible defence against powdery mildew infection. Aust. J. Grape Wine Res. 1998, 4, 14–22. [Google Scholar] [CrossRef]
- Mestre, P.; Arista, G.; Piron, M.-C.; Rustenholz, C.; Ritzenthaler, C.; Merdinoglu, D.; Chich, J.-F. Identification of a Vitis vinifera endo-β-1,3-glucanase with antimicrobial activity against Plasmopara viticola. Mol. Plant Pathol. 2017, 18, 708–719. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vitis sp. Variety | Biological Material | Xf Inoc. | Method | Peptide Spectra Analysis | Total Proteins | Molecular Weight (kDa) | Matched Peptides | Signal Peptide | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Chardonnay | Xylem sap | No | 2D-PAGE MALDI-TOF MS/MS | GPM | 10 | 25–150 | 1 | No | [21] |
| PD tolerant/susceptible varieties | Xylem sap | No | 2D-PAGE LC-MS/MS | Mascot | 100 * | 20–75 | 1–4 | No | [22] |
| PD tolerant/susceptible varieties | Stem | Yes | 2D-PAGE nano-LC-MS/MS | Bioworks | 200 * | 14.4–45 | 2–32 | No | [23] |
| Chardonnay | Leaf and apoplastic fluid | No | 2D-PAGE MALDI-TOF MS/MS | Mascot | 227 | 15–120 | NA | No | [24] |
| PD tolerant/susceptible varieties | Xylem tissue | No | 2D-PAGE MALDI-TOF MS/MS | Mascot | 200 * | 20–75 | NA | No | [25] |
| Thompson Seedless | Xylem sap | Yes | LC-MS/MS | Scaffold | 91 | 10–114 | 2–23 | Yes | [26] |
| Thompson Seedless | Vascular leaf sap | Yes | LC-MS/MS | ScaffoldDIA (GPF) | 145 | 12–217 | 2–22 | Yes | This study |
| Thompson Seedless | Vascular leaf sap | Yes | LC-MS/MS | ScaffoldDIA+Prosit | 360 | 8–217 | 2–31 | Yes | This study |
| Accession Number | Arabidopsis Best Match | Protein Name | GPF DIA | DIA + Prosit | ||||
|---|---|---|---|---|---|---|---|---|
| Matched Peptide | Ratio * (log2) | FDR | Matched Peptide | Ratio * (log2) | FDR | |||
| Pathogenesis-related (PR1) | ||||||||
| VIT_03s0088g00810 | AT2G14610.1 | pathogenesis-related gene 1 | 2 | 6.85 | 9.37 × 10−12 | 2 | 8.76 | 8.77 × 10−13 |
| Beta-1,3-glucanases (PR2) | ||||||||
| VIT_08s0007g06060 | AT3G57240.1 | beta-1,3-glucanase 3 | 11 | 8.92 | 9.73 × 10−17 | 10 | 9.09 | 1.43 × 10−9 |
| VIT_08s0007g06040 | AT3G57240.1 | beta-1,3-glucanase 3 | 9 | 5.36 | 8.31 × 10−10 | 6 | 5.50 | 2.82 × 10−7 |
| VIT_06s0061g00120 | AT3G57240.1 | beta-1,3-glucanase 3 | 13 | 4.79 | 1.08 × 10−10 | 11 | 5.37 | 1.48 × 10−9 |
| Pathogenesis-related (PR3) | ||||||||
| VIT_04s0008g00120 | AT3G12500.1 | basic chitinase | 6 | 5.80 | 3.09 × 10−13 | 10 | 6.57 | 2.33 × 10−12 |
| Pathogenesis-related (PR4) | ||||||||
| VIT_14s0081g00030 | AT3G04720.1 | pathogenesis-related 4 | 3 | 4.96 | 4.05 × 10−11 | 3 | 5.44 | 1.07 × 10−8 |
| Chitinases | ||||||||
| VIT_16s0050g02230 | AT5G24090.1 | chitinase A | 7 | 4.37 | 3.63 × 10−4 | 9 | 8.41 | 1.06 × 10−12 |
| VIT_15s0046g01570 | AT5G24090.1 | chitinase A | 3 | 3.54 | 1.36 × 10−6 | 3 | 3.44 | 3.72 × 10−5 |
| VIT_11s0149g00380 | AT4G19810.1 | Glycosyl hydrolase ** | 5 | −2.94 | 2.26 × 10−4 | 3 | −2.47 | 9.29 × 10−3 |
| VIT_11s0206g00030 | AT4G19810.1 | Glycosyl hydrolase ** | 4 | −4.94 | 2.62 × 10−7 | 3 | −4.44 | 1.60 × 10−4 |
| VIT_16s0050g02210 | AT5G24090.1 | chitinase A | 6 | 6.03 | 2.88 × 10−8 | |||
| Peroxidases | ||||||||
| VIT_06s0004g07740 | AT5G05340.1 | Peroxidase superfamily | 8 | 3.82 | 3.10 × 10−7 | 6 | 4.15 | 1.36 × 10−6 |
| VIT_07s0191g00050 | AT2G22420.1 | Peroxidase superfamily | 6 | −3.53 | 1.46 × 10−5 | 7 | −2.72 | 1.20 × 10−3 |
| VIT_12s0055g01000 | AT5G64120.1 | Peroxidase superfamily | ND | ND | ND | 7 | −4.13 | 2.04 × 10−3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Helena Duarte Sagawa, C.; Zaini, P.A.; de A. B. Assis, R.; Saxe, H.; Salemi, M.; Jacobson, A.; Wilmarth, P.A.; Phinney, B.S.; M. Dandekar, A. Deep Learning Neural Network Prediction Method Improves Proteome Profiling of Vascular Sap of Grapevines during Pierce’s Disease Development. Biology 2020, 9, 261. https://doi.org/10.3390/biology9090261
Helena Duarte Sagawa C, Zaini PA, de A. B. Assis R, Saxe H, Salemi M, Jacobson A, Wilmarth PA, Phinney BS, M. Dandekar A. Deep Learning Neural Network Prediction Method Improves Proteome Profiling of Vascular Sap of Grapevines during Pierce’s Disease Development. Biology. 2020; 9(9):261. https://doi.org/10.3390/biology9090261
Chicago/Turabian StyleHelena Duarte Sagawa, Cíntia, Paulo A. Zaini, Renata de A. B. Assis, Houston Saxe, Michelle Salemi, Aaron Jacobson, Phillip A. Wilmarth, Brett S. Phinney, and Abhaya M. Dandekar. 2020. "Deep Learning Neural Network Prediction Method Improves Proteome Profiling of Vascular Sap of Grapevines during Pierce’s Disease Development" Biology 9, no. 9: 261. https://doi.org/10.3390/biology9090261