
Structural and Dynamics Perspectives on the Binding of Substrate and Inhibitors in Mycobacterium tuberculosis DHFR


Abstract
:1. Introduction
2. Method of Calculations
2.1. Molecular Structure Preparations
2.2. Molecular Modeling Studies
2.3. Thermodynamics Quantities Calculations
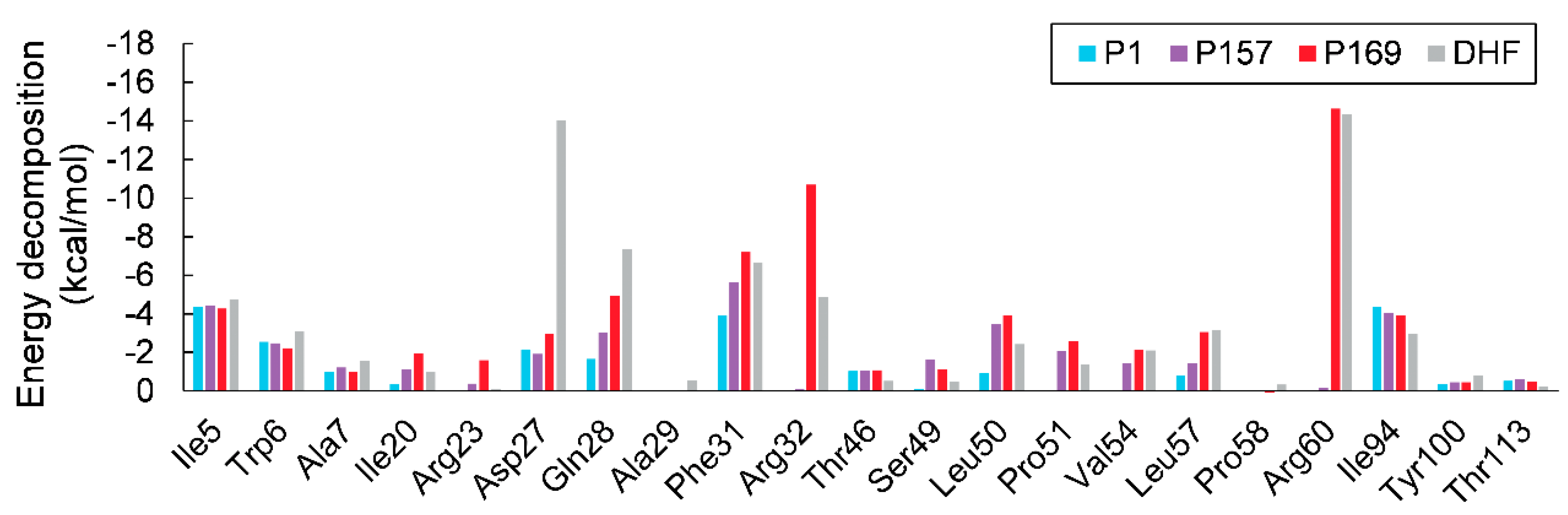
3. Results and Discussion
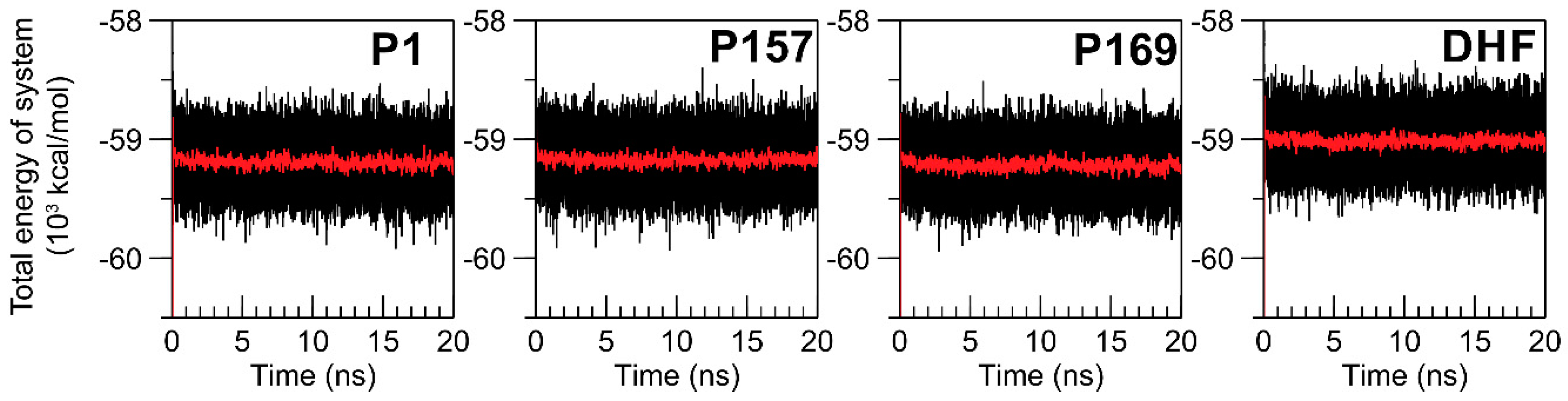
3.1. Equilibration of MD Simulations
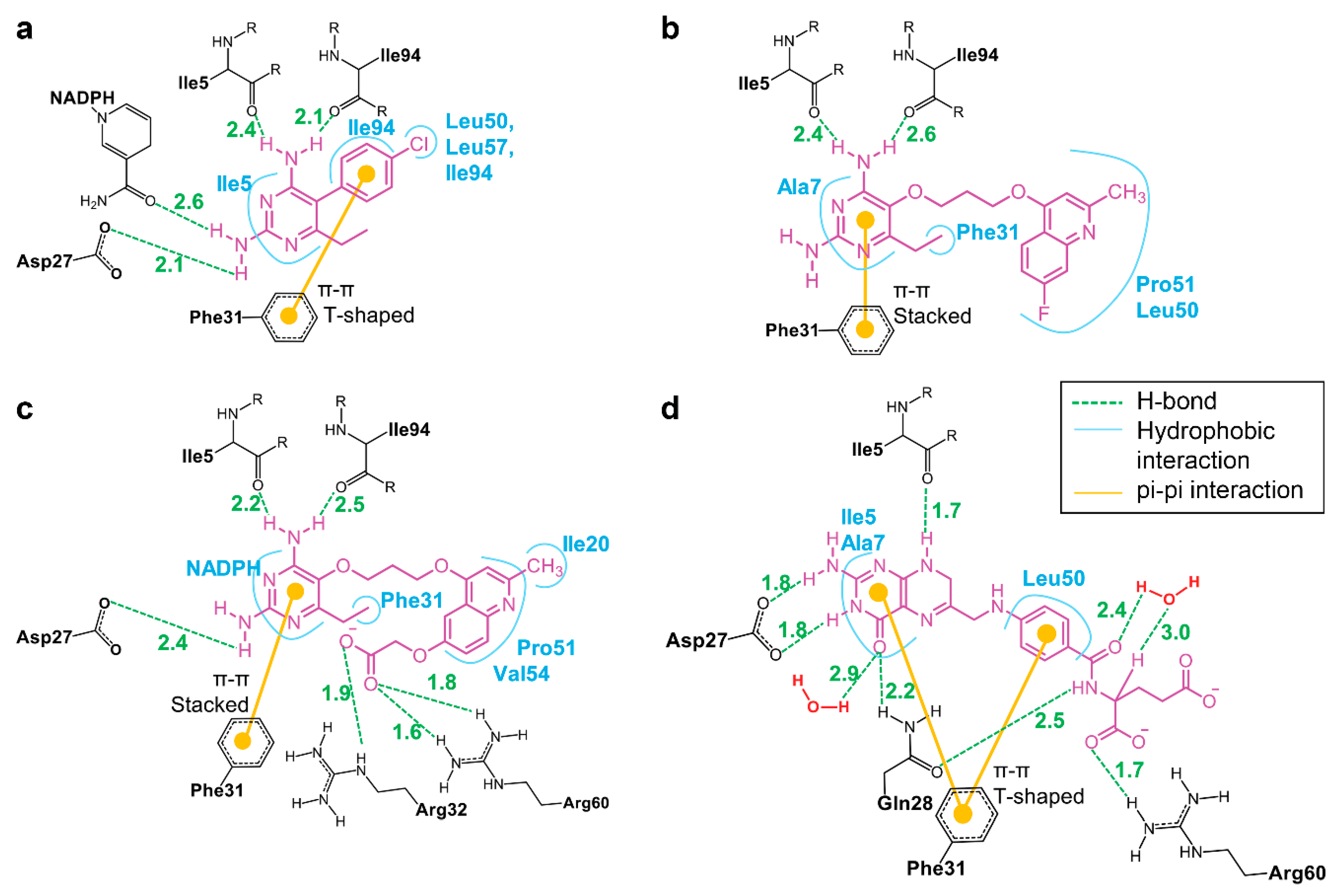
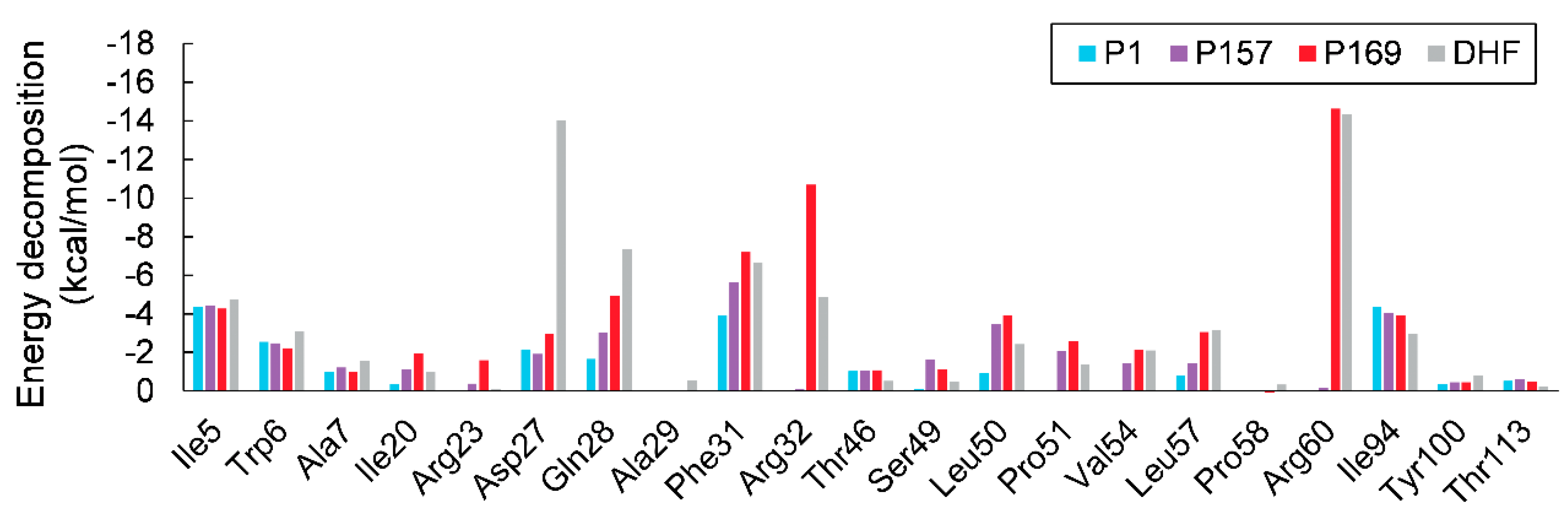
3.2. MM/GBSA Calculations
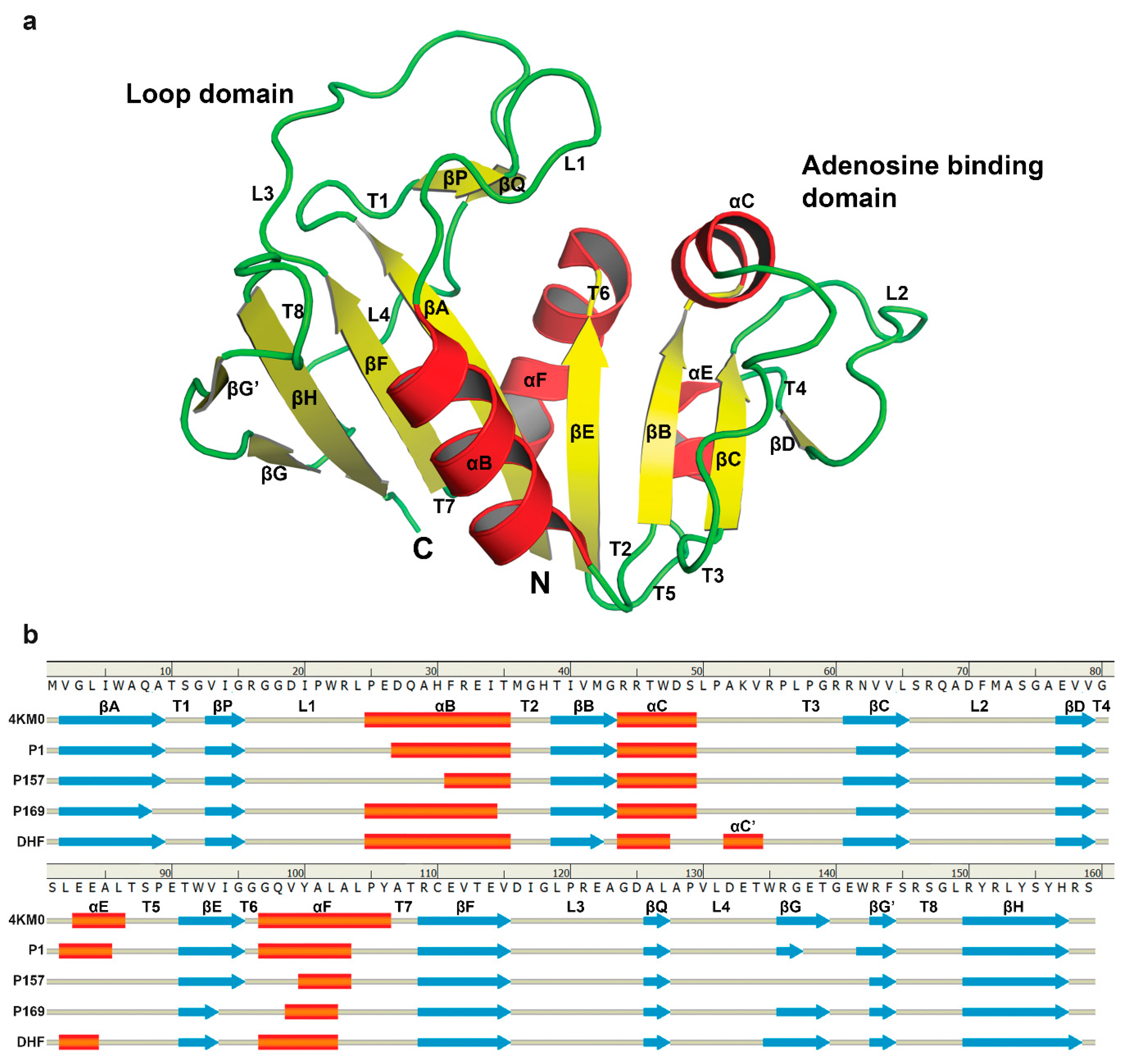
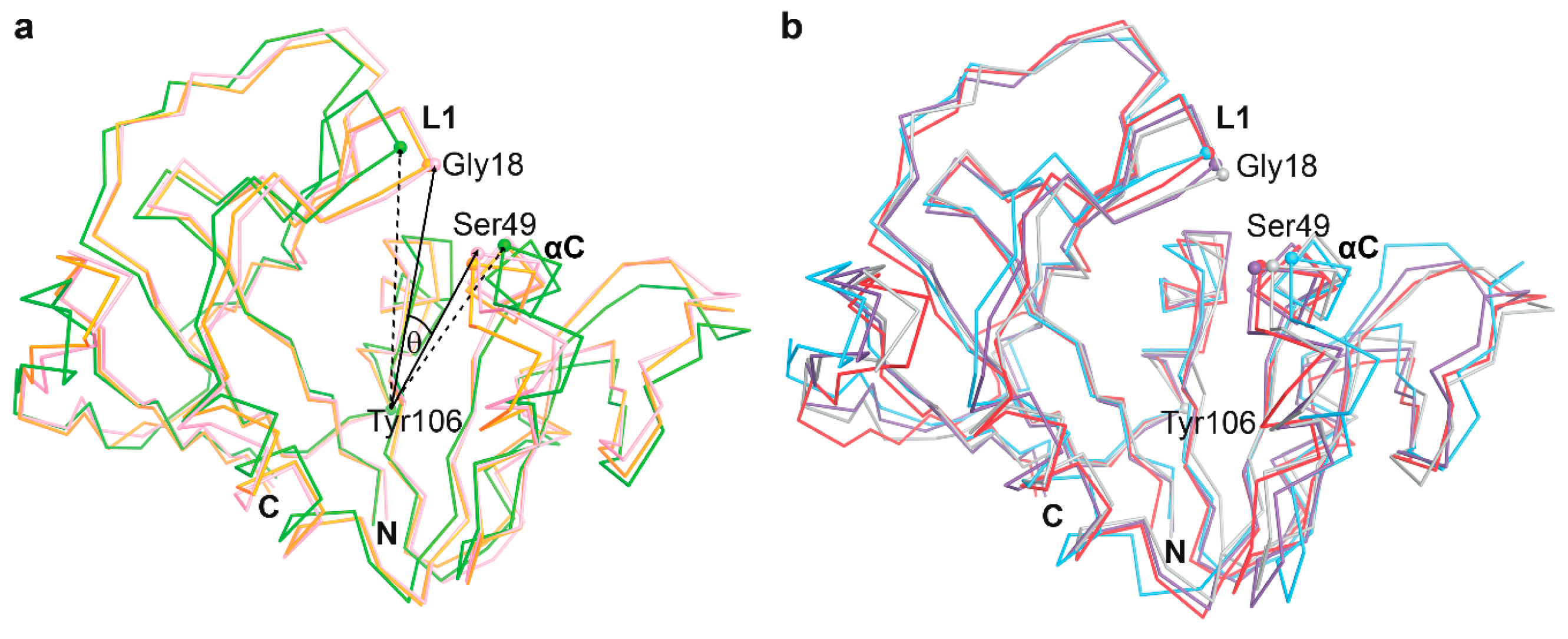
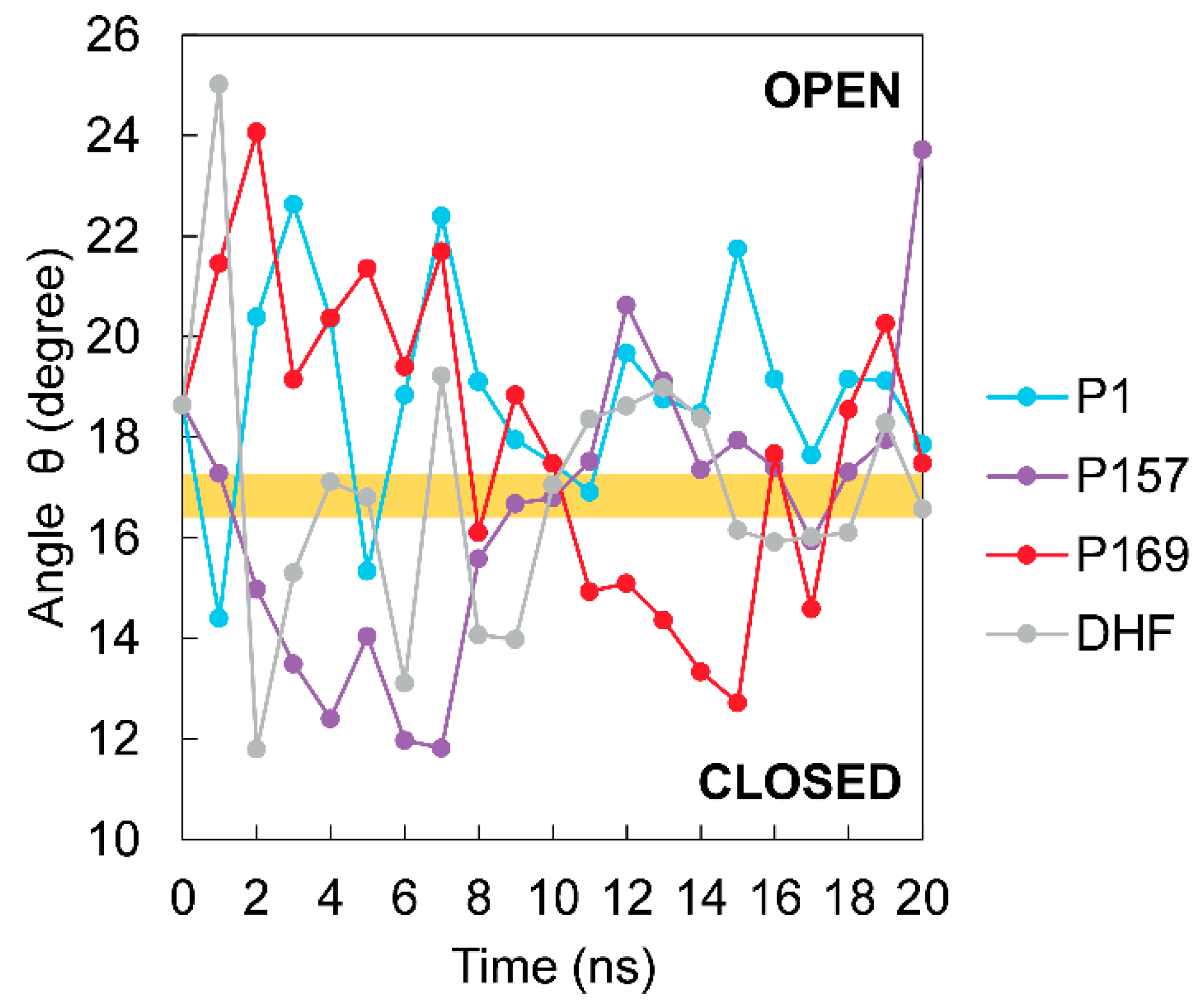
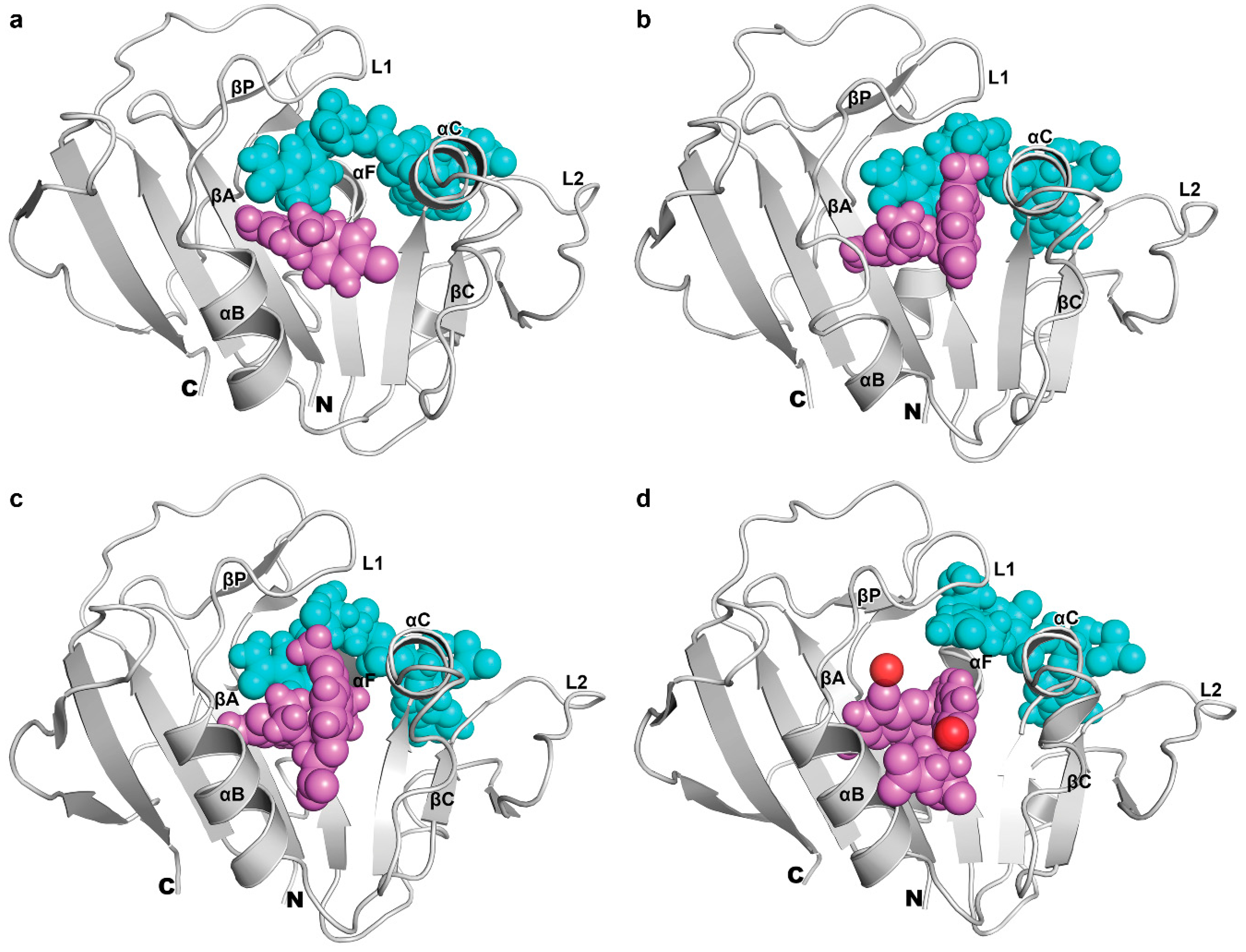
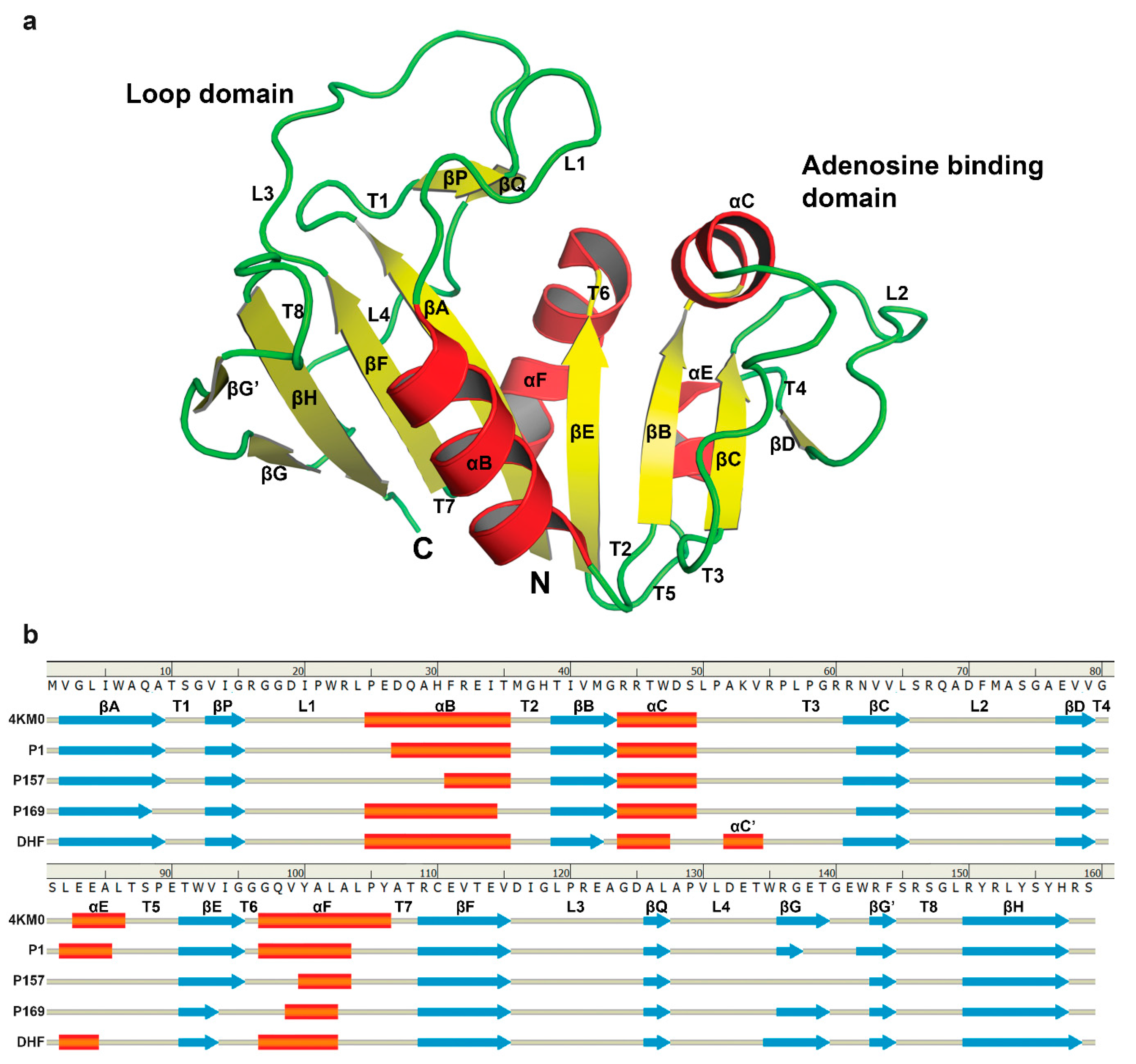
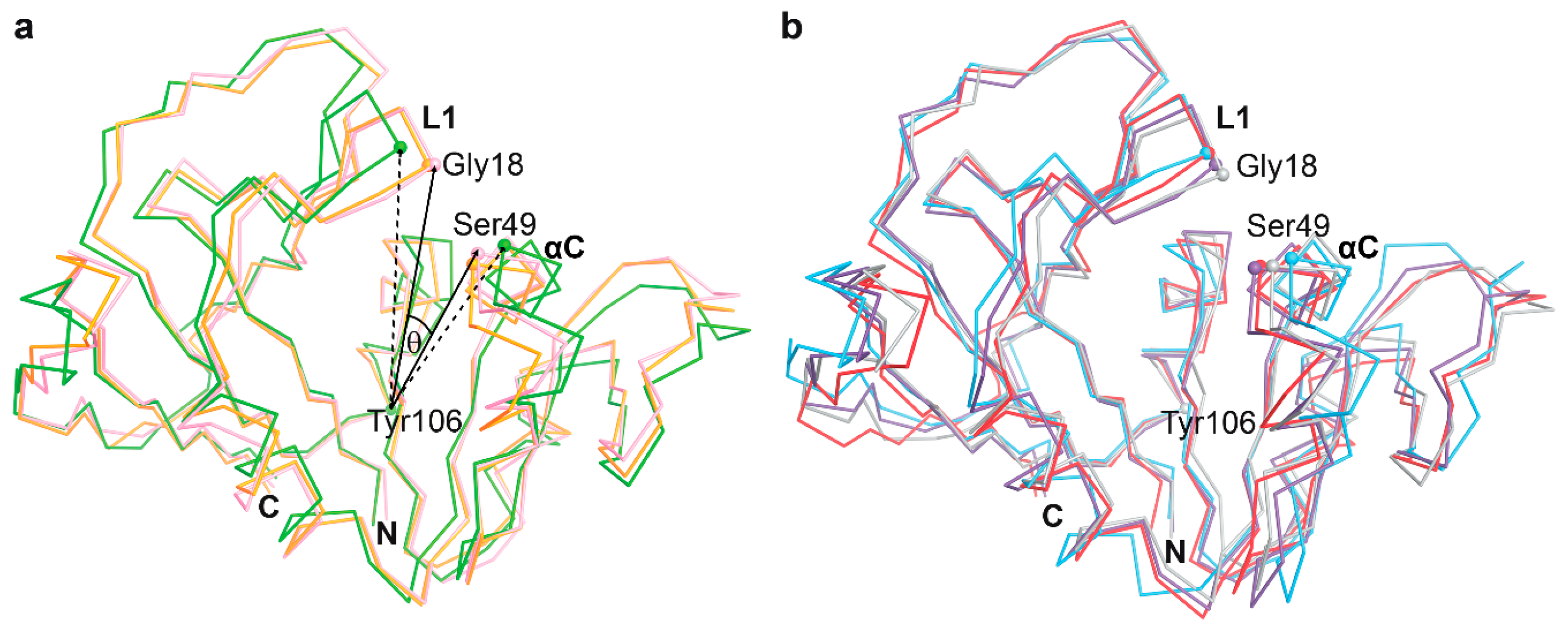
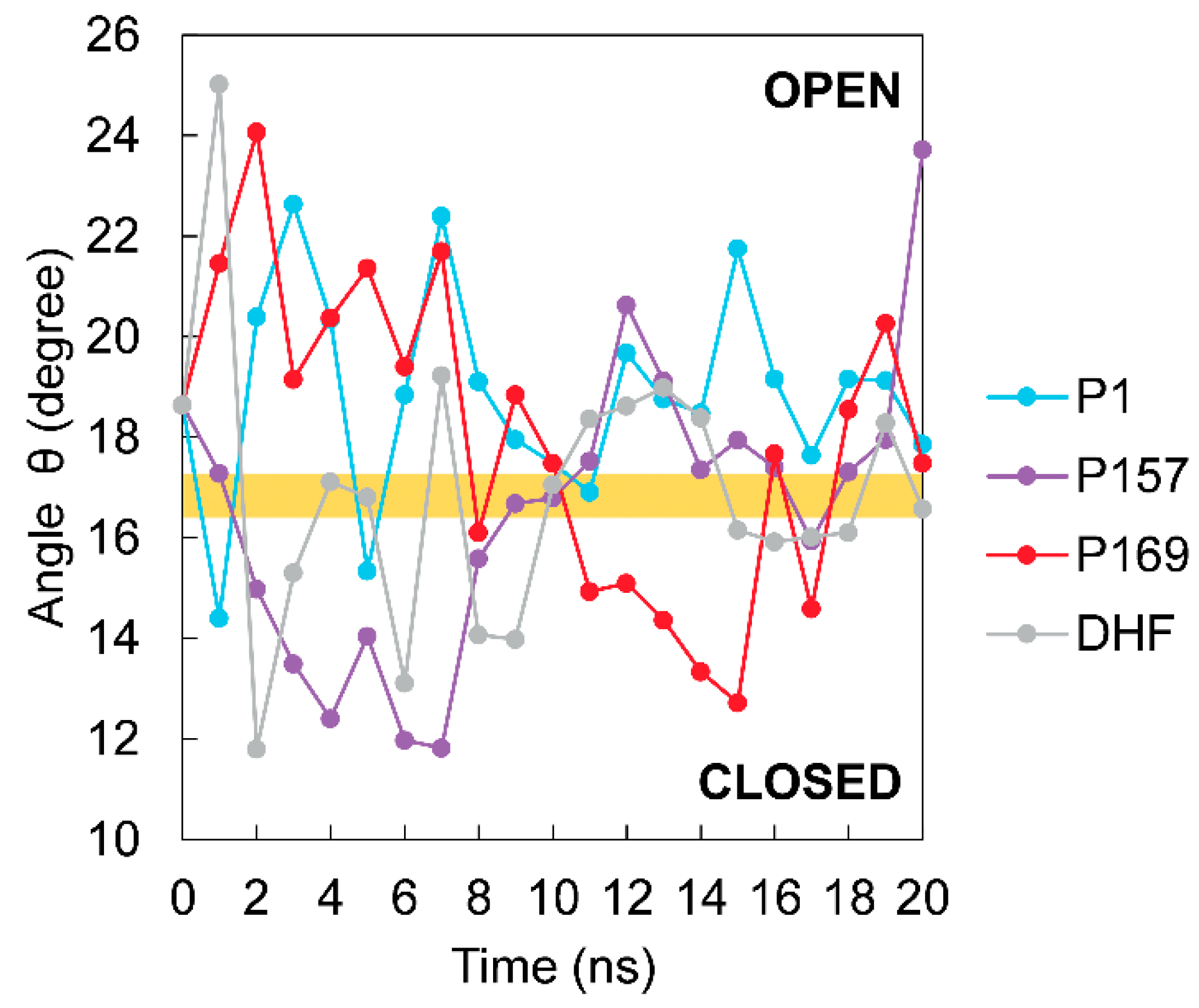
3.3. The Dynamics of Loops and Domains
3.4. The Binding of NADPH on Mycobacterium tuberculosis Dihydrofolate Reductase
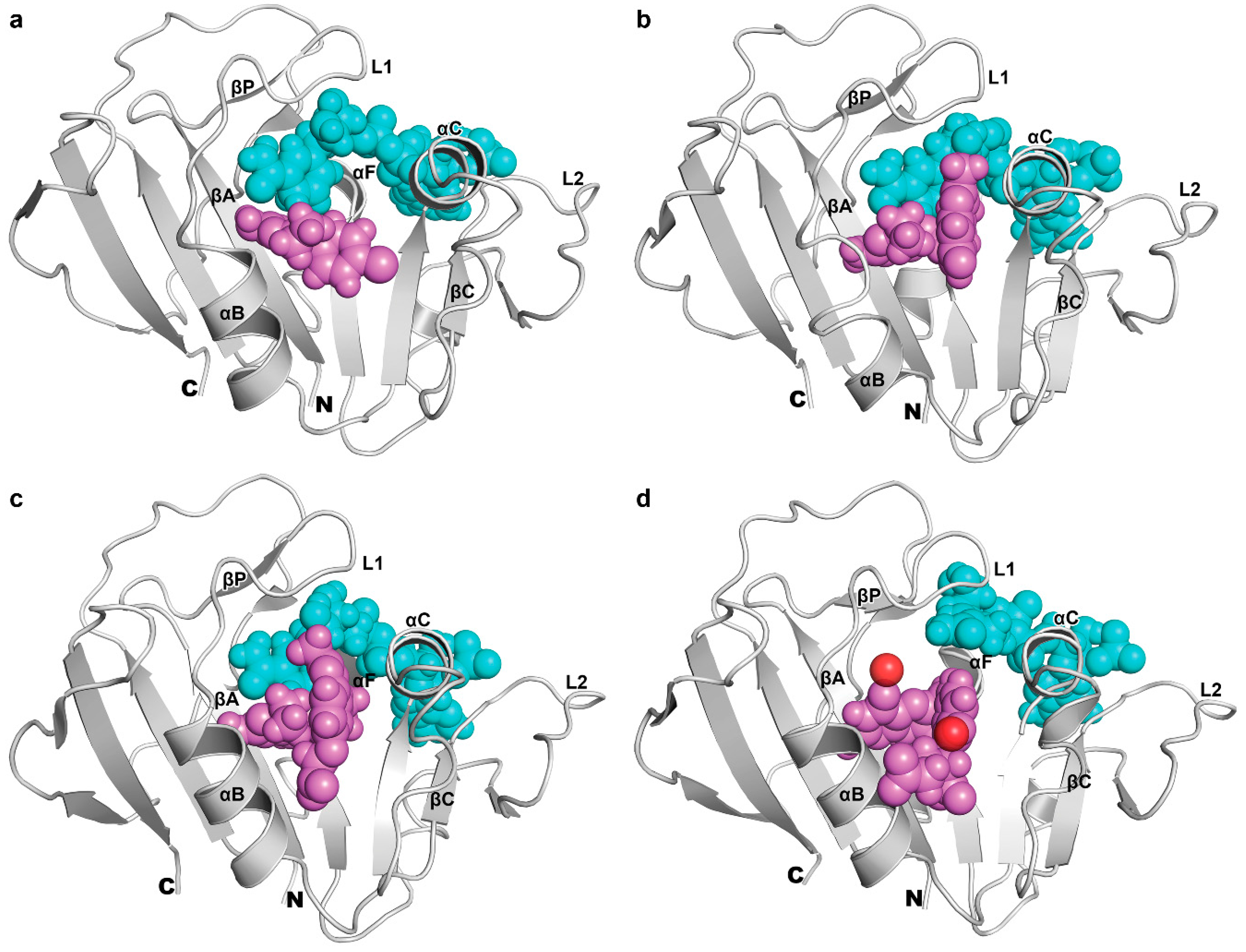
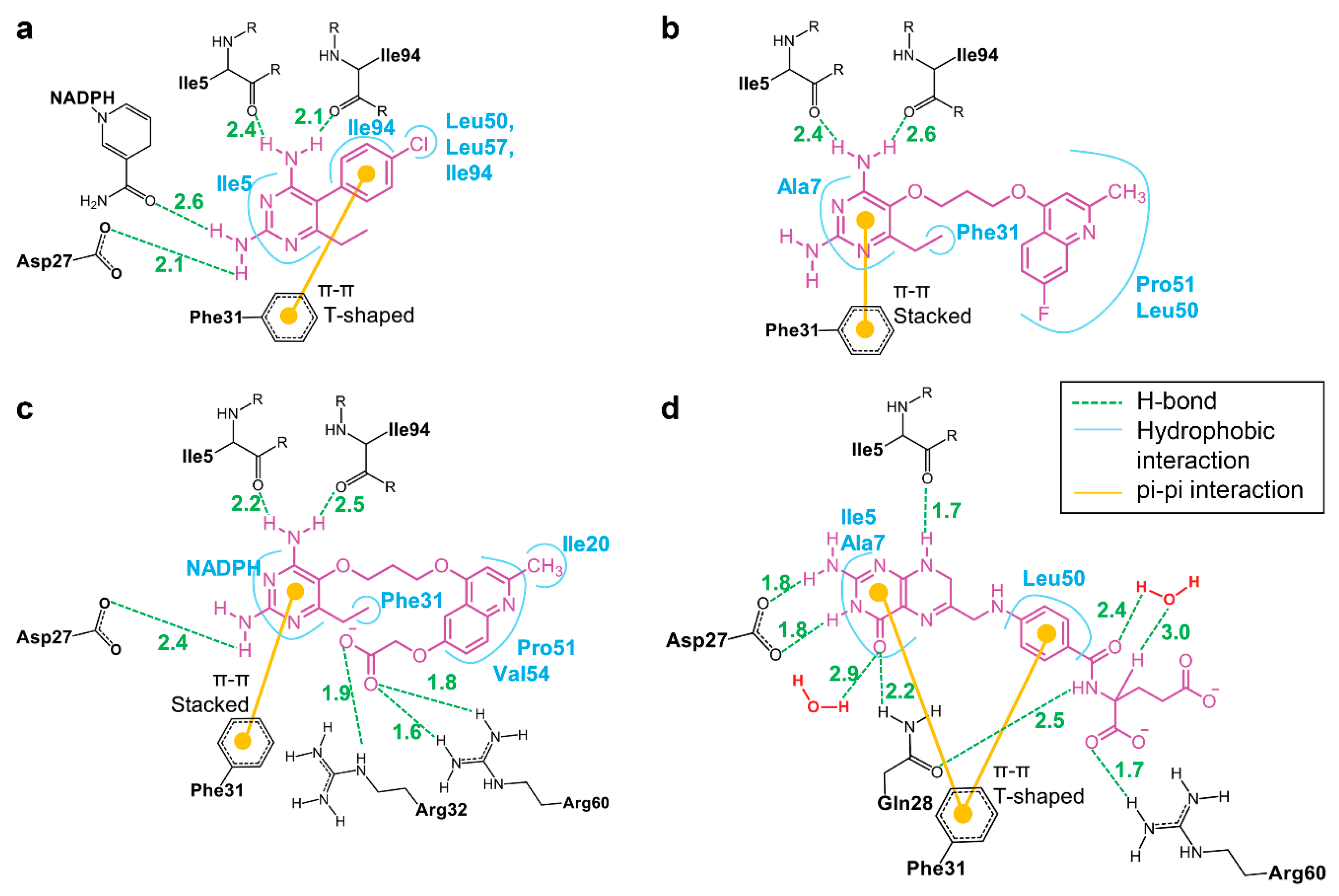
3.5. Binding of Substrate and Inhibitor to Mycobacterium tuberculosis Dihydrofolate Reductase
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization (WHO). Global Tuberculosis Report 2016; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- Anderson, A.C. Targeting DHFR in parasitic protozoa. Drug Discov. Today 2005, 10, 121–128. [Google Scholar] [CrossRef]
- Matthews, D.A.; Bolin, J.T.; Burridge, J.M.; Filman, D.J.; Volz, K.W.; Kaufman, B.T.; Beddell, C.R.; Champness, J.N.; Stammers, D.K.; Kraut, J. Refined crystal structures of Escherichia coli and chicken liver dihydrofolate reductase containing bound trimethoprim. J. Biol. Chem. 1985, 260, 381–391. [Google Scholar] [PubMed]
- Rajagopalan, P.T.R.; Zhang, Z.; McCourt, L.; Dwyer, M.; Benkovic, S.J.; Hammes, G.G. Interaction of dihydrofolate reductase with methotrexate: Ensemble and single-molecule kinetics. Proc. Natl. Acad. Sci. USA 2002, 99, 13481–13486. [Google Scholar] [CrossRef] [PubMed]
- Blakley, R.L. Dihydrofolate Reductase; Blakley, R.L., Benkovic, S.J., Eds.; Wiley: New York, NY, USA, 1984; Volume 1. [Google Scholar]
- Blakley, R.L. Eukaryotic dihydrofolate reductase. Adv. Enzymol. Relat. Subj. Biochem. 1995, 70, 23–102. [Google Scholar]
- Kuyper, L.F.; Garvey, J.M.; Baccanari, D.P.; Champness, J.N.; Stammers, D.K.; Beddell, C.R. Pyrrolo[2,3-d]pyrimidines and pyrido[2,3-d]pyrimidines as conformationally restricted analogues of the antibacterial agent trimethoprim. Biorg. Med. Chem. 1996, 4, 593–602. [Google Scholar] [CrossRef]
- Kuyper, L.F.; Baccanari, D.P.; Jones, M.L.; Hunter, R.N.; Tansik, R.L.; Joyner, S.S.; Boytos, C.M.; Rudolph, S.K.; Knick, V.; Wilson, H.R.; et al. High-affinity inhibitors of dihydrofolate reductase: Antimicrobial and anticancer activities of 7,8-dialkyl-1,3-diaminopyrrolo[3,2-f]quinazolines with small molecular size. J. Med. Chem. 1996, 39, 892–903. [Google Scholar] [CrossRef] [PubMed]
- Mdluli, K.; Spigelman, M. Novel targets for tuberculosis drug discovery. Curr. Opin. Pharm. 2006, 6, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, B.; Rodrigues, J.V.; Tonddast-Navaei, S.; Shakhnovich, E.; Skolnick, J. Rational design of novel allosteric dihydrofolate reductase inhibitors showing antibacterial effects on drug-resistant Eescherichia coli escape variants. ACS Chem. Biol. 2017, 12, 1848–1857. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Srinivasan, B.; Skolnick, J. Poli: A virtual screening pipeline based on template pocket and ligand similarity. J. Chem. Inf. Model. 2015, 55, 1757–1770. [Google Scholar] [CrossRef] [PubMed]
- Dias, M.V.; Tyrakis, P.; Domingues, R.R.; Paes Leme, A.F.; Blundell, T.L. Mycobacterium tuberculosis dihydrofolate reductase reveals two conformational states and a possible low affinity mechanism to antifolate drugs. Structure 2014, 22, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Sittikornpaiboon, P.; Toochinda, P.; Thongpanchang, C.; Leartsakulpanich, U.; Lawtrakul, L. Molecular docking study of Mycobacterium tuberculosis dihydrofolate reductase in complex with 2,4-diaminopyrimidines analogues. Chiang Mai J. Sci. 2015, 43, 931–945. [Google Scholar]
- Schnell, J.R.; Dyson, H.J.; Wright, P.E. Structure, dynamics, and catalytic function of dihydrofolate reductase. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 119–140. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, M.R.; Kraut, J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: Crystallographic evidence. Biochemistry 1997, 36, 586–603. [Google Scholar] [CrossRef] [PubMed]
- Cannon, W.R.; Garrison, B.J.; Benkovic, S.J. Electrostatic characterization of enzyme complexes: Evaluation of the mechanism of catalysis of dihydrofolate reductase. JACS 1997, 119, 2386–2395. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The mm/pbsa and mm/gbsa methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Accelrys. Discovery Studio Modeling Environment, 4.0; Accelrys Software Inc.: San Diego, CA, USA, 2013. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision a.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and autodocktools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Darden, T.A.; Cheatham, T.E.; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. AMBER12; University of California: San Francisco, CA, USA, 2012. [Google Scholar]
- Wang, J.; Cieplak, P.; Kollman, P.A. How well does a restrained electrostatic potential (resp) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general AMBER force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the Cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Li, R.; Sirawaraporn, R.; Chitnumsub, P.; Sirawaraporn, W.; Wooden, J.; Athappilly, F.; Turley, S.; Hol, W.G. Three-dimensional structure of M. tuberculosis dihydrofolate reductase reveals opportunities for the design of novel tuberculosis drugs. J. Mol. Biol. 2000, 295, 307–323. [Google Scholar] [CrossRef] [PubMed]
- Arora, K.; Brooks, C.L. Functionally important conformations of the met20 loop in dihydrofolate reductase are populated by rapid thermal fluctuations. JACS 2009, 131, 5642–5647. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, B.; Skolnick, J. Insights into the slow-onset tight-binding inhibition of Escherichia coli dihydrofolate reductase: Detailed mechanistic characterization of pyrrolo [3,2-f] quinazoline-1,3-diamine and its derivatives as novel tight-binding inhibitors. FEBS J. 2015, 282, 1922–1938. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, B.; Tonddast-Navaei, S.; Skolnick, J. Ligand binding studies, preliminary structure–activity relationship and detailed mechanistic characterization of 1-phenyl-6,6-dimethyl-1,3,5-triazine-2,4-diamine derivatives as inhibitors of Escherichia coli dihydrofolate reductase. Eur. J. Med. Chem. 2015, 103, 600–614. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Energy (kcal/mol) | P1 | P157 | P169 | DHF |
|---|---|---|---|---|
| ΔEvdw | −31.93 | −46.23 | −52.60 | −46.65 |
| ΔEele | −6.77 | −14.98 | −85.70 | −127.65 |
| ΔGGB | 19.83 | 32.78 | 86.52 | 128.08 |
| ΔGSA | −3.96 | −5.51 | −7.09 | −6.44 |
| ΔEMM | −38.71 | −61.21 | −138.31 | −174.30 |
| ΔGsolv | 15.87 | 27.27 | 79.44 | 121.63 |
| TΔS | −21.48 | −26.04 | −30.27 | −30.17 |
| ΔGbind | −1.36 | −7.90 | −28.60 | −22.50 |
| Complex | PDB ID | Conformation | θ Angel (Degrees) |
|---|---|---|---|
| mtbDHFR-NADPH | 4KLX | open | 18.34 |
| mtbDHFR-NADPH | 4KL9 | closed | 13.57 |
| mtbDHFR-NADPH | 1DG8 | closed | 15.07 |
| mtbDHFR-NADPH-Methotrexate | 1DF7 | closed | 15.81 |
| mtbDHFR-NADPH-Br-WR99210 | 1DG7 | closed | 14.72 |
| mtbDHFR-NADPH-Trimethoprim | 1DG5 | closed | 14.54 |
| mtbDHFR-NADPH-Trimethoprim | 4KM2 | open | 18.79 |
| mtbDHFR-NADPH-Cycloguanil | 4KNE | open | 17.88 |
| mtbDHFR-NADPH-Pyrimethamine | 4KM0 | open | 18.64 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sittikornpaiboon, P.; Toochinda, P.; Lawtrakul, L. Structural and Dynamics Perspectives on the Binding of Substrate and Inhibitors in Mycobacterium tuberculosis DHFR. Sci. Pharm. 2017, 85, 31. https://doi.org/10.3390/scipharm85030031
Sittikornpaiboon P, Toochinda P, Lawtrakul L. Structural and Dynamics Perspectives on the Binding of Substrate and Inhibitors in Mycobacterium tuberculosis DHFR. Scientia Pharmaceutica. 2017; 85(3):31. https://doi.org/10.3390/scipharm85030031
Chicago/Turabian StyleSittikornpaiboon, Pimonluck, Pisanu Toochinda, and Luckhana Lawtrakul. 2017. "Structural and Dynamics Perspectives on the Binding of Substrate and Inhibitors in Mycobacterium tuberculosis DHFR" Scientia Pharmaceutica 85, no. 3: 31. https://doi.org/10.3390/scipharm85030031