Synthesis, Crystal Structure, and Biological Activity of Ethyl 4-Methyl-2,2-dioxo-1H-2λ6,1-benzothiazine-3-carboxylate Polymorphic Forms

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
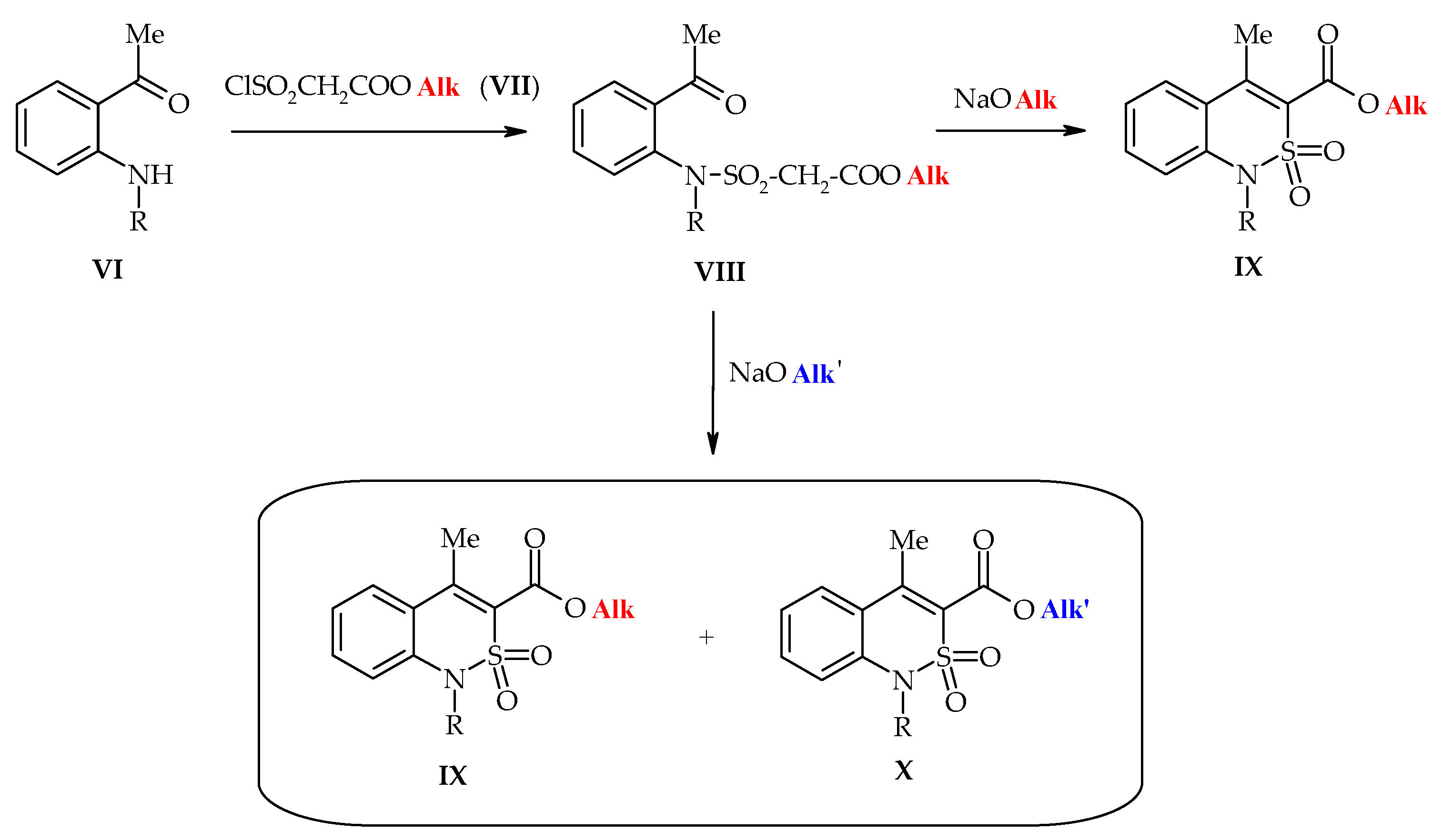
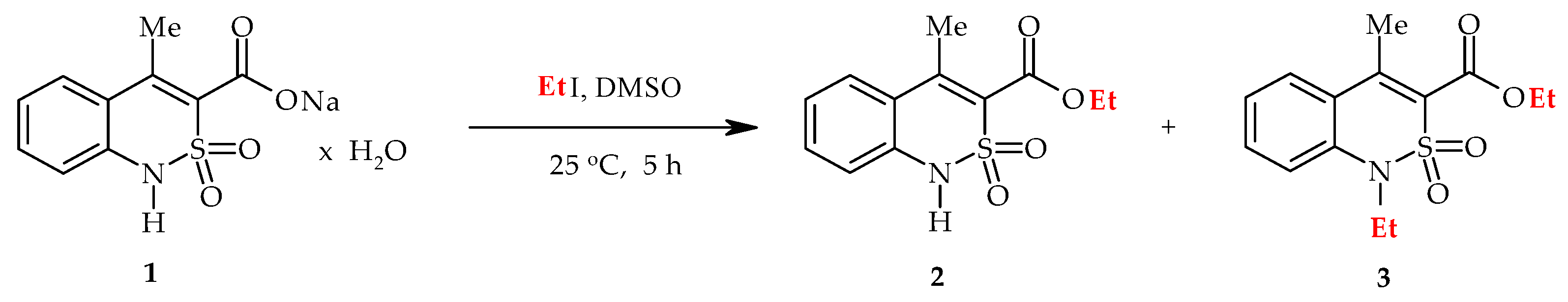
2.1. Chemistry
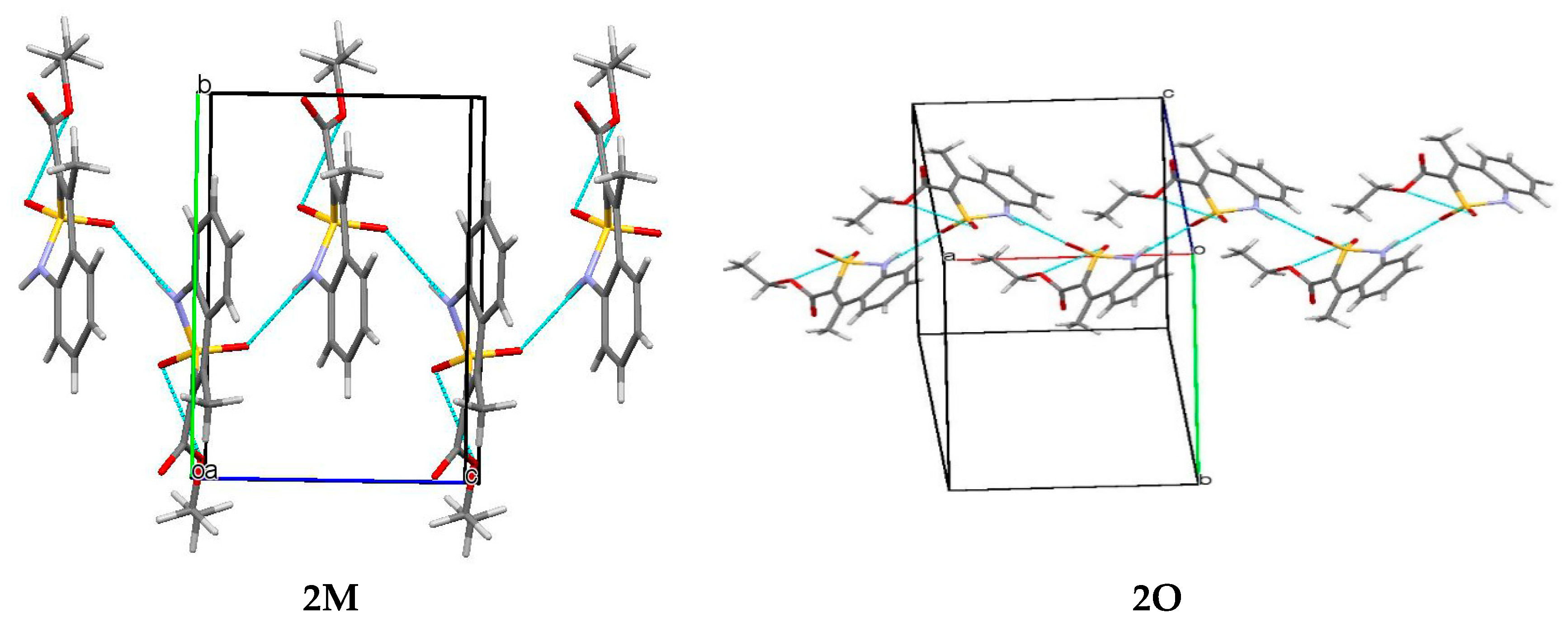
2.2. Monoclinic Form of Ethyl 4-Methyl-2,2-dioxo-1H-2λ6,1-benzothiazine-3-carboxylate (2M)
2.3. Orthorhombic Form of Ethyl 4-Methyl-2,2-dioxo-1H-2λ6,1-benzothiazine-3-carboxylate (2O)
2.4. Ethyl 1-Ethyl-4-methyl-2,2-dioxo-1H-2λ6,1-benzothiazine-3-carboxylate (3)
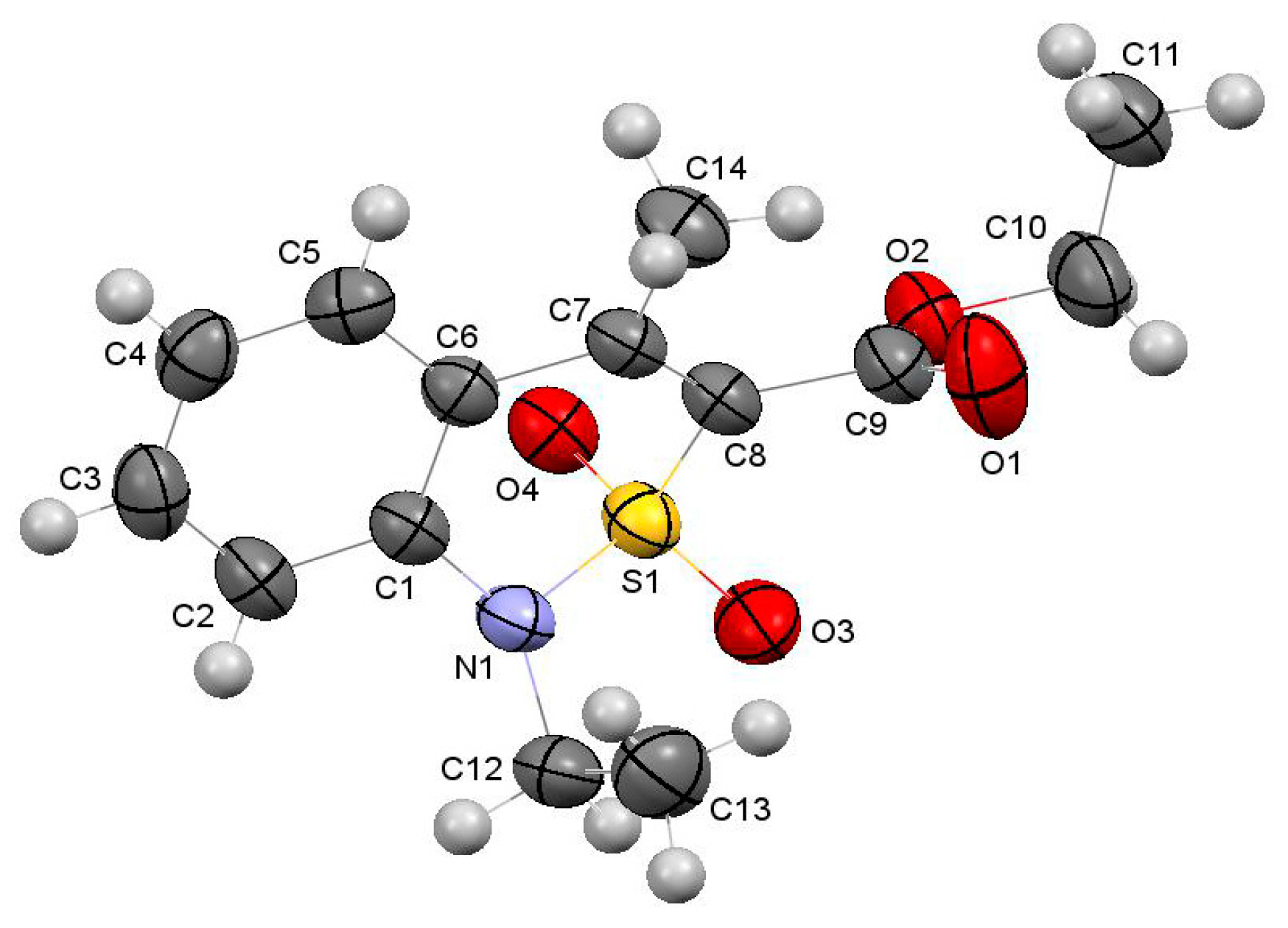
2.5. X-ray Structural Analysis of Ethyl 4-Methyl-2,2-dioxo-1H-2λ6,1-benzothiazine-3-carboxylate Monoclinic Form (2M)
2.6. X-ray Structural Analysis of Ethyl 4-Methyl-2,2-dioxo-1H-2λ6,1-benzothiazine-3-carboxylate Orthorhombic Form (2O)
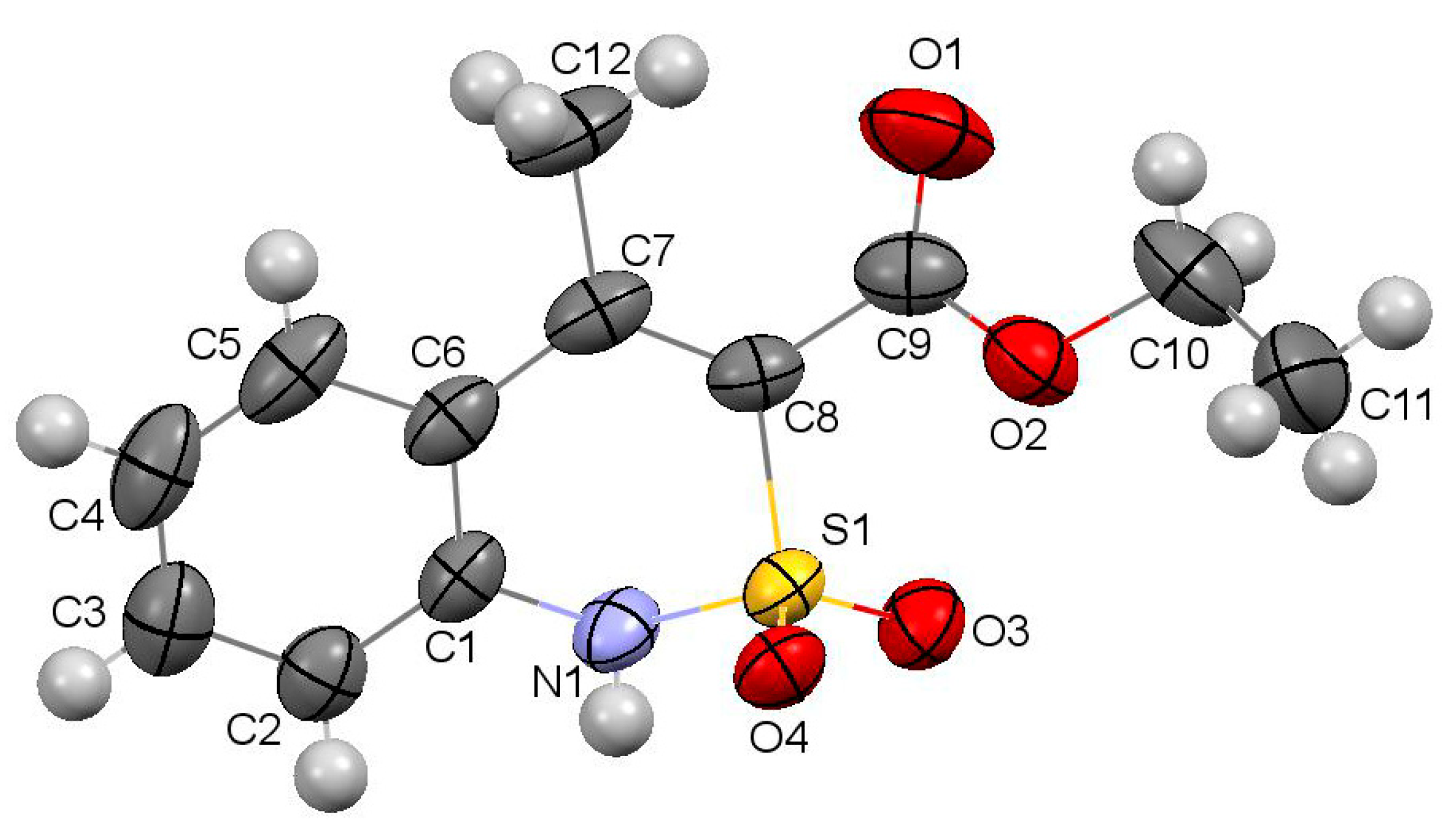
2.7. X-ray Structural Analysis of Ethyl 1-Ethyl-4-methyl-2,2-dioxo-1H-2λ6,1-benzothiazine-3-carboxylate (3)
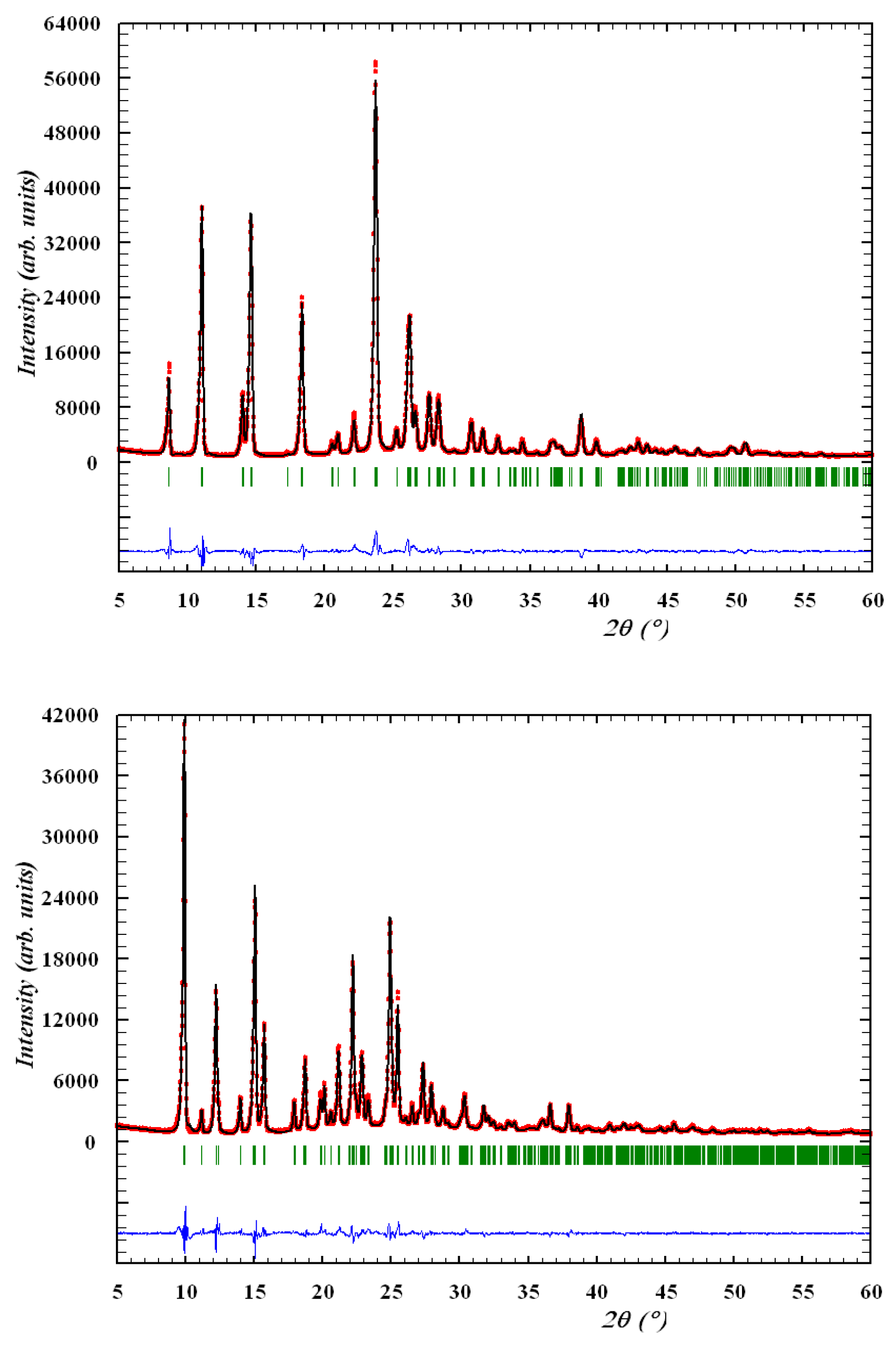
2.8. Powder Diffraction Study of Ethyl 4-Methyl-2,2-dioxo-1H-2λ6,1-benzothiazine-3-carboxylate Monoclinic and Orthorhombic Form (2M and 2O)
2.9. Pharmacology
Anti-Inflammatory and Analgesic Tests
2.10. Quantum-Chemical Calculations
3. Results and Discussion
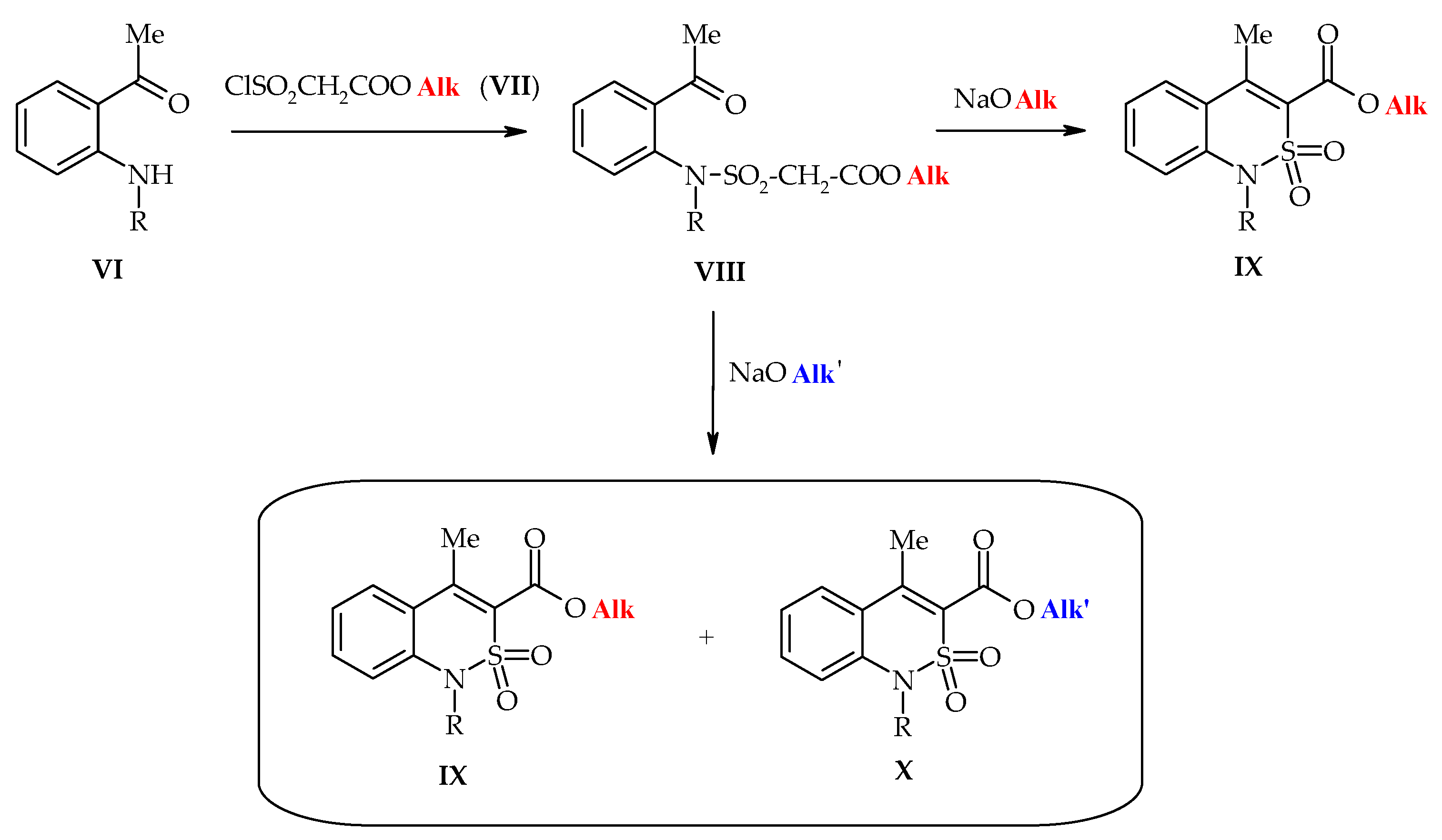
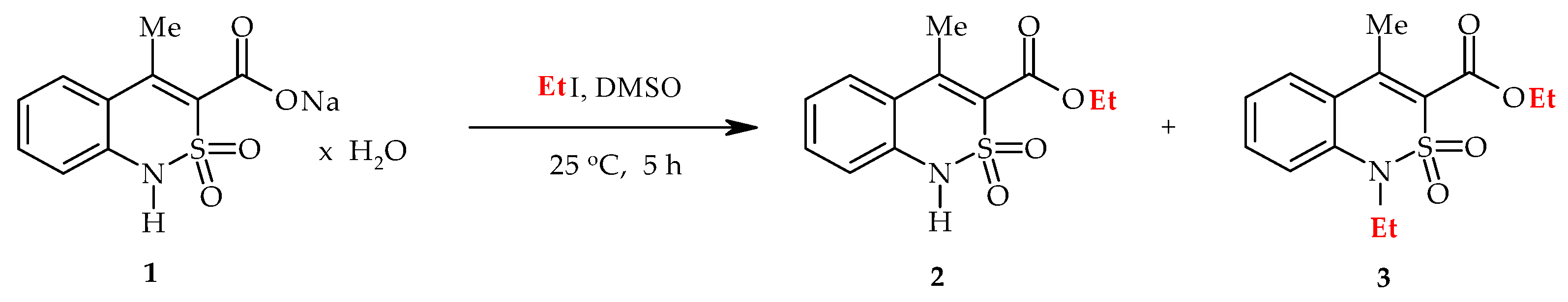
3.1. Chemistry
3.2. Evaluation of the Anti-Inflammatory and Analgesic Activity
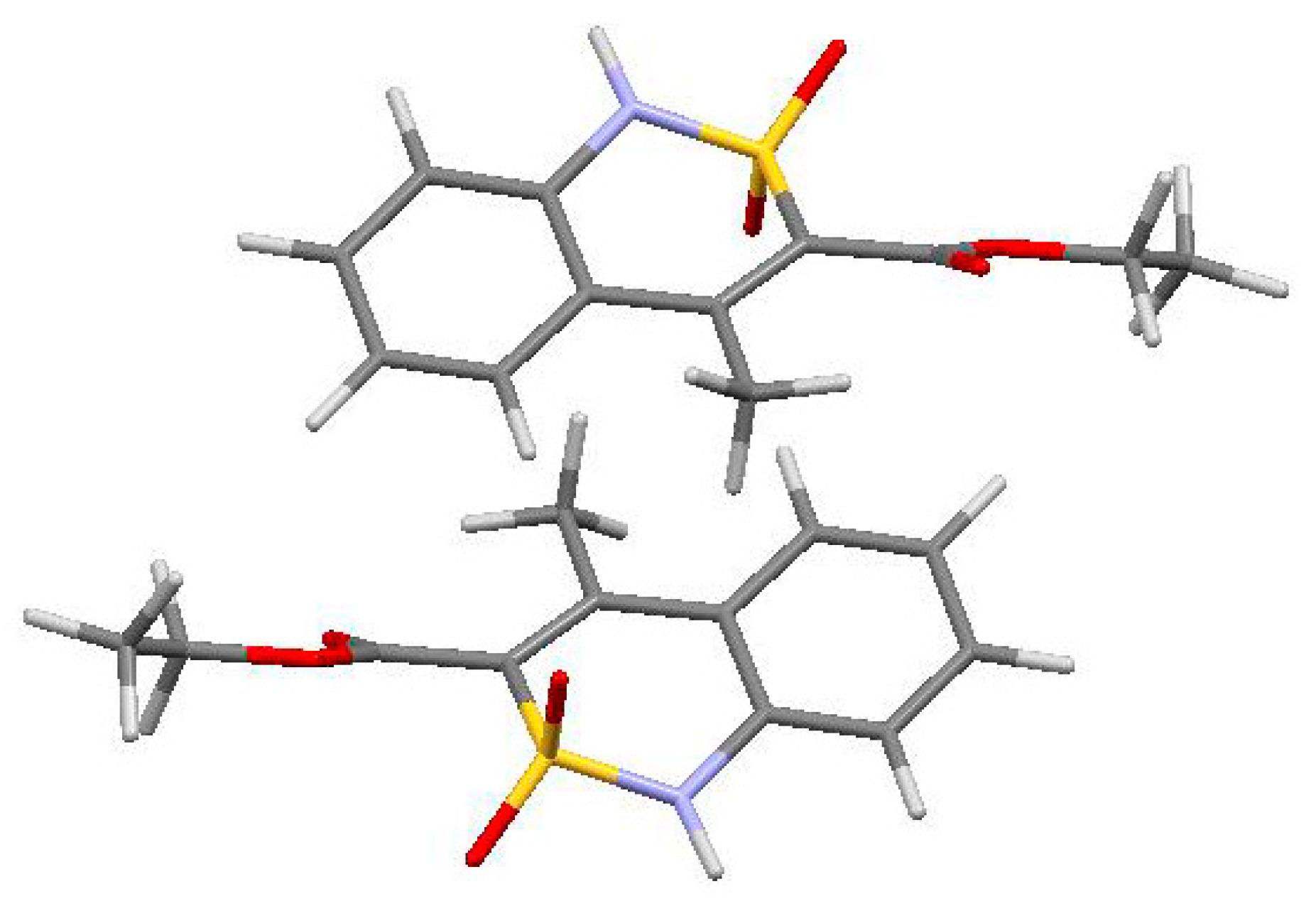
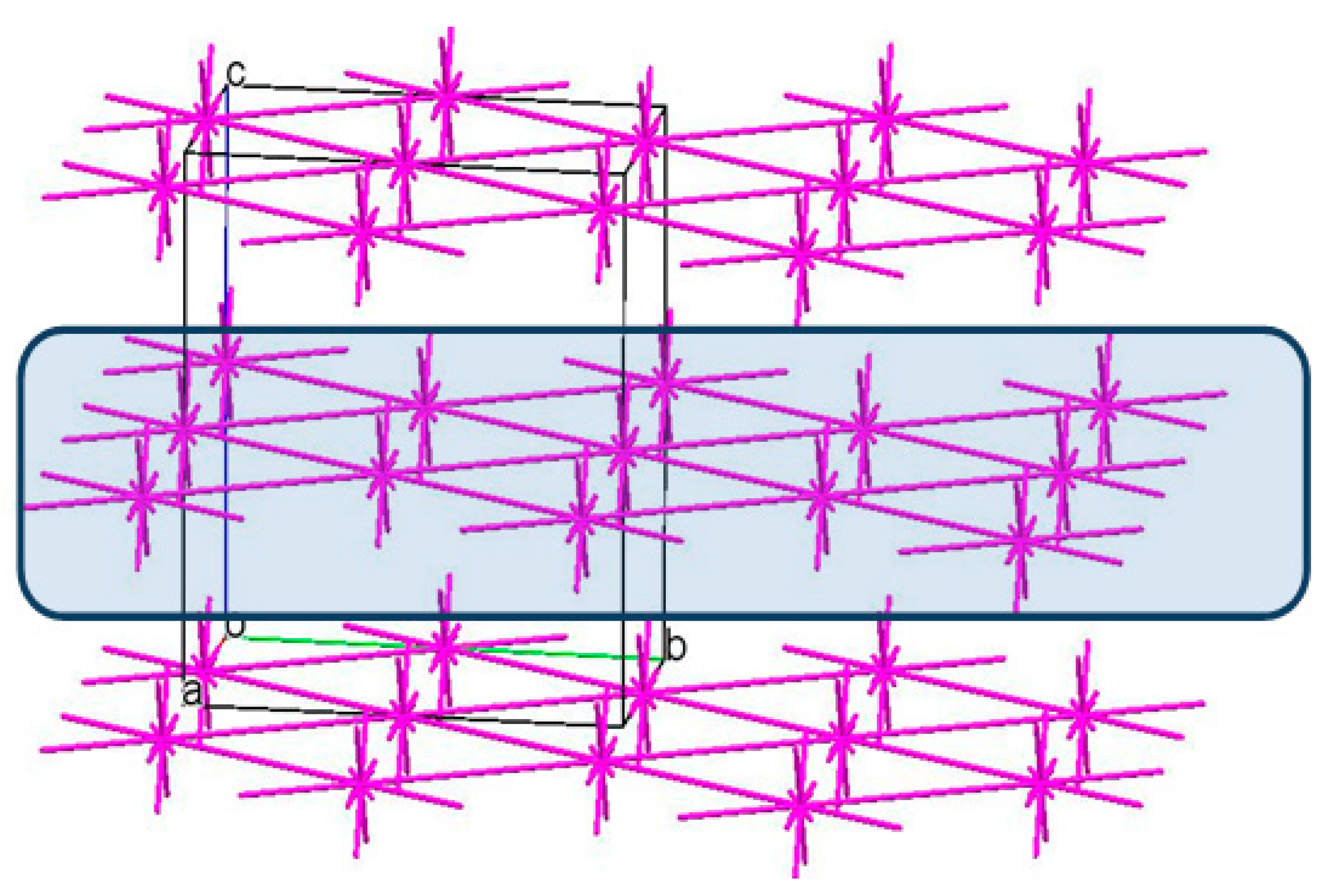
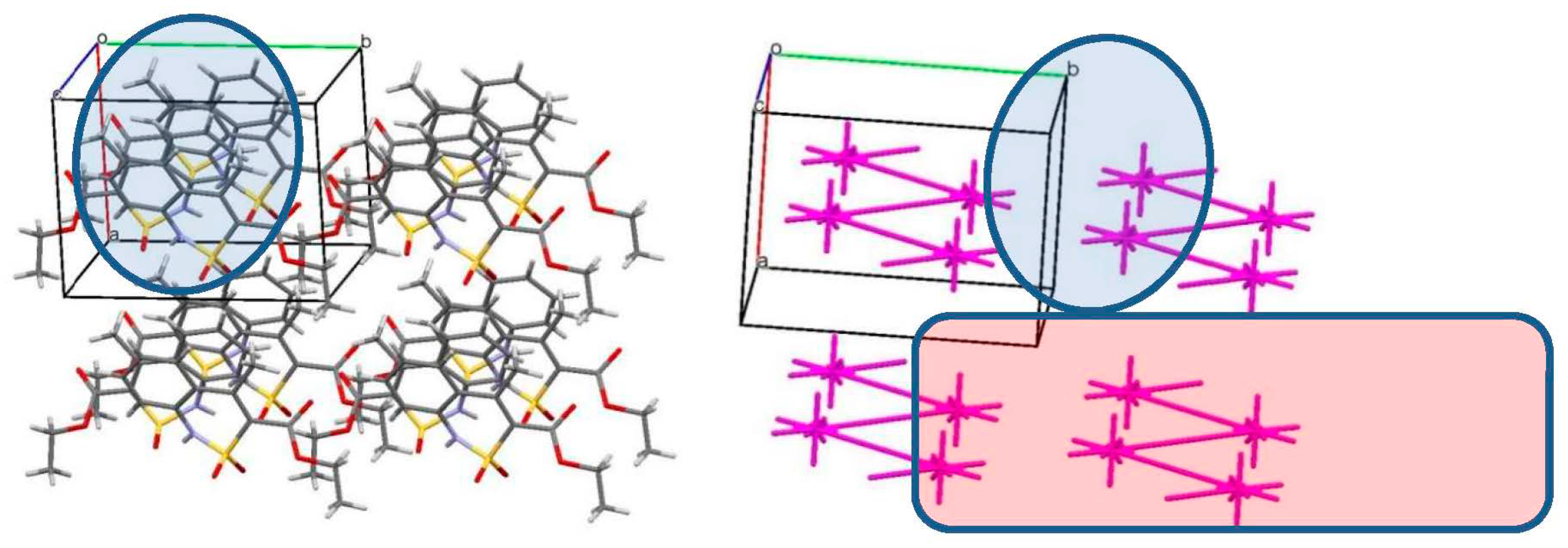
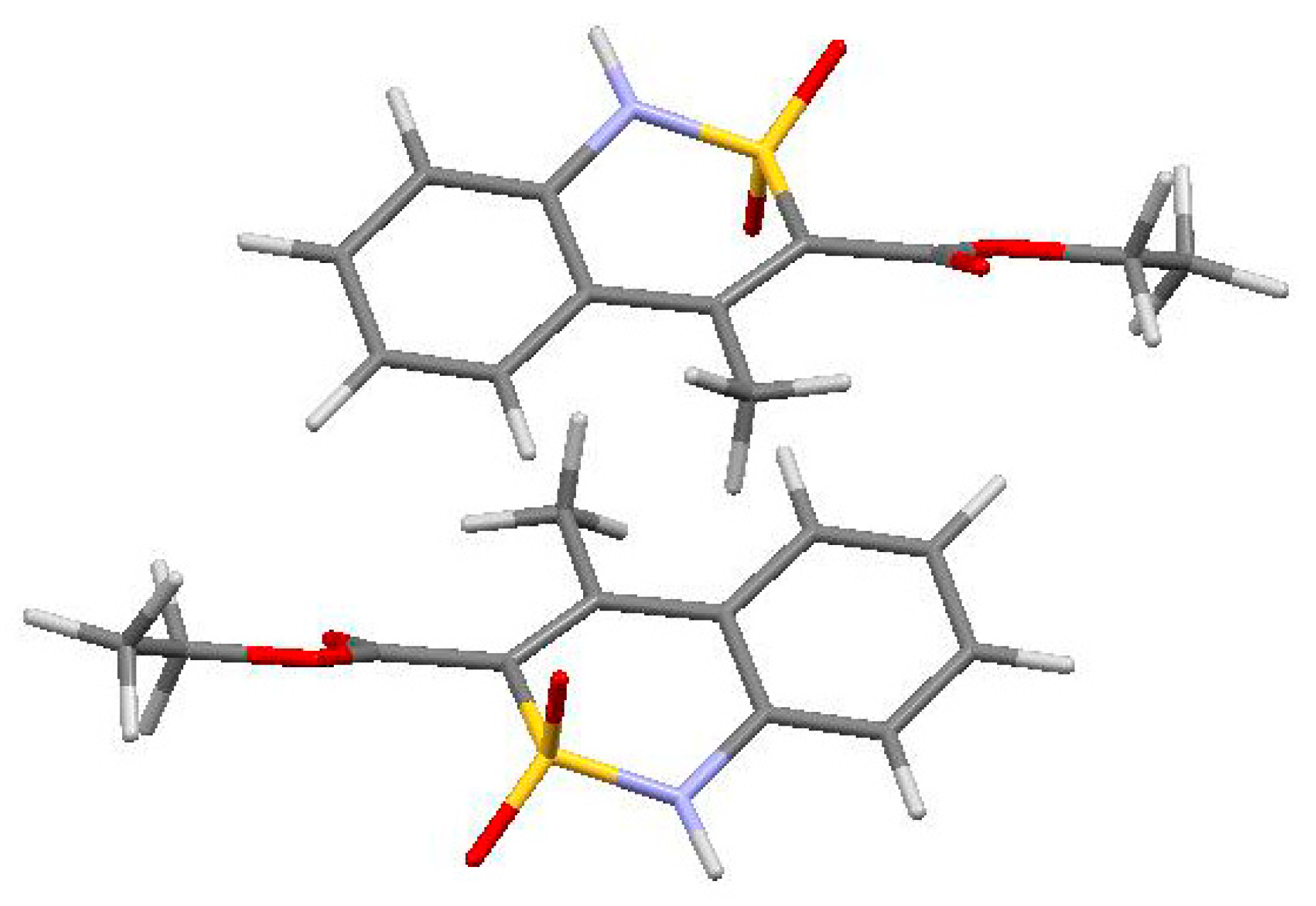
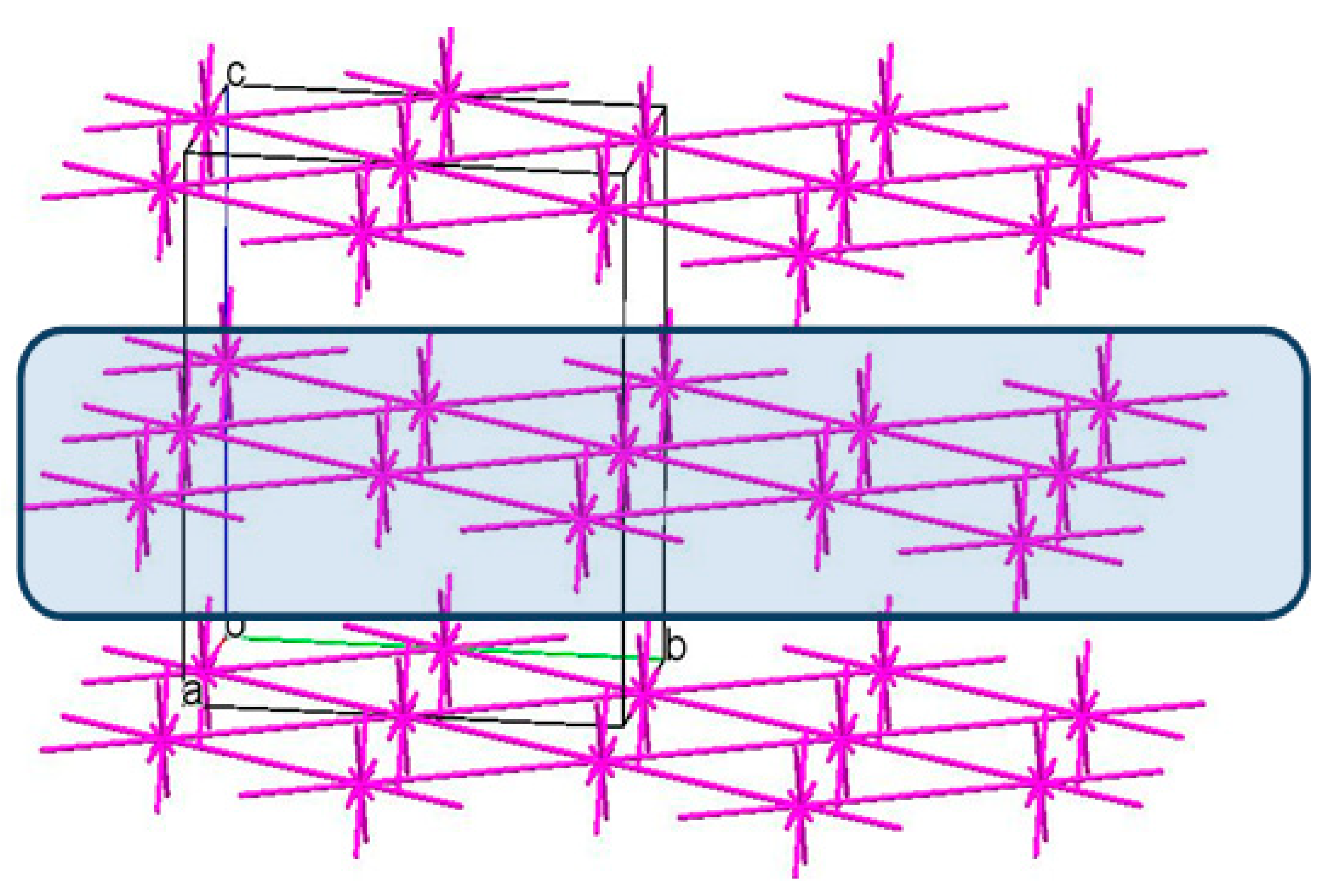
3.3. The Crystal Structure Study
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Kleemann, A.; Engel, J.; Kutscher, B.; Reichert, D. Pharmaceutical Substances: Syntheses, Patents, Applications of the Most Relevant APIs, 5th ed.; Thieme: Stuttgart, Germany, 2008. [Google Scholar]
- O’Neil, M.J.; Heckelman, P.E.; Koch, C.B.; Roman, K.J. The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals, 14th ed.; Merck and Co., Inc.: Whitehouse Station, NJ, USA, 2006. [Google Scholar]
- Kuznetsov, S.G.; Chigareva, S.M.; Ramsh, S.M. Prodrugs: Chemical aspect. Results Sci. Technol. VINITI Ser. Org. Chem. 1981, 19, 1–176. [Google Scholar]
- Fukami, T.; Yokoi, T. The emerging role of human esterases. Drug Metab. Pharmacokinet. 2012, 27, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Sakakura, A.; Kawajiri, K.; Ohkubo, T.; Kosugi, Y.; Ishihara, K. Widely useful DMAP-catalyzed esterification under auxiliary base- and solvent-free conditions. J. Am. Chem. Soc. 2007, 129, 14775–14779. [Google Scholar] [CrossRef] [PubMed]
- Pittelkow, M.; Kamounah, F.S.; Boas, U.; Pedersen, B.; Christensen, J.B. TFFH as an excellent reagent for acylation of alcohols, thiols and dithiocarbamates. Synthesis 2004, 2485–2492. [Google Scholar] [CrossRef]
- Dhimitruka, I.; Santa Lucia, J. Investigation of the Yamaguchi esterification mechanism. Synthesis of a Lux-S enzyme inhibitor using an improved esterification method. Org. Lett. 2006, 8, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, G.; Bosco, M.; Carlone, A.; Dalpozzo, R.; Marcantoni, E.; Melchiorre, P.; Sambri, L. Reaction of dicarbonates with carboxylic acids catalyzed by weak Lewis acids: General method for the synthesis of anhydrides and esters. Synthesis 2007, 22, 3489–3496. [Google Scholar] [CrossRef]
- Dev, D.; Palakurthy, N.B.; Thalluri, K.; Chandra, J.; Mandal, B. Ethyl 2-cyano-2-(2-nitrobenzene-sulfonyloxyimino)acetate (o-NosylOXY): A recyclable coupling reagent for racemization-free synthesis of peptide, amide, hydroxamate, and ester. J. Org. Chem. 2014, 79, 5420–5431. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wen, Y.; Fu, Y.; Chen, H.; Ye, M.; Luo, G. Graphene oxide: An efficient acid catalyst for the construction of esters from acids and alcohols. Synlett 2017, 28, 981–985. [Google Scholar] [CrossRef]
- Minakawa, M.; Baek, H.; Yamada, Y.M.A.; Han, J.W.; Uozumi, Y. Direct dehydrative esterification of alcohols and carboxylic acids with a macroporous polymeric acid catalyst. Org. Lett. 2013, 15, 5798–5801. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-H.; Han, J.; Jung, B.Y.; Baek, H.; Yamada, Y.M.A.; Uozumi, Y.; Lee, Y.-S. Production of valuable esters from oleic acid with a porous polymeric acid catalyst without water removal. Synlett 2016, 27, 29–32. [Google Scholar] [CrossRef]
- Jaita, S.; Phakhodee, W.; Pattarawarapan, M. Ultrasound-assisted methyl esterification of carboxylic acids catalyzed by polymer-supported triphenylphosphine. Synlett 2015, 26, 2006–2008. [Google Scholar] [CrossRef]
- Huang, Z.; Reilly, J.R.; Buckle, R.N. An efficient synthesis of amides and esters via triacyloxyboranes. Synlett 2007, 1026–1030. [Google Scholar] [CrossRef]
- Chakraborti, A.K.; Singh, B.; Chankeshwara, S.V.; Patel, A.R. Protic acid immobilized on solid support as an extremely efficient recyclable catalyst system for a direct and atom economical esterification of carboxylic acids with alcohols. J. Org. Chem. 2009, 74, 5967–5974. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, K.V.N.S.; Mahender, I.; Das, B. Silica Chloride: A versatile heterogeneous catalyst for esterification and transesterification. Synthesis 2003, 2479–2482. [Google Scholar] [CrossRef]
- Wang, Y.; Aleiwi, B.A.; Wang, Q.; Kurosu, M. Selective esterifications of primary alcohols in a water-containing solvent. Org. Lett. 2012, 14, 4910–4913. [Google Scholar] [CrossRef] [PubMed]
- Twibanire, J.K.; Grindley, T.B. Efficient and controllably selective preparation of esters using uronium-based coupling agents. Org. Lett. 2011, 13, 2988–2991. [Google Scholar] [CrossRef] [PubMed]
- Heller, S.T.; Sarpong, R. Chemoselective esterification and amidation of carboxylic acids with imidazole carbamates and ureas. Org. Lett. 2010, 12, 4572–4575. [Google Scholar] [CrossRef] [PubMed]
- Velusamy, S.; Borpuzari, S.; Punniyamurthy, T. Cobalt(II)-catalyzed direct acetylation of alcohols with acetic acid. Tetrahedron 2005, 61, 2011–2015. [Google Scholar] [CrossRef]
- Sharghi, H.; Hosseini Sarvari, M. Al2O3/MeSO3H (AMA) as a new reagent with high selective ability for monoesterification of diols. Tetrahedron 2003, 59, 3627–3633. [Google Scholar] [CrossRef]
- Hong, X.; Harmata, M. Recent Progress in the Chemistry of 2,1-Benzothiazines. In Progress in Heterocyclic Chemistry; Gribble, G.W., Joule, J.A., Eds.; Elsevier Science Ltd.: Oxford, UK, 2008; Volume 19, pp. 1–43. [Google Scholar]
- Azotla-Cruz, L.; Lijanova, I.V.; Ukrainets, I.V.; Likhanova, N.V.; Olivares-Xometl, O.; Bereznyakova, N.L. New synthesis, structure and analgesic properties of methyl 1-R-4-methyl-2,2-dioxo-1H-2λ6,1- benzothiazine-3-carboxylates. Sci. Pharm. 2017, 85, 2. [Google Scholar] [CrossRef] [PubMed]
- Ukrainets, I.V.; Petrushova, L.A.; Dzyubenko, S.P. 2,1-Benzothiazine 2,2-dioxides. 1. Synthesis, structure, and analgesic activity of 1-R-4-hydroxy-2,2-dioxo-1H-2λ6,1-benzothiazine-3-carboxylic acid esters. Chem. Heterocycl. Compd. 2013, 49, 1378–1383. [Google Scholar] [CrossRef]
- Ukrainets, I.V.; Petrushova, L.A.; Sim, G.; Grinevich, L.A.S.P. Synthesis and molecular structure of ethyl-4-hydroxy-1-phenyl-2,2-dioxo-1H-2λ6,1-benzothiazine-3-carboxylate. Pharm. Chem. J. 2017, 51, 482–485. [Google Scholar] [CrossRef]
- Ukrainets, I.V.; Hamza, G.M.; Burian, A.A.; Shishkina, S.V.; Voloshchuk, N.I.; Malchenko, O.V. 4-Methyl-2,2-dioxo-1H-2λ6,1-benzothiazine-3-carboxylic acid. Peculiarities of preparation, structure, and biological properties. Sci. Pharm. 2018, 86, 9. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Cambridge Crystallographic Data Center. Request Quoting via: CCDC1835802. Available online: www.ccdc.cam.ac.uk/conts/retrieving.html (accessed on 9 April 2018).
- Cambridge Crystallographic Data Center. Request Quoting via: CCDC1835803. Available online: www.ccdc.cam.ac.uk/conts/retrieving.html (accessed on 9 April 2018).
- Cambridge Crystallographic Data Center. Request Quoting via: CCDC1835804. Available online: www.ccdc.cam.ac.uk/conts/retrieving.html (accessed on 9 April 2018).
- Rodriguez-Carvajal, J. FullProf; Version 4.80; ILL: Grenoble, France, 2010. [Google Scholar]
- Rodriguez-Carvajal, J.; Roisnel, T. WinPLOTR; Trans Tech Publications: Zurich, Switzerland, 2004; pp. 123–126. [Google Scholar]
- Rodriguez-Carvajal, J.; Roisnel, T. FullProf.98 and WinPLOTR New Windows 99/NT Applications for Diffraction, Commission for Powder Diffraction; Newsletter; International Union of Crystallography: Chester, UK, 1998; Volume 20, pp. 35–36. [Google Scholar]
- Ukrainian Law No. 3447-IV. On Protection of Animals from Severe Treatment. Available online: http://zakon2.rada.gov.ua/laws/show/3447-15 (accessed on 4 August 2017).
- Vogel, H.G. Drug Discovery and Evaluation: Pharmacological Assays, 2nd ed.; Springer: Berlin, Germany, 2008; pp. 1103–1106. [Google Scholar]
- Gregory, N.S.; Harris, A.L.; Robinson, C.R.; Dougherty, P.M.; Fuchs, P.N.; Sluka, K.A. An overview of animal models of pain: Disease models and outcome measures. J. Pain. 2013, 14, 1255–1269. [Google Scholar] [CrossRef] [PubMed]
- Shishkin, O.V.; Dyakonenko, V.V.; Maleev, A.V. Supramolecular architecture of crystals of fused hydrocarbons based on topology of intermolecular interactions. CrystEngComm 2012, 14, 1795–1804. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriquez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Coppens, P. The use of a polarized hydrogen atom in X-ray structure refinement. Acta Cryst. B 1972, 28, 1638–1640. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Zefirov, N.S.; Palyulin, V.A.; Dashevskaya, E.E. Stereochemical studies. XXXIV. Quantitative description of ring puckering via torsional angles. The case of six-membered rings. J. Phys. Org. Chem. 1990, 3, 147–158. [Google Scholar] [CrossRef]
- Orpen, A.G.; Brammer, L.; Allen, F.H.; Kennard, O.; Watson, D.G.; Taylor, R. Typical interatomic distances in organic compounds and organometallic compounds and coordination complexes of the d- and f-block metals. In Structure Correlation; Burgi, H.-B., Dunitz, J.D., Eds.; Wiley-VCH: Weinheim, Germany, 1994. [Google Scholar]
- Zefirov, Y.V. Reduced intermolecular contacts and specific interactions in molecular crystals. Crystallogr. Rep. 1997, 42, 865–886. [Google Scholar]
- Bernstein, J. Polymorphism in Molecular Crystals; Oxford University Press: New York, NY, USA, 2002. [Google Scholar]
- Ukrainets, I.V.; Petrushova, L.A.; Shishkina, S.V.; Grinevich, L.A.; Sim, G. Synthesis, spatial structure and analgesic activity of sodium 3-benzylaminocarbonyl-1-methyl-2,2-dioxo-1H-2λ6,1-benzothiazin-4-olate solvates. Sci. Pharm. 2016, 84, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Shishkin, O.V.; Zubatyuk, R.I.; Shishkina, S.V.; Dyakonenko, V.V.; Medviediev, V.V. Role of supramolecular synthons in the formation of the supramolecular architecture of molecular crystals revisited from an energetic viewpoint. PhysChemChemPhys 2014, 16, 6773–6786. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Parameter | Monoclinic (2M) | Orthorhombic (2O) |
|---|---|---|---|
| 1 | S | 0.58 | 0.63 |
| 2 | Θ, deg | 49.8 | 54.2 |
| 3 | Ψ, deg | 37.5 | 31.4 |
| 4 | S(1), Å | 0.78 | 0.89 |
| 5 | C(8), Å | 0.22 | 0.33 |
| 6 | C(7)–C(8), Å | 1.361(4) | 1.362(4) |
| 7 | N(1)–C(1), Å | 1.412(4) | 1.398(3) |
| 8 | C(7)–C(8)–C(9)–O(1), deg | −36.1(5) | −37.8(5) |
| 9 | C(8)–C(9)–O(2)–C(10), deg | 177.9(3) | −179.8(2) |
| 10 | C(9)–O(2)–C(10)–C(11), deg | −160.4(4) | 173.3(3) |
| 11 | C(12)…O(1), Å | 2.94 | 2.96 |

| Entry | Product | R | Volume of Damaged Extremity (mm3) | Volume of Non-Damaged Extremity (mm3) | Δ Volume (Volume Increase) | Anti-Inflammatory Activity, Compared to Control (%) |
|---|---|---|---|---|---|---|
| 1 | 2M | H | 377.1 ± 29.9 | 318.3 ± 20.4 | 58.8 ± 5.8 1,2,3 | 85.8 |
| 2 | 2O | H | 736.2 ± 23.3 | 449.5 ± 23.9 | 286.7 ± 8.6 1,2,3 | 30.7 |
| 3 | 3 | Et | 743.5 ± 32.0 | 455.2 ± 21.2 | 288.2 ± 8.0 1,2,3 | 30.3 |
| 4 | Piroxicam | − | 566.7 ± 160.4 | 342.8 ± 19.8 | 223.8 ± 6.1 1 | 45.9 |
| 5 | Meloxicam | − | 492.6 ± 39.3 | 361.2 ± 26.1 | 131.3 ± 8.0 1 | 68.3 |
| 6 | Control | − | 768.7 ± 27.3 | 354.9 ± 11.6 | 413.7 ± 32.2 | 0 |
| Entry | Product | R | Pain Threshold on Damaged Extremity (g/mm2) | Pain Threshold on Non-Damaged Extremity (g/mm2) | Δ Pain Threshold | Analgesic Activity, Compared to Control (%) |
|---|---|---|---|---|---|---|
| 1 | 2M | H | 370.0 ± 15.2 | 340.0 ± 17.0 | 30.0 ± 4.5 1,2,3 | 90.6 |
| 2 | 2O | H | 344.0 ± 28.8 | 236.0 ± 11.4 | 108.0 ± 8.6 1,2,3 | 66.0 |
| 3 | 3 | Et | 380.0 ± 10.5 | 220.0 ± 13.4 | 160.0 ± 10.5 1,2,3 | 49.7 |
| 4 | Piroxicam | − | 504.0 ± 18.1 | 340.0 ± 15.2 | 164.0 ± 8.1 1 | 48.4 |
| 5 | Meloxicam | − | 414.0 ± 19.6 | 326.0 ± 26.4 | 88.0 ± 11.6 1 | 72.3 |
| 6 | Control | − | 593.0 ± 56.3 | 275.0 ± 32.1 | 318.0 ± 34.9 | 0 |
| Dimer | Symmetry Operation | Eint (kcal · mol−1) | Contribution to the Total Interaction Energy (%) | Interaction Type |
|---|---|---|---|---|
| Crystals of 2M | ||||
| 2M_1 | x, 1 − y, 0.5 + z | −17.95 | 22.98 | N–H…O, stacking |
| 2M_2 | x, 1 − y, −0.5 + z | −17.95 | 22.98 | N–H…O, stacking |
| 2M_3 | x, 2 − y, 0.5 + z | −8.78 | 11.24 | non-specific |
| 2M_4 | x, 2 − y, −0.5 + z | −8.78 | 11.24 | non-specific |
| 2M_5 | 1 + x, y, z | −5.86 | 7.50 | non-specific |
| 2M_6 | −1 + x, y, z | −5.86 | 7.50 | non-specific |
| Crystals of 2O, building unit is a molecule | ||||
| 2O_1 | 1 − x, 1 − y, 1 − z | −16.56 | 22.41 | C–H…O, C–H…π |
| 2O_2 | 0.5 + x, 0.5 − y, 1 − z | −11.51 | 15.58 | N–H…O |
| 2O_3 | −0.5 + x, 0.5 − y, 1 − z | −11.51 | 15.58 | N–H…O |
| 2O_4 | −x, 1 − y, 1 − z | −7.98 | 10.81 | stacking |
| 2O_5 | 0.5 + x, y, 0.5 − z | −5.03 | 6.81 | C–H…π |
| 2O_6 | −0.5 + x, y, 0.5 − z | −5.03 | 6.81 | C–H…π |
| Crystals of 2O, building unit is a dimer | ||||
| 2O_d_1 | 1/2 + x, 1/2 − y, 1 − z | −16.11 | 13.25 | N–H…O |
| 2O_d_2 | 1/2 + x, 3/2 − y, 1 − z | −16.11 | 13.25 | N–H…O |
| 2O_d_3 | −1/2 + x, 1/2 − y, 1 − z | −16.11 | 13.25 | N–H…O |
| 2O_d_4 | −1/2 + x, 3/2 − y, 1 − z | −16.11 | 13.25 | N–H…O |
| 2O_d_5 | 1 + x, y, z | −10.47 | 8.61 | stacking |
| 2O_d_6 | −1 + x, y, z | −10.47 | 8.61 | stacking |
| 2O_d_7 | 0.5 − x, 1 − y, 0.5 + z | −7.52 | 6.18 | C–H…O, C–H…π |
| 2O_d_8 | 1/2 − x, 1 − y, −1/2 + z | −7.52 | 6.18 | C–H…O, C–H…π |
| 2O_d_9 | 3/2 − x, 1 − y, 1/2 + z | −7.52 | 6.18 | C–H…O, C–H…π |
| 2O_d_10 | 3/2 − x, 1 − y, −1/2 + z | −7.52 | 6.18 | C–H…O, C–H…π |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ukrainets, I.V.; Burian, A.A.; Baumer, V.N.; Shishkina, S.V.; Sidorenko, L.V.; Tugaibei, I.A.; Voloshchuk, N.I.; Bondarenko, P.S. Synthesis, Crystal Structure, and Biological Activity of Ethyl 4-Methyl-2,2-dioxo-1H-2λ6,1-benzothiazine-3-carboxylate Polymorphic Forms. Sci. Pharm. 2018, 86, 21. https://doi.org/10.3390/scipharm86020021
Ukrainets IV, Burian AA, Baumer VN, Shishkina SV, Sidorenko LV, Tugaibei IA, Voloshchuk NI, Bondarenko PS. Synthesis, Crystal Structure, and Biological Activity of Ethyl 4-Methyl-2,2-dioxo-1H-2λ6,1-benzothiazine-3-carboxylate Polymorphic Forms. Scientia Pharmaceutica. 2018; 86(2):21. https://doi.org/10.3390/scipharm86020021
Chicago/Turabian StyleUkrainets, Igor V., Anna A. Burian, Vyacheslav N. Baumer, Svitlana V. Shishkina, Lyudmila V. Sidorenko, Igor A. Tugaibei, Natali I. Voloshchuk, and Pavlo S. Bondarenko. 2018. "Synthesis, Crystal Structure, and Biological Activity of Ethyl 4-Methyl-2,2-dioxo-1H-2λ6,1-benzothiazine-3-carboxylate Polymorphic Forms" Scientia Pharmaceutica 86, no. 2: 21. https://doi.org/10.3390/scipharm86020021
APA StyleUkrainets, I. V., Burian, A. A., Baumer, V. N., Shishkina, S. V., Sidorenko, L. V., Tugaibei, I. A., Voloshchuk, N. I., & Bondarenko, P. S. (2018). Synthesis, Crystal Structure, and Biological Activity of Ethyl 4-Methyl-2,2-dioxo-1H-2λ6,1-benzothiazine-3-carboxylate Polymorphic Forms. Scientia Pharmaceutica, 86(2), 21. https://doi.org/10.3390/scipharm86020021