Synthesis and Cytotoxicity Evaluation of Novel Asymmetrical Mono-Carbonyl Analogs of Curcumin (AMACs) against Vero, HeLa, and MCF7 Cell Lines


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemistry
2.1.1. General Procedures
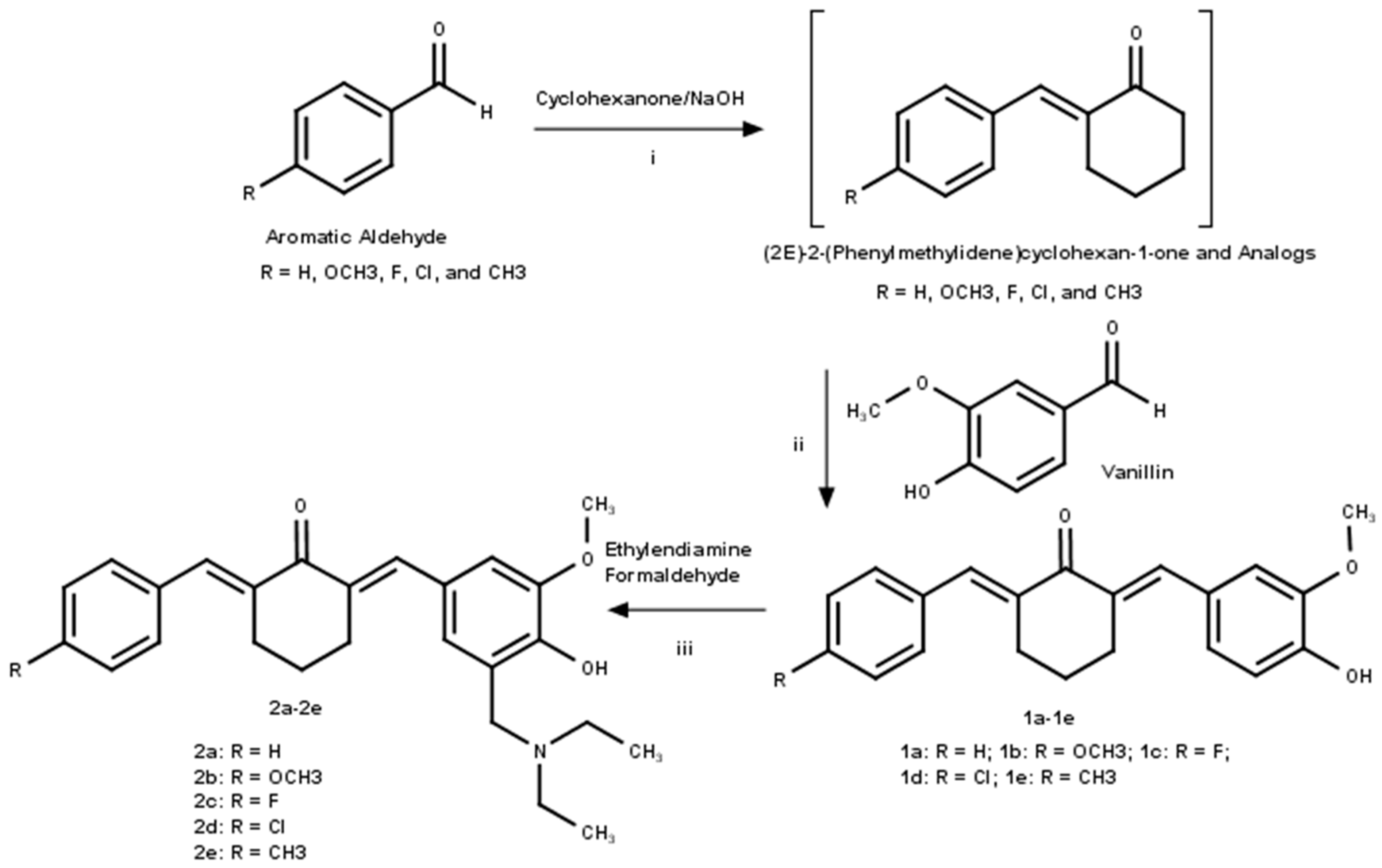
2.1.2. Synthesis of (2E)-2-(phenylmethylidene)cyclohexan-1-one and Analogs
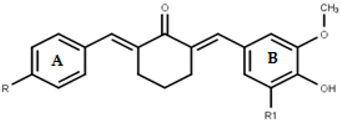
2.1.3. Synthesis of Asymmetrical Mono-Carbonyl Analogs of Curcumin (AMACs) (1a–1e)
2.1.4. Synthesis of Diethylamine Mannich Base of AMACs (2a–2e)
2.2. Cytotoxicity Test
2.2.1. Brine Shrimp Lethality Test
2.2.2. MTT Proliferation Assay
3. Results and Discussion
3.1. Chemistry
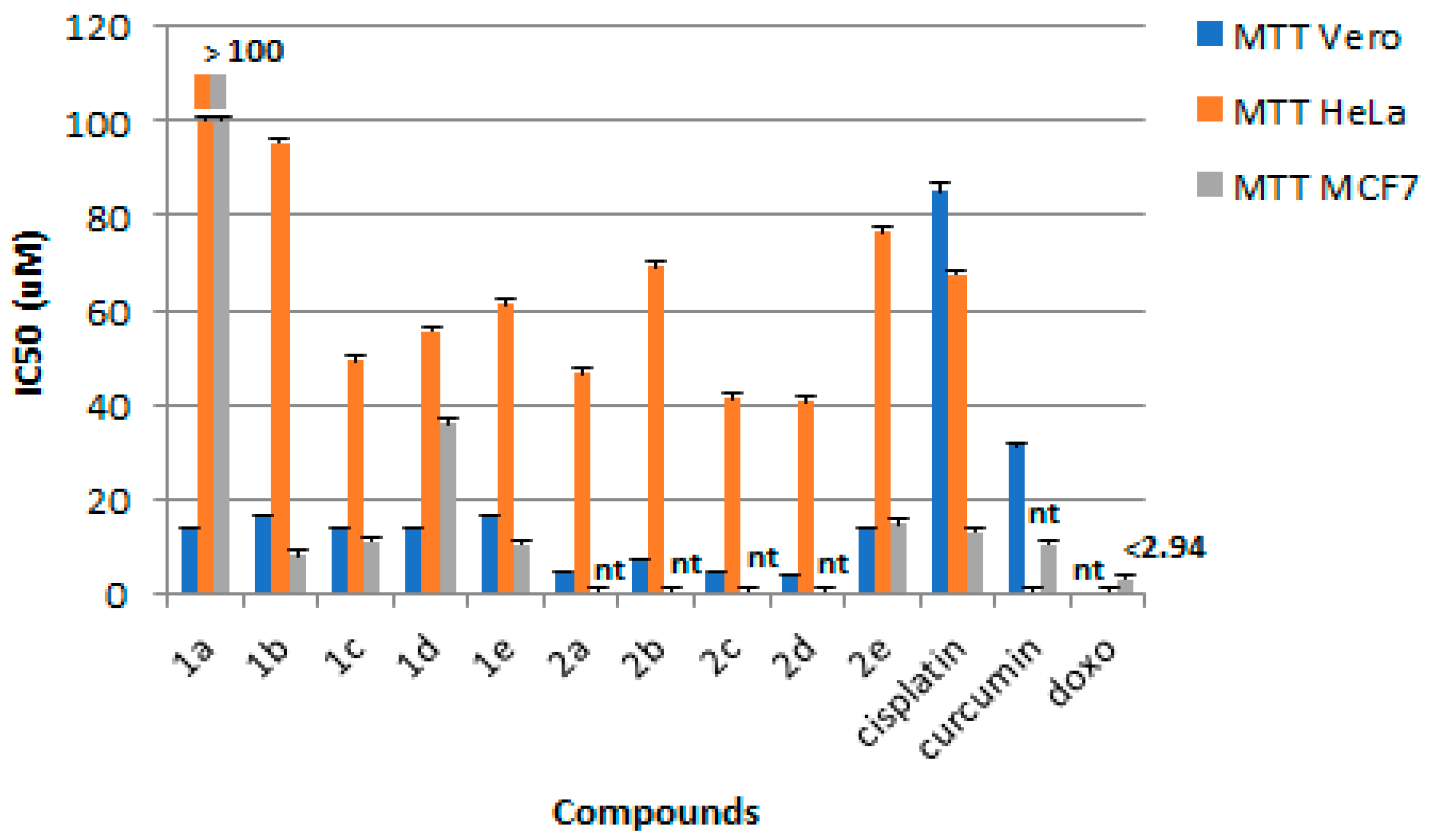
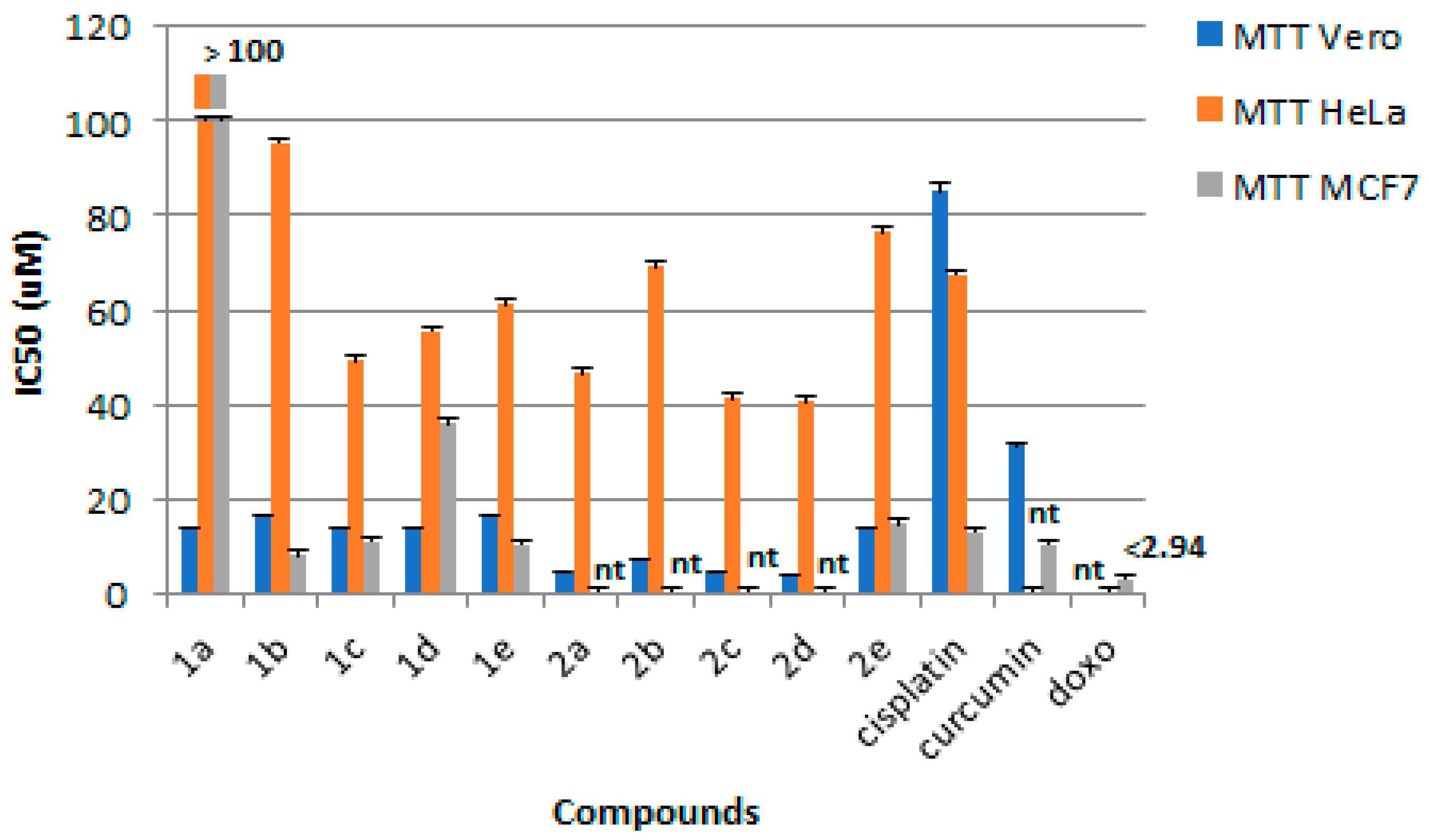
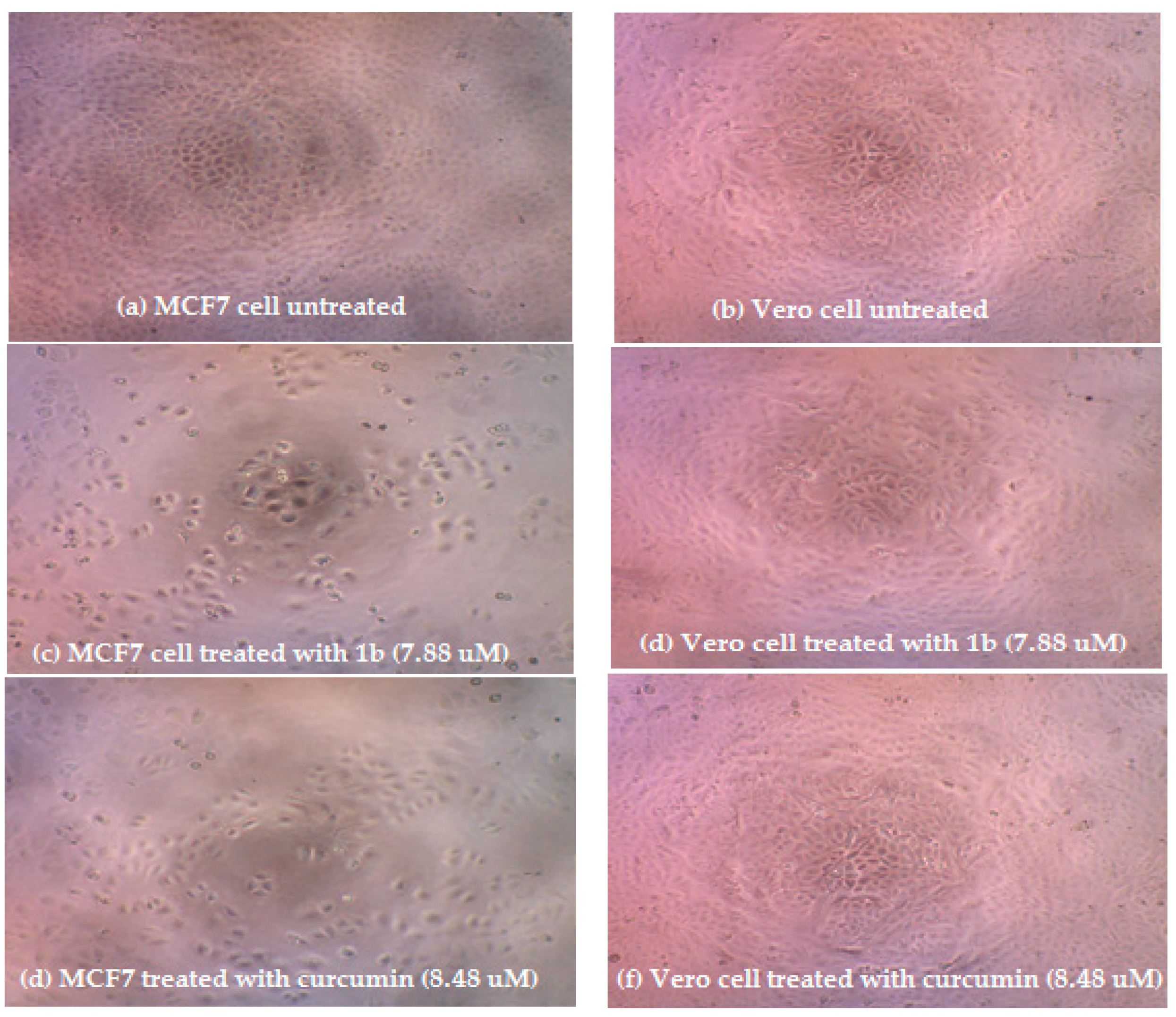
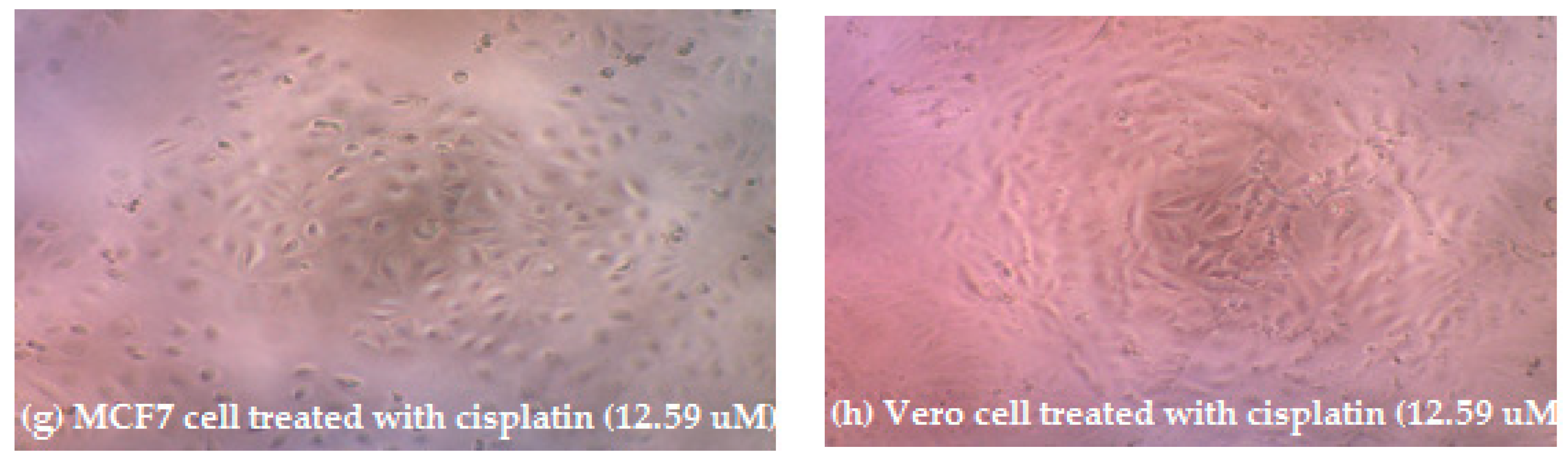
3.2. Cytotoxic Activity
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- The World Health Organization. Cancer Country Profile, 2014. Available online: http://www.who.int/cancer/country-profiles/idn_en.pdf?ua (accessed on 11 December 2017).
- Roche, V.F. Cancer and chemotherapy. In Foye’s Principles of Medicinal Chemistry, 11th ed.; Lemke, T.L., Williams, D.A., Roche, V.F., Zito, S.W., Eds.; Lippincott Wiliams and Wilkins: Baltimore, MD, USA, 2016; pp. 1199–1266. [Google Scholar]
- Basnet, P.; Skalko-Basne, N. Curcumin: An Anti-Inflammatory Molecule from a Curry Spice on the Path to Cancer Treatment. Molecules 2011, 16, 4567–4598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, P.; Thomas, S.G.; Kunnumakkara, A.B.; Sundaram, C.; Harikumar, K.B.; Sung, B.; Tharakan, S.T.; Misra, K.; Priyadarsini, I.K.; Rajasekharan, K.N.; et al. Biological activities of curcumin and its analogues (Congeners) made by man and Mother Nature. Biochem. Pharmacol. 2008, 76, 1590–1611. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavalibility of curcumin: Problems and promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef]
- Sharma, R.A.; Euden, S.A.; Platton, S.L.; Cooke, D.N.; Shafayat, A.; Hewitt, H.R.; Marczylo, T.H.; Morgan, B.; Hemingway, D.; Plummer, S.M.; et al. Phase I clinical trial of oral curcumin: Biomarkers of systemic activity and compliance. Clin. Cancer Res. 2004, 10, 6847–6854. [Google Scholar] [CrossRef] [PubMed]
- Garcea, G.; Berry, D.P.; Jones, D.J.; Singh, R.; Dennison, A.R.; Farmer, P.B.; Sharma, R.A.; Steward, W.P.; Gescher, A.J. Consumption of the putative chemopreventive agent curcumin by cancer patients: Assessment of curcumin levels in the colorectum and their pharmacodynamic consequences. Cancer Epidemiol. Biomark. Prev. 2005, 14, 120–125. [Google Scholar]
- Shetty, D.; Kim, Y.J.; Shim, H.; Snyder, J.P. Eliminating the Heart from the Curcumin Molecule: Monocarbonyl Curcumin Mimics (MACs). Molecules 2015, 20, 249–292. [Google Scholar] [CrossRef] [PubMed]
- Ohori, H.; Yamakoshi, H.; Tomizawa, M.; Shibuya, M.; Kakudo, Y.; Takahashi, A.; Takahashi, S.; Kato, S.; Suzuki, T.; Ishioka, C.; et al. Synthesis and biological analysis of new curcumin analogues bearing an enhanced potential for the medicinal treatment of cancer. Mol. Cancer Ther. 2006, 5, 2563–2571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, S.; Zheng, X.; Yao, X.; Wang, Y.; Liao, D. Synthesis and Anticancer Activity of Mono-Carbonyl Analogues of Curcumin. J. Cancer Ther. 2013, 4, 113–123. [Google Scholar] [CrossRef]
- Adams, B.K.; Ferstl, E.M.; Davis, M.C.; Herold, M.; Kurtkaya, S.; Camalier, R.F.; Hollingshead, M.G.; Kaur, G.; Sausville, E.A.; Rickles, F.R.; et al. Synthesis and biological evaluation of novel curcumin analogs as anti-cancer and anti-angiogenesis agents. Bioorg. Med. Chem. 2004, 12, 3871–3883. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Shao, L.; Wang, Y.; Zhao, C.; Chu, Y.; Xiao, J.; Zhao, Y.; Li, X.; Yang, S. Exploration and synthesis of curcumin analogues with improved structural stability both in vitro and in vivo as cytotoxic agents. Bioorg. Med. Chem. 2009, 17, 2623–2631. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Liu, Z.; Liang, G. Promising curcumin-based drug design: Monocarbonyl analogues of curcumin (MACs). Curr. Pharm. Res. 2013, 19, 2114–2135. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, L.; Wu, J.; Jiang, X.; Dong, L.; Xu, F.; Liang, G. Synthesis and evaluation of a series of novel asymmetrical curcumin analogs for the treatment of inflammation. Molecules 2014, 19, 7287–7307. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jiang, X.; Peng, K.; Chen, C.; Fi, L.; Wang, Z.; Feng, J.; Liu, Z.; Zhang, H.; Liang, G.; et al. Discovery and evaluation of novel anti-inflammatory derivatives of natural bioactive curcumin. Drug Des. Dev. Ther. 2014, 8, 2161–2171. [Google Scholar] [CrossRef]
- Aluwi, M.F.M.F.; Rullah, K.; Yamin, B.M.; Leong, S.W.; Bahari, M.N.A.; Lim, S.J.; Lam, K.W. Synthesis of unsymmetrical monocarbonyl curcumin analogues with potent inhibition on prostaglandin E2 production in LPS-induced murine and human macrophages cell lines. Bioorg. Med. Chem. Lett. 2016, 26, 2531–2538. [Google Scholar] [CrossRef] [PubMed]
- Bandgar, B.P.; Kinkar, S.N.; Chavan, H.V.; Jalde, S.S.; Shaikh, R.U.; Gacche, R.N. Synthesis and biological evaluation of asymmetric indole curcumin analogs as potential anti-inflammatory and antioxidant agents. J. Enzym. Inhib. Med. Chem. 2014, 29, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Chen, J.; Luo, S.; Xu, J.; Huang, Q.; Liu, T. Synthesis and assessment of the antioxidant and antitumor properties of asymmetric curcumin analogues. Eur. J. Med. Chem. 2015, 93, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Furniss, B.S.; Hannaford, A.J.; Smith, P.W.G.; Tatchell, A.R. Vogel’s Textbook of Practical Organic Chemistry, 5th ed.; Longman Group Ltd.: London, UK, 1989; pp. 1032–1033. [Google Scholar]
- Silverstein, R.M.; Webster, F.X.; Kiemle, D.J. Spectrometric Identification of Organic Compounds, 7th ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2005. [Google Scholar]
- Geschickter, C.F.; Meadow, J.R. Curcumin Derivatives. U.S. Patent 3,479,345, 18 November 1969. [Google Scholar]
- Untung, J.; Iskandarsyah, I.; Hayun, H. 2-[(2,6-Dimethylmorpholin-4-yl)methyl]-4-[(E)-2-{3-[(E)-2-{3-[(2,6-dimethylmorpholin-4-yl)methyl]-4-hydroxy-5-methoxyphenyl}ethenyl]-1H-pyrazol-5-yl}ethenyl]-6-methoxyphenol. Molbank 2017, 3, M949. [Google Scholar] [CrossRef]
- Meyer, B.N.; Ferrigni, N.R.; Putnam, J.E.; Jacobson, L.B.; Nichols, D.E.; McLaughlin, J.L. Brine shrim.p.: A convenient general bioassay for active plant constituents. Planta Med. 1982, 45, 31–34. [Google Scholar] [CrossRef] [PubMed]
- MTT Cell Proliferation Assay. Available online: https://www.atcc.org/~/media/DA5285A1F52C414E864C966FD78C9A79.ashx (accessed on 5 October 2017).
- GraphPad Software, Inc. Available online: www.graphpad.com (accessed on 13 December 2017).
- Badisa, R.B.; Darling-Reed, S.F.; Joseph, P.; Cooperwood, J.S.; Latinwo, L.M.; Goodman, C.B. Selective Cytotoxic Activities of Two Novel synthetic Drugs on Hyman Breast Carcinoma MCF-7 Cells. Anticancer Res. 2009, 29, 2993–2996. [Google Scholar] [PubMed]
- Chisalberti, E.L. Detection and Isolation of Bioactive Natural Products. In Bioactive Natural Products, Detection, Isolation, and Structur Determination, 2nd ed.; Colegate, S.M., Molyneux, R.J., Eds.; Taylor and Francis Groups, LLC: Boca Raton, FL, USA, 2008; p. 18. [Google Scholar]
- Aydin, S.; Becit, M.; Basaran, A.; Basaran, N. Effects of curcumin on cisplatin cytotoxicity in HeLa cells. Toxicol. Lett. 2016, 258, S259–S260. [Google Scholar] [CrossRef]
- Bala, S.; Sharma, N.; Kajal, A.; Kamboj, S.; Saini, V. Mannich Bases: An Important Pharmacophore in Present Scenario. Int. J. Med. Chem. 2014, 2014, 191072. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Dong, L.; Wang, S.; Wang, Q. Synthesis and antiproliferative activity of pterostilbene and 3′-methoxy pterostilbene Mannich base derivatives against Hela cells. Mol. Divers. 2015, 19, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Yerdelen, K.O.; Gul, H.I.; Sakagami, H.; Umemura, N. Synthesis and biological evaluation of 1,5-bis(4-hydroxy-3-methoxyphenyl)penta-1,4-dien-3-one and its aminomethyl derivatives. J. Enzym. Inhib. Med. Chem. 2015, 30, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.V.B.; Su, C.-R.; Chiou, W.-F.; Liu, Y.-N.; Chen, R.Y.; Bastow, K.F.; Lee, K.-H.; Wu, T.-S. Design, synthesis, and biological evaluation of Mannich bases of heterocyclic chalcone analogs as cytotoxic agents. Bioorg. Med. Chem. 2008, 16, 7358–7370. [Google Scholar] [CrossRef] [PubMed]
- Roman, G. Mannich bases in medicinal chemistry and drug design. Eur. J. Med. Chem. 2015, 89, 743–816. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, F.J.; Bourden, B.F.; Toth, S.I. Antitumor and Cytotoxic Compounds from Marine Organisms. In Marine Biotechnology, Pharmaceutical and Bioactive Natural Products; Attaway, D.H., Zaborsky, O.R., Eds.; Plenum Press: New York, NY, USA, 1993; Volume 1, p. 198. [Google Scholar]
- Burger, A.M.; Fiebig, H.H. Preclinical Screening for New Anticancer Agents. In Handbook of Anticancer Pharmacokinetics and Pharmacodynamics, Cancer Drug Discovery and Development; Figg, W.D., McLeod, H.L., Eds.; Humana Press Inc.: Totowa, NJ, USA, 2004; pp. 36–37. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Compd | Substuents | LC50 (µM) | IC50 (µM) (1) | SI (2) | ||||
|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | BSLT | Vero | Hela | MCF7 | Hela | MCF7 | ||
| 1 | 1a | H | H | 196.63 | 13.98 ± 0.04 | >100 | >100 | <1 | <1 |
| 2 | 1b | OCH3 | H | 322.63 | 15.43 ± 0.34 | 95.55 ± 7.19 | 7.86 ± 1.05 | <1 | 1.96 |
| 3 | 1c | F | H | 177.36 | 13.39 ± 0.39 | 49.15 ± 1.17 | 10.94 ± 0.79 | <1 | 1.28 |
| 4 | 1d | Cl | H | 204.09 | 14.06 ± 0.18 | 55.60 ± 1.49 | 35.88 ± 4.57 | <1 | <1 |
| 5 | 1e | CH3 | H | 78.71 | 16.15 ± 0.18 | 61.19 ± 2.86 | 10.39 ± 0.36 | <1 | 1.55 |
| 6 | 2a | H | X | 921.08 | 4.14 ± 0.21 | 46.61 ± 1.54 | nt | <1 | - |
| 7 | 2b | OCH3 | X | 80.21 | 7.29 ± 0.12 | 69.29 ± 3.17 | nt | <1 | - |
| 8 | 2c | F | X | 88.37 | 4.23 ± 0.32 | 41.10 ± 0.16 | nt | <1 | - |
| 9 | 2d | Cl | X | 29.80 | 3.94 ± 0.07 | 40.65 ± 0.98 | nt | <1 | - |
| 10 | 2e | CH3 | X | 1704.23 | 15.02 ± 0.14 | 76.61 ± 4.27 | 14.55 ± 1.96 | <1 | 1.03 |
| 11 | Curcumin | - | - | nt | 31.41 ± 0.41 | nt | 10.47 ± 1.10 | - | 3.00 |
| 12 | Cisplatin | - | - | nt | 84.66 ± 2.09 | 67.59 ± 2.04 | 12.85 ± 1.35 | 1.26 | 6.61 |
| 13 | Doxorubicin | - | - | nt | nt | nt | < 2.94 | - | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiji Prasetyaningrum, P.; Bahtiar, A.; Hayun, H. Synthesis and Cytotoxicity Evaluation of Novel Asymmetrical Mono-Carbonyl Analogs of Curcumin (AMACs) against Vero, HeLa, and MCF7 Cell Lines. Sci. Pharm. 2018, 86, 25. https://doi.org/10.3390/scipharm86020025
Wiji Prasetyaningrum P, Bahtiar A, Hayun H. Synthesis and Cytotoxicity Evaluation of Novel Asymmetrical Mono-Carbonyl Analogs of Curcumin (AMACs) against Vero, HeLa, and MCF7 Cell Lines. Scientia Pharmaceutica. 2018; 86(2):25. https://doi.org/10.3390/scipharm86020025
Chicago/Turabian StyleWiji Prasetyaningrum, Pekik, Anton Bahtiar, and Hayun Hayun. 2018. "Synthesis and Cytotoxicity Evaluation of Novel Asymmetrical Mono-Carbonyl Analogs of Curcumin (AMACs) against Vero, HeLa, and MCF7 Cell Lines" Scientia Pharmaceutica 86, no. 2: 25. https://doi.org/10.3390/scipharm86020025
APA StyleWiji Prasetyaningrum, P., Bahtiar, A., & Hayun, H. (2018). Synthesis and Cytotoxicity Evaluation of Novel Asymmetrical Mono-Carbonyl Analogs of Curcumin (AMACs) against Vero, HeLa, and MCF7 Cell Lines. Scientia Pharmaceutica, 86(2), 25. https://doi.org/10.3390/scipharm86020025