Anticancer Activity Evaluation of New Thieno[2,3-d]pyrimidin-4(3H)-ones and Thieno[3,2-d]pyrimidin-4(3H)-one Derivatives

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemistry
2.2. Pharmacology
2.2.1. Anticancer Assay via NCI Protocol
2.2.2. Cell Proliferation (MTT) Assay
2.2.3. Statistical Analysis
3. Results and Discussion
3.1. Chemistry
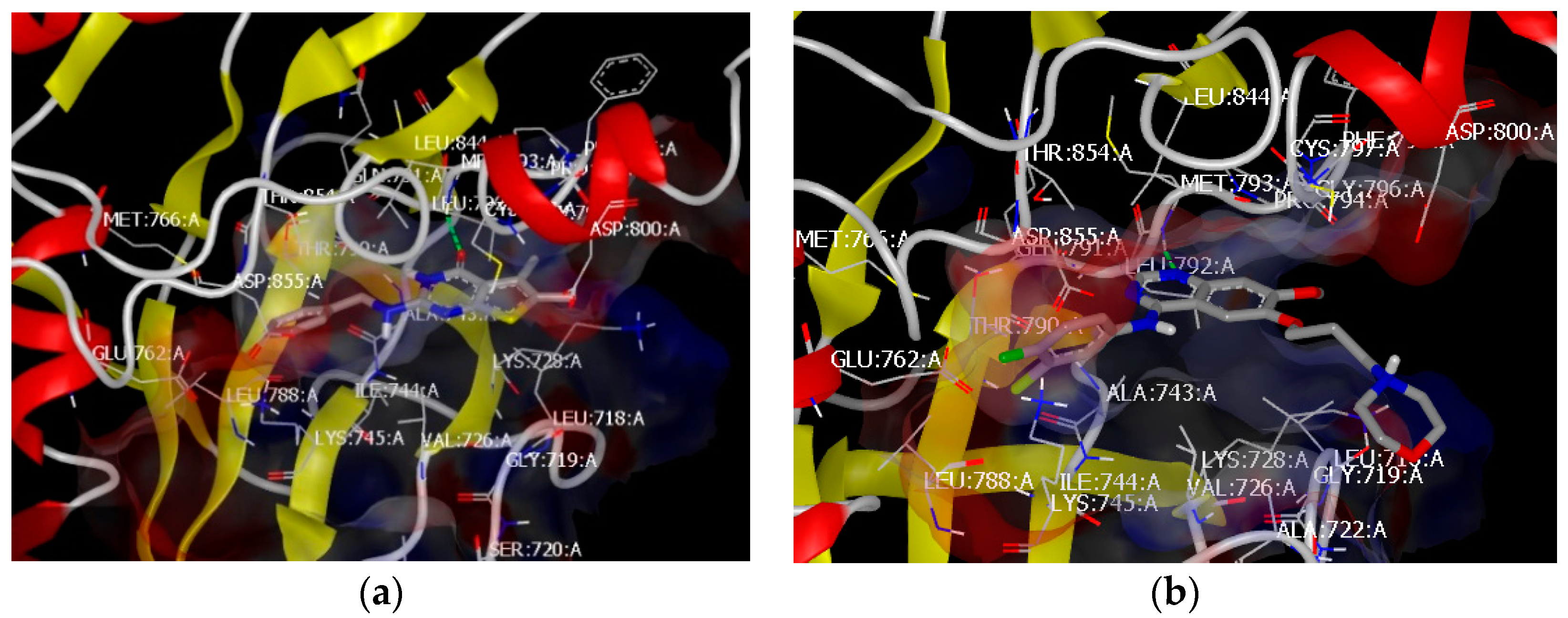
3.2. Molecular Docking
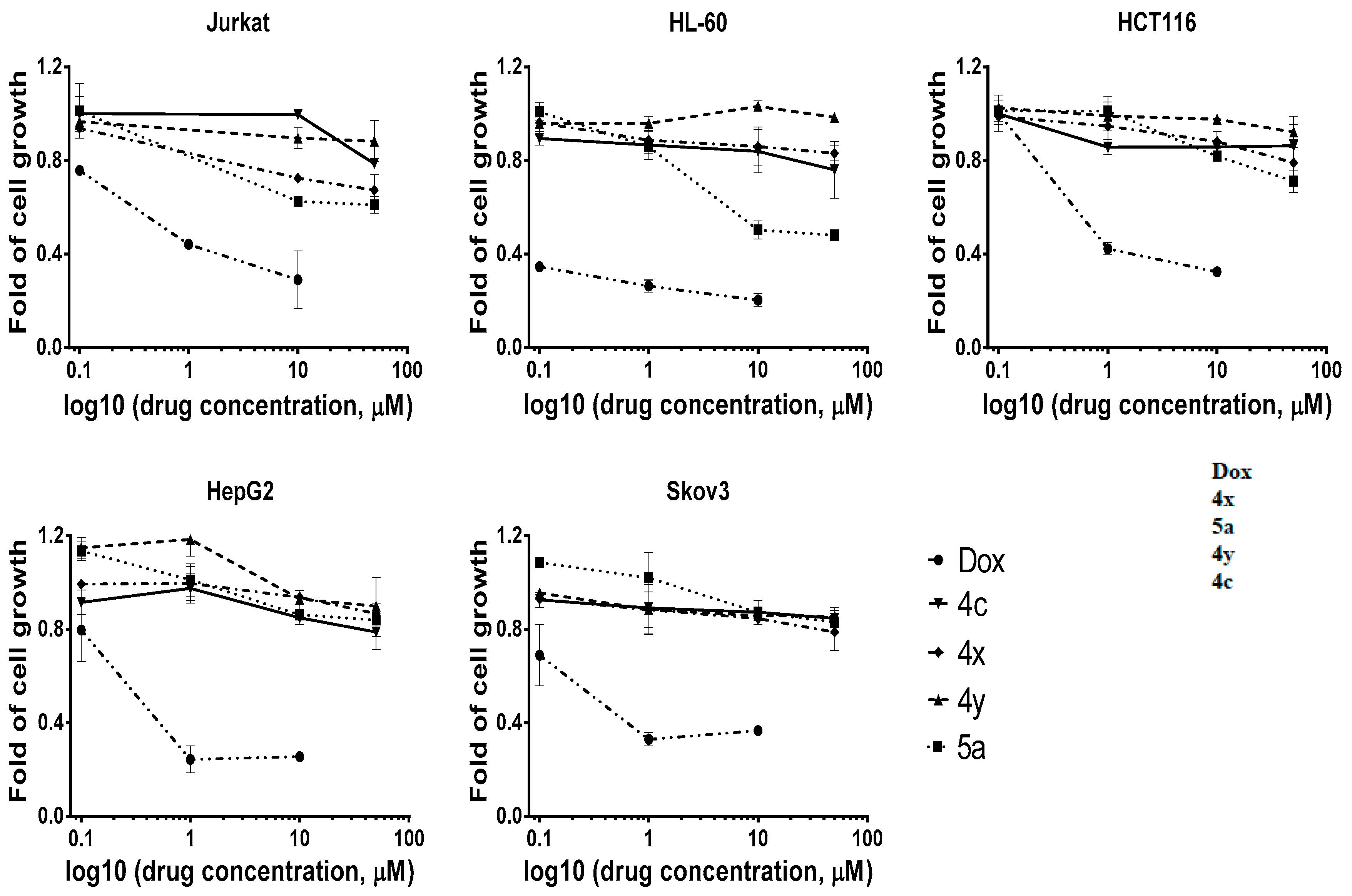
3.3. Evaluation of Anticancer Activity In Vitro
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wilding, B.; Klempier, N. Newest developments in the preparation of thieno[2,3-d]pyrimidines. Org. Prep. Proced. Int. 2017, 49, 183–215. [Google Scholar] [CrossRef]
- El-Sharkawy, L.Y.; El-Sakhawy, R.A.; Abdel-Halim, M.; Lee, K.; Piazza, G.A.; Ducho, C.; Hartmann, R.W.; Abadi, A.H. Design and synthesis of novel annulated thienopyrimidines as phosphodiesterase 5 (PDE5) inhibitors. Arch. Pharm. Chem. Life Sci. 2018, 351, e1800018. [Google Scholar] [CrossRef] [PubMed]
- Bysting, F.; Bugge, S.; Sundby, E.; Hoff, H. Investigation of Heck coupling on 6-bromo[2,3-d]thienopyrimidines for construction of new EGFR inhibitor lead structures. RSC Adv. 2017, 7, 18569–18577. [Google Scholar] [CrossRef]
- Pao, W.; Chmielecki, J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat. Rev. Cancer 2010, 10, 760–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Red, B.M.; Yun, C.H.; Lai, D.; Lemmon, M.A.; Eck, M.J.; Pao, W. Mechanism for activation of mutated epidermal growth factor receptors in lung cancer. Proc. Natl. Acad. Sci. USA 2013, 110, E3595–E3604. [Google Scholar] [CrossRef]
- Lee, H.J.; Seo, A.N.; Kim, E.J.; Jang, M.H.; Kim, Y.J.; Kim, J.H.; Kim, S.W.; Ryu, H.S.; Park, I.A.; Im, S.A.; et al. Prognostic and predictive values of EGFR overexpression and EGFR copy number alteration in HER2-positive breast cancer. Br. J. Cancer 2015, 112, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Cook, N.; Frese, K.K.; Moore, M. Assessing the role of the EGF receptor in the development and progression of pancreatic cancer. Gastrointest. Cancer Targets Ther. 2014, 4, 23–37. [Google Scholar] [Green Version]
- Makker, V.; Recio, F.O.; Ma, L.; Matulonis, U.A.; Lauchle, J.O.; Parmar, H.; Gilbert, H.N.; Ware, J.A.; Zhu, R.; Lu, S.; et al. A multicenter, single-arm, open-label, phase 2 study of apitolisib (GDC-0980) for the treatment of recurrent or persistent endometrial carcinoma (MAGGIE study). Cancer 2016, 122, 3519–3528. [Google Scholar] [CrossRef] [PubMed]
- Drug Profile, SNS 314. Available online: https://adisinsight.springer.com/drugs/800019470 (accessed on 11 April 2018).
- Sarker, D.; Ang, J.E.; Baird, R.; Kristeleit, R.; Shah, K.; Moreno, V.; Clarke, P.A.; Raynaud, F.I.; Levy, G.; Ware, J.A.; et al. First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2015, 21, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Gewald, K.; Schinke, E.; Bottcher, H. Heterocyclen aus CH-aciden Nitrilen, VIII. 2-Amino-thiophene aus methylenaktiven Nitrilen, Carbonylverbindungen und Schwefel. Chem. Banner 1966, 99, 94–100. [Google Scholar] [CrossRef]
- Gewald, K. Reaktion von methylenaktiven Nitrilen mit Senfolen und Schwefel. J. Prakt. Chem. 1966, 32, 26–30. [Google Scholar] [CrossRef]
- Liu, X.; Zu, Y.; Fu, Y.; Yao, L.; Gu, C.; Wang, W.; Efferth, T. Antimicrobial activity and cytotoxicity towards cancer cells of Melaleuca alternifolia (tea tree) oil. Eur. Food Res. Technol. 2009, 229, 247–253. [Google Scholar] [CrossRef]
- Pokhodylo, N.T.; Matiychuk, V.S.; Obushak, M.D. New convenient synthesis of 2,3-diaminothieno[2,3-d]pyrimidin-4(3H)-one derivates from substituted alkyl 2-(1H-tetrazol-1-yl)thiophene-3-carboxylates. Tetrahedron 2008, 64, 1430–1434. [Google Scholar] [CrossRef]
- Pokhodylo, N.T.; Shyyka, O.Y.; Matiychuk, V.S.; Obushak, M.D. New convenient strategy for annulation of pyrimidines to thiophenes or furans via the one-pot multistep cascade reaction of 1H-tetrazoles with aliphatic amines. ACS Comb. Sci. 2015, 17, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Shyyka, O.Y.; Pokhodylo, N.T.; Slyvka, Y.I.; Goreshnik, E.A.; Obushak, M.D. Understanding the tetrazole ring cleavage reaction with hydrazines: Structural determination and mechanistic insight. Tetrahedron Lett. 2018, 59, 1112–1115. [Google Scholar] [CrossRef]
- Developmental Therapeutics Program. Available online: http://dtp.nci.nih.gov.
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Boyd, M.R. The NCI In Vitro Anticancer Drug Discovery Screen. In Anticancer Drug Development Guide. Cancer Drug Discovery and Development; Teicher, B.A., Ed.; Humana Press: Totowa, NJ, USA, 1997; ISBN 978-1-4615-8154-3. [Google Scholar]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 10, 813–823. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Compound | Mean Growth % | Range of Growth % | The Most Sensitive Cell Lines | Growth of the Most Sensitive Cell Lines |
|---|---|---|---|---|---|
| 5a |  | 51.01 | −31.02 to 91.62 | MDA-MB-435 (Melanoma) | −31.02 |
| K-562 (Leukemia) | 12.90 | ||||
| HT 29 (Colon Cancer) | 13.28 | ||||
| SR (Leukemia) | 14.44 | ||||
| HL-60 (TB) (Leukemia) | 22.45 | ||||
| MDA-MB-468 (Breast Cancer) | 23.78 | ||||
| HCT 116 (Colon Cancer) | 24.50 | ||||
| HT SW-620 (Colon Cancer) | 25.96 | ||||
| NCI-H460 (Non-Small Cell Lung Cancer) | 26.58 | ||||
| 4x |  | 85.98 | 58.71 to 105.80 | A549/ATCC (Non-Small Cell Lung Cancer) | 58.71 |
| HCT-15 (Colon Cancer) | 59.57 | ||||
| UACC-62 (Melanoma) | 63.06 | ||||
| HOP-92 (Non-Small Cell Lung Cancer) | 68.44 | ||||
| A498 (Renal Cancer) | 70.69 | ||||
| PC-3 (Prostate Cancer) | 71.86 | ||||
| 4s |  | 88.10 | 51.19 to 105.87 | HOP-92 (Non-Small Cell Lung Cancer) | 51.19 |
| SNB-75 (CNS Cancer) | 70.21 | ||||
| U251 (CNS Cancer) | 71.86 | ||||
| MOLT-4 (Leukemia) | 72.91 | ||||
| PC-3 (Prostate Cancer) | 73.10 | ||||
| MDA-MB-468 (Breast Cancer) | 74.47 | ||||
| 6a |  | 91.38 | 58.12 to 110.00 | HOP-92 (Non-Small Cell Lung Cancer) | 58.12 |
| MALME-3M (Melanoma) | 74.98 | ||||
| NCI/ADR-RES (Ovarian Cancer) | 78.18 | ||||
| NCI-H322M (Non-Small Cell Lung Cancer) | 78.19 | ||||
| UO-31 (Renal Cancer) | 79.54 | ||||
| 4r |  | 92.97 | 66.09 to 113.41 | SNB-75 (CNS Cancer) | 66.09 |
| HOP-92 (Non-Small Cell Lung Cancer) | 67.24 | ||||
| MALME-3M (Melanoma) | 73.78 | ||||
| BT-549 (Breast Cancer) | 76.08 | ||||
| UO-31 (Renal Cancer) | 77.53 | ||||
| 4c |  | 93.98 | 67.36 to 117.18 | SNB-75 (CNS Cancer) | 67.36 |
| COLO 205 (Colon Cancer) | 76.97 | ||||
| MDA-MB-468 (Breast Cancer) | 80.04 | ||||
| 4m |  | 95.06 | 53.30 to 110.21 | EKVX (Non-Small Cell Lung Cancer) | 53.30 |
| U251 (CNS Cancer) | 69.49 | ||||
| NCI-H522 (Non-Small Cell Lung Cancer) | 76.95 | ||||
| HOP-92 (Non-Small Cell Lung Cancer) | 79.72 | ||||
| 4d |  | 97.43 | 59.72 to 129.11 | EKVX (Non-Small Cell Lung Cancer) | 59.72 |
| SNB-75 (CNS Cancer) | 63.87 | ||||
| HOP-92 (Non-Small Cell Lung Cancer) | 74.21 | ||||
| MDA-MB-468 (Breast Cancer) | 78.04 | ||||
| 4b |  | 99.06 | 43.50 to 126.21 | EKVX (Non-Small Cell Lung Cancer) | 43.50 |
| MDA-MB-468 (Breast Cancer) | 84.23 | ||||
| SNB-75 (CNS Cancer) | 85.73 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shyyka, O.; Pokhodylo, N.; Finiuk, N.; Matiychuk, V.; Stoika, R.; Obushak, M. Anticancer Activity Evaluation of New Thieno[2,3-d]pyrimidin-4(3H)-ones and Thieno[3,2-d]pyrimidin-4(3H)-one Derivatives. Sci. Pharm. 2018, 86, 28. https://doi.org/10.3390/scipharm86030028
Shyyka O, Pokhodylo N, Finiuk N, Matiychuk V, Stoika R, Obushak M. Anticancer Activity Evaluation of New Thieno[2,3-d]pyrimidin-4(3H)-ones and Thieno[3,2-d]pyrimidin-4(3H)-one Derivatives. Scientia Pharmaceutica. 2018; 86(3):28. https://doi.org/10.3390/scipharm86030028
Chicago/Turabian StyleShyyka, Olga, Nazariy Pokhodylo, Nataliya Finiuk, Vasyl Matiychuk, Rostyslav Stoika, and Mykola Obushak. 2018. "Anticancer Activity Evaluation of New Thieno[2,3-d]pyrimidin-4(3H)-ones and Thieno[3,2-d]pyrimidin-4(3H)-one Derivatives" Scientia Pharmaceutica 86, no. 3: 28. https://doi.org/10.3390/scipharm86030028