Metformin: On Ongoing Journey across Diabetes, Cancer Therapy and Prevention

Abstract
:1. Introduction: The Evolution of Metformin
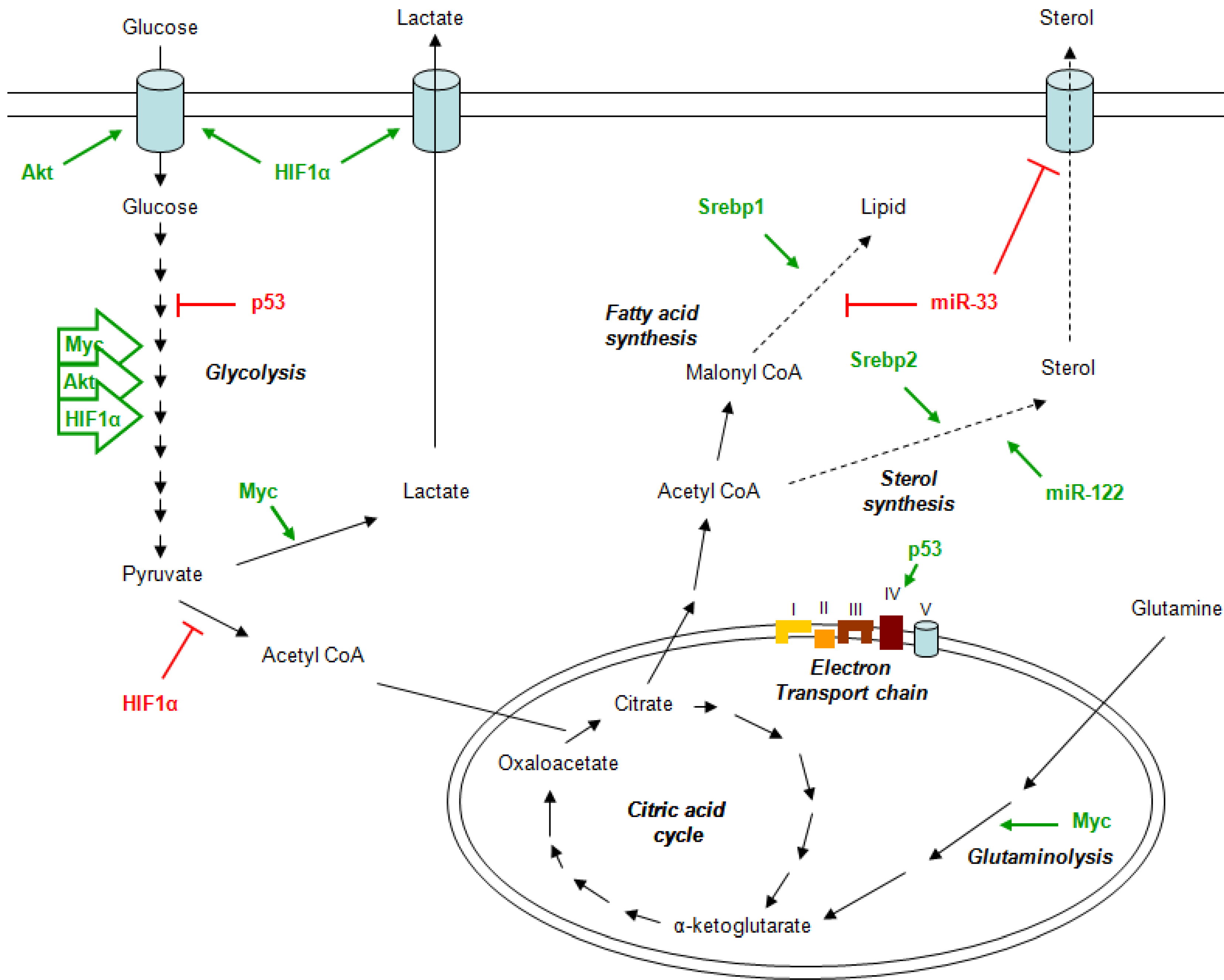
2. Metabolism and Cancer

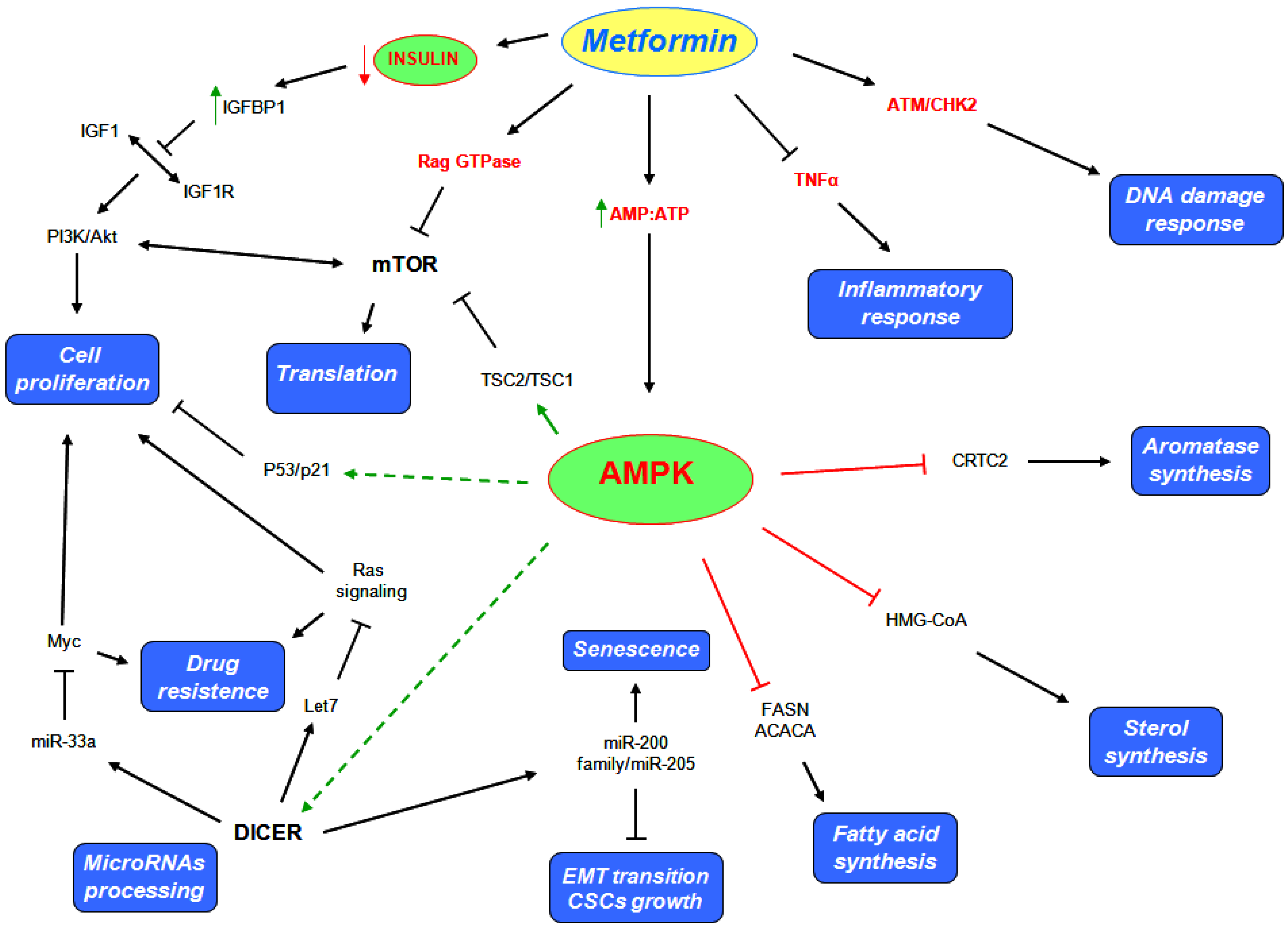
3. Metformin Mechanism of Action
4. The Pharmacokinetics of Metformin
5. Metformin Anticancer Properties
5.1. AMPK Dependent Mechanisms

5.2. AMPK Independent Mechanisms
6. Metformin and Cancer Therapy
7. Role of Metformin as a Chemoprevention Agent
7.1. All Cancers

{kind=link}
{kind=link}
| Study | Study Types | Inclusion | Results |
|---|---|---|---|
| Franciosi, M 2013 PLoS One | Meta analysis 41 observational 12 RCT | Observational studies which examined association between metformin exposure and cancer. RCTs which compared metformin and other hypoglycaemic agents | Observational studies, metformin associated with: Decreased risk of death (6 studies, OR 0.65, 95% CI 0.53–0.80) Decreased all malignancies (18 studies, OR 0.73, 95% CI 0.61–0.88) Decreased liver cancer (8 studies, OR 0.34 95% CI 0.19–0.60) Decreased colorectal cancer (12 studies, OR 0.83, 95% CI 0.74–0.92) Decreased pancreas cancer (9 studies, OR 0.56, 95% CI 0.36–0.86) Decreased stomach cancer (2 studies, OR 0.83, 95% CI 0.76–0.91) Decrased esophageal cancer (2 studies OR 0.90 95% CI 0.83–0.98) In RCTs: No significant effect of metformin on death or malignancies |
| Thakkar, B 2013 Metabolism | Meta analysis 13 case contro l9 cohort 2 RCTs | Studies that assess metformin and/or sulfonylurea effects on cancer risk in diabetic patients | In observational studies, metformin use was associated with reduced risk of developing cancer: Cohort studies (RR 0.70, 95% CI 0.67–0.73) Case-control studies (OR 0.90 95% CI 0.84–0.98) In RCTs: No effect on developing cancer (RR 1.01 95% CI 0.81–1.26) |
| Stevens RJ 2012 Diabetologia | Meta analysis 13 RCTs | RCTs which compared metformin to another diabetic treatment or placebo/usual care, with minimum 500 participants and 1 year follow-up | In RCTs looking at cancer outcomes (11 studies): RR 1.02 (95% CI 0.82–1.26) (11 studies) In RCTs looking at all-cause mortality (13 studies): RR 0.94 (95% CI 0.79–1.12) |
| Soranna D., 2012 Oncologist | Meta analysis 17 observational | Observational studies of diabetic patients treated with metformin and/or sulfonylurea where risk of all cancers and specific cancer mortality was investigated | Metformin associated with decreased RR of: All cancers (RR 0.61, 95% CI 0.54–0.70) Colorectal cancer (RR 0.64, 95% CI 0.54–0.76) Pancreatic ca. (RR 0.38, 95% CI 0.14–0.91) Sulfonylurea not associated with reduced RR |
| Noto H., 2012 PLoS ONE | Meta analysis Cancer Mortality: 4 cohort 2 RCTs Cancer Incidence: 6 cohort 2 case control 2 RCTs | Studies of diabetic patients taking metformin compared to those not taking metformin | Metformin users compared to non users had: Lower cancer mortality (pooled RR 0.66, 95% CI 0.49–0.88) Lower cancer incidence (pooled RR 0.67, 95% CI 0.53–0.85) Lower colorectal ca. incidence (pooled RR 0.68, 95% CI 0.53–0.88) Lower HCC (pooled RR 0.20, 95% CI 0.07–0.59) Lower lung cancer (pooled RR 0.67, 95% CI 0.45–0.99) |
7.2. Colorectal Cancer
7.3. Lung Cancer
7.4. Hepatocellular Carcinoma
7.5. Breast Cancer
7.6. Ongoing Studies
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Bailey, C.J.; Day, C. Traditional plant medicines as treatments for diabetes. Diabetes Care 1989, 12, 553–564. [Google Scholar]
- Witters, L.A. The blooming of the french lilac. J. Clin. Invest. 2001, 108, 1105–1107. [Google Scholar]
- Parturier, G.; Hugnot, G. Le galega dans le traitement du diabete; (in French). Massons: Paris, France, 1935. [Google Scholar]
- Watanabe, C. Studies in the metabolic changes induced by administration of guanidine bases. J. Biol. Chem. 1918, 33, 253–265. [Google Scholar]
- Muller, H.; Rheinwein, H. Pharmacology of galegin. Arch. Exp. Path. Pharmacol. 1927, 125, 212–228. [Google Scholar] [CrossRef]
- Hesse, G.; Taubmann, G. Die wirkung des biguanids und seiner derivate aud den zuckerstoffwechsel. (in German). Arch. Exp. Path. Pharmacol. 1929, 142, 290–308. [Google Scholar] [CrossRef]
- Bailey, C.J.; Day, C. Metformin: Its botanical background. Pract. Diab. Int. 2004, 21, 115–117. [Google Scholar] [CrossRef]
- Nattrass, M.; Alberti, K.G. Biguanides. Diabetologia 1978, 14, 71–74. [Google Scholar] [CrossRef]
- Hundal, R.S.; Krssak, M.; Dufour, S.; Laurent, D.; Lebon, V.; Chandramouli, V.; Inzucchi, S.E.; Schumann, W.C.; Petersen, K.F.; Landau, B.R.; et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes 2000, 49, 2063–2069. [Google Scholar] [CrossRef]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Averet, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex i. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar]
- Emami Riedmaier, A.; Fisel, P.; Nies, A.T.; Schaeffeler, E.; Schwab, M. Metformin and cancer: From the old medicine cabinet to pharmacological pitfalls and prospects. Trends Pharmacol. Sci. 2013, 34, 126–135. [Google Scholar] [CrossRef]
- Hardie, D.G. Amp-activated protein kinase: A master switch in glucose and lipid metabolism. Rev. Endocr. Metab. Disord. 2004, 5, 119–125. [Google Scholar] [CrossRef]
- Lee, J.O.; Lee, S.K.; Jung, J.H.; Kim, J.H.; You, G.Y.; Kim, S.J.; Park, S.H.; Uhm, K.O.; Kim, H.S. Metformin induces rab4 through ampk and modulates glut4 translocation in skeletal muscle cells. J. Cell. Physiol. 2011, 226, 974–981. [Google Scholar] [CrossRef]
- Stephenne, X.; Foretz, M.; Taleux, N.; van der Zon, G.C.; Sokal, E.; Hue, L.; Viollet, B.; Guigas, B. Metformin activates amp-activated protein kinase in primary human hepatocytes by decreasing cellular energy status. Diabetologia 2011, 54, 3101–3110. [Google Scholar] [CrossRef]
- Hasan, J.A.; Memon, G.U. Impact of metformin therapy in patients with polycystic ovarian syndrome. J. Coll. Physicians Surg. Pak. 2005, 15, 712–715. [Google Scholar]
- Moghetti, P.; Castello, R.; Negri, C.; Tosi, F.; Perrone, F.; Caputo, M.; Zanolin, E.; Muggeo, M. Metformin effects on clinical features, endocrine and metabolic profiles, and insulin sensitivity in polycystic ovary syndrome: A randomized, double-blind, placebo-controlled 6-month trial, followed by open, long-term clinical evaluation. J. Clin. Endocrinol. Metab. 2000, 85, 139–146. [Google Scholar] [CrossRef]
- Bianchi, C.; Penno, G.; Romero, F.; Del Prato, S.; Miccoli, R. Treating the metabolic syndrome. Expert Rev. Cardiovasc. Ther. 2007, 5, 491–506. [Google Scholar] [CrossRef]
- Kourelis, T.V.; Siegel, R.D. Metformin and cancer: New applications for an old drug. Med Oncol 2012, 29, 1314–1327. [Google Scholar] [CrossRef]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. Br. Med. J. 2005, 330, 1304–1305. [Google Scholar] [CrossRef]
- Pollak, M.N. Investigating metformin for cancer prevention and treatment: The end of the beginning. Cancer Discov. 2012, 2, 778–790. [Google Scholar] [CrossRef]
- Dowling, R.J.; Niraula, S.; Stambolic, V.; Goodwin, P.J. Metformin in cancer: Translational challenges. J. Mol. Endocrinol. 2012, 48, R31–R43. [Google Scholar] [CrossRef]
- Shackelford, D.B.; Abt, E.; Gerken, L.; Vasquez, D.S.; Seki, A.; Leblanc, M.; Wei, L.; Fishbein, M.C.; Czernin, J.; Mischel, P.S.; et al. Lkb1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 2013, 23, 143–158. [Google Scholar] [CrossRef]
- Algire, C.; Amrein, L.; Bazile, M.; David, S.; Zakikhani, M.; Pollak, M. Diet and tumor lkb1 expression interact to determine sensitivity to anti-neoplastic effects of metformin in vivo. Oncogene 2011, 30, 1174–1182. [Google Scholar] [CrossRef]
- Buzzai, M.; Jones, R.G.; Amaravadi, R.K.; Lum, J.J.; DeBerardinis, R.J.; Zhao, F.; Viollet, B.; Thompson, C.B. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007, 67, 6745–6752. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Semenza, G.L.; Roth, P.H.; Fang, H.M.; Wang, G.L. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 1994, 269, 23757–23763. [Google Scholar]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. Hif-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. Hif-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef]
- Miyamoto, S.; Murphy, A.N.; Brown, J.H. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-ii. Cell Death Differ 2008, 15, 521–529. [Google Scholar] [CrossRef]
- Lewis, B.C.; Shim, H.; Li, Q.; Wu, C.S.; Lee, L.A.; Maity, A.; Dang, C.V. Identification of putative c-myc-responsive genes: Characterization of rcl, a novel growth-related gene. Mol. Cell. Biol. 1997, 17, 4967–4978. [Google Scholar]
- Menssen, A.; Hermeking, H. Characterization of the c-myc-regulated transcriptome by sage: Identification and analysis of c-myc target genes. Proc. Natl. Acad. Sci. USA 2002, 99, 6274–6279. [Google Scholar] [CrossRef]
- Dang, C.V. Rethinking the warburg effect with myc micromanaging glutamine metabolism. Cancer Res. 2010, 70, 859–862. [Google Scholar] [CrossRef]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. Tigar, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. P53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef]
- Milgraum, L.Z.; Witters, L.A.; Pasternack, G.R.; Kuhajda, F.P. Enzymes of the fatty acid synthesis pathway are highly expressed in in situ breast carcinoma. Clin. Cancer Res. 1997, 3, 2115–2120. [Google Scholar]
- Alo, P.L.; Visca, P.; Marci, A.; Mangoni, A.; Botti, C.; Di Tondo, U. Expression of fatty acid synthase (fas) as a predictor of recurrence in stage i breast carcinoma patients. Cancer 1996, 77, 474–482. [Google Scholar] [CrossRef]
- Visca, P.; Sebastiani, V.; Botti, C.; Diodoro, M.G.; Lasagni, R.P.; Romagnoli, F.; Brenna, A.; de Joannon, B.C.; Donnorso, R.P.; Lombardi, G.; et al. Fatty acid synthase (fas) is a marker of increased risk of recurrence in lung carcinoma. Anticancer Res. 2004, 24, 4169–4173. [Google Scholar]
- Sebastiani, V.; Visca, P.; Botti, C.; Santeusanio, G.; Galati, G.M.; Piccini, V.; Capezzone de Joannon, B.; Di Tondo, U.; Alo, P.L. Fatty acid synthase is a marker of increased risk of recurrence in endometrial carcinoma. Gynecol. Oncol. 2004, 92, 101–105. [Google Scholar] [CrossRef]
- Alo, P.L.; Visca, P.; Trombetta, G.; Mangoni, A.; Lenti, L.; Monaco, S.; Botti, C.; Serpieri, D.E.; Di Tondo, U. Fatty acid synthase (fas) predictive strength in poorly differentiated early breast carcinomas. Tumori 1999, 85, 35–40. [Google Scholar]
- Gansler, T.S.; Hardman, W., III; Hunt, D.A.; Schaffel, S.; Hennigar, R.A. Increased expression of fatty acid synthase (oa-519) in ovarian neoplasms predicts shorter survival. Hum. Pathol. 1997, 28, 686–692. [Google Scholar] [CrossRef]
- Mashima, T.; Seimiya, H.; Tsuruo, T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br. J. Cancer 2009, 100, 1369–1372. [Google Scholar] [CrossRef]
- Farazi, T.A.; Spitzer, J.I.; Morozov, P.; Tuschl, T. Mirnas in human cancer. J. Pathol. 2011, 223, 102–115. [Google Scholar] [CrossRef]
- Martello, G.; Rosato, A.; Ferrari, F.; Manfrin, A.; Cordenonsi, M.; Dupont, S.; Enzo, E.; Guzzardo, V.; Rondina, M.; Spruce, T.; et al. A microrna targeting dicer for metastasis control. Cell 2010, 141, 1195–1207. [Google Scholar] [CrossRef]
- Karube, Y.; Tanaka, H.; Osada, H.; Tomida, S.; Tatematsu, Y.; Yanagisawa, K.; Yatabe, Y.; Takamizawa, J.; Miyoshi, S.; Mitsudomi, T.; et al. Reduced expression of dicer associated with poor prognosis in lung cancer patients. Cancer Sci. 2005, 96, 111–115. [Google Scholar] [CrossRef]
- Merritt, W.M.; Lin, Y.G.; Han, L.Y.; Kamat, A.A.; Spannuth, W.A.; Schmandt, R.; Urbauer, D.; Pennacchio, L.A.; Cheng, J.F.; Nick, A.M.; et al. Dicer, drosha, and outcomes in patients with ovarian cancer. New Engl. J. Med. 2008, 359, 2641–2650. [Google Scholar] [CrossRef]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. Mir-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef]
- Krutzfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of micrornas in vivo with ‘antagomirs’. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef]
- Marquart, T.J.; Allen, R.M.; Ory, D.S.; Baldan, A. Mir-33 links srebp-2 induction to repression of sterol transporters. Proc. Natl. Acad. Sci. USA 2010, 107, 12228–12232. [Google Scholar] [CrossRef]
- Najafi-Shoushtari, S.H.; Kristo, F.; Li, Y.; Shioda, T.; Cohen, D.E.; Gerszten, R.E.; Naar, A.M. Microrna-33 and the srebp host genes cooperate to control cholesterol homeostasis. Science 2010, 328, 1566–1569. [Google Scholar] [CrossRef]
- Rayner, K.J.; Suarez, Y.; Davalos, A.; Parathath, S.; Fitzgerald, M.L.; Tamehiro, N.; Fisher, E.A.; Moore, K.J.; Fernandez-Hernando, C. Mir-33 contributes to the regulation of cholesterol homeostasis. Science 2010, 328, 1570–1573. [Google Scholar] [CrossRef]
- Davalos, A.; Goedeke, L.; Smibert, P.; Ramirez, C.M.; Warrier, N.P.; Andreo, U.; Cirera-Salinas, D.; Rayner, K.; Suresh, U.; Pastor-Pareja, J.C.; et al. Mir-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 9232–9237. [Google Scholar] [CrossRef]
- Gerin, I.; Clerbaux, L.A.; Haumont, O.; Lanthier, N.; Das, A.K.; Burant, C.F.; Leclercq, I.A.; MacDougald, O.A.; Bommer, G.T. Expression of mir-33 from an srebp2 intron inhibits cholesterol export and fatty acid oxidation. J. Biol. Chem. 2010, 285, 33652–33661. [Google Scholar] [CrossRef]
- Rayner, K.J.; Esau, C.C.; Hussain, F.N.; McDaniel, A.L.; Marshall, S.M.; van Gils, J.M.; Ray, T.D.; Sheedy, F.J.; Goedeke, L.; Liu, X.; et al. Inhibition of mir-33a/b in non-human primates raises plasma hdl and lowers vldl triglycerides. Nature 2011, 478, 404–407. [Google Scholar] [CrossRef]
- Rottiers, V.; Najafi-Shoushtari, S.H.; Kristo, F.; Gurumurthy, S.; Zhong, L.; Li, Y.; Cohen, D.E.; Gerszten, R.E.; Bardeesy, N.; Mostoslavsky, R.; et al. Micrornas in metabolism and metabolic diseases. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 225–233. [Google Scholar] [CrossRef]
- Kim, H.S.; Xiao, C.; Wang, R.H.; Lahusen, T.; Xu, X.; Vassilopoulos, A.; Vazquez-Ortiz, G.; Jeong, W.I.; Park, O.; Ki, S.H.; et al. Hepatic-specific disruption of sirt6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 2010, 12, 224–236. [Google Scholar] [CrossRef]
- Blandino, G.; Valerio, M.; Cioce, M.; Mori, F.; Casadei, L.; Pulito, C.; Sacconi, A.; Biagioni, F.; Cortese, G.; Galanti, S.; et al. Metformin elicits anticancer effects through the sequential modulation of dicer and c-myc. Nat. Commun. 2012, 3, Article 865. [Google Scholar]
- Fogarty, S.; Hardie, D.G. Development of protein kinase activators: Ampk as a target in metabolic disorders and cancer. Biochim. Biophys. Acta 2010, 1804, 581–591. [Google Scholar] [CrossRef]
- Foretz, M.; Hebrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the lkb1/ampk pathway via a decrease in hepatic energy state. J. Clin. Invest. 2010, 120, 2355–2369. [Google Scholar] [CrossRef] [Green Version]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef]
- Kemp, B.E.; Stapleton, D.; Campbell, D.J.; Chen, Z.P.; Murthy, S.; Walter, M.; Gupta, A.; Adams, J.J.; Katsis, F.; van Denderen, B.; et al. Amp-activated protein kinase, super metabolic regulator. Biochem. Soc. Trans. 2003, 31, 162–168. [Google Scholar]
- Sanders, M.J.; Grondin, P.O.; Hegarty, B.D.; Snowden, M.A.; Carling, D. Investigating the mechanism for amp activation of the amp-activated protein kinase cascade. Biochem. J. 2007, 403, 139–148. [Google Scholar] [CrossRef]
- Xiao, B.; Heath, R.; Saiu, P.; Leiper, F.C.; Leone, P.; Jing, C.; Walker, P.A.; Haire, L.; Eccleston, J.F.; Davis, C.T.; et al. Structural basis for amp binding to mammalian amp-activated protein kinase. Nature 2007, 449, 496–500. [Google Scholar] [CrossRef]
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.H.; Bardeesy, N.; Depinho, R.A.; Montminy, M.; Cantley, L.C. The kinase lkb1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005, 310, 1642–1646. [Google Scholar] [CrossRef]
- Legro, R.S.; Barnhart, H.X.; Schlaff, W.D.; Carr, B.R.; Diamond, M.P.; Carson, S.A.; Steinkampf, M.P.; Coutifaris, C.; McGovern, P.G.; Cataldo, N.A.; et al. Ovulatory response to treatment of polycystic ovary syndrome is associated with a polymorphism in the stk11 gene. J. Clin. Endocrinol. Metab. 2008, 93, 792–800. [Google Scholar]
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brule, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J.; et al. Metformin, independent of ampk, inhibits mtorc1 in a rag gtpase-dependent manner. Cell Metab. 2010, 11, 390–401. [Google Scholar] [CrossRef]
- Sun, Y.; Connors, K.E.; Yang, D.Q. Aicar induces phosphorylation of ampk in an atm-dependent, lkb1-independent manner. Mol. Cell. Biochem. 2007, 306, 239–245. [Google Scholar] [CrossRef]
- Suzuki, A.; Kusakai, G.; Kishimoto, A.; Shimojo, Y.; Ogura, T.; Lavin, M.F.; Esumi, H. Igf-1 phosphorylates ampk-alpha subunit in atm-dependent and lkb1-independent manner. Biochem. Biophys. Res. Commun. 2004, 324, 986–992. [Google Scholar] [CrossRef]
- Viollet, B.; Guigas, B.; Sanz Garcia, N.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 2012, 122, 253–270. [Google Scholar] [CrossRef] [Green Version]
- Graham, G.G.; Punt, J.; Arora, M.; Day, R.O.; Doogue, M.P.; Duong, J.K.; Furlong, T.J.; Greenfield, J.R.; Greenup, L.C.; Kirkpatrick, C.M.; et al. Clinical pharmacokinetics of metformin. Clin. Pharmacokinet. 2011, 50, 81–98. [Google Scholar] [CrossRef]
- Shu, Y.; Sheardown, S.A.; Brown, C.; Owen, R.P.; Zhang, S.; Castro, R.A.; Ianculescu, A.G.; Yue, L.; Lo, J.C.; Burchard, E.G.; et al. Effect of genetic variation in the organic cation transporter 1 (oct1) on metformin action. J. Clin. Invest. 2007, 117, 1422–1431. [Google Scholar] [CrossRef]
- Schaeffeler, E.; Hellerbrand, C.; Nies, A.T.; Winter, S.; Kruck, S.; Hofmann, U.; van der Kuip, H.; Zanger, U.M.; Koepsell, H.; Schwab, M. DNA methylation is associated with downregulation of the organic cation transporter oct1 (slc22a1) in human hepatocellular carcinoma. Genome Med. 2011, 3. [Google Scholar] [CrossRef]
- Kimura, N.; Masuda, S.; Tanihara, Y.; Ueo, H.; Okuda, M.; Katsura, T.; Inui, K. Metformin is a superior substrate for renal organic cation transporter oct2 rather than hepatic oct1. Drug Metab. Pharmacokinet. 2005, 20, 379–386. [Google Scholar] [CrossRef]
- Umehara, K.I.; Iwatsubo, T.; Noguchi, K.; Kamimura, H. Functional involvement of organic cation transporter1 (oct1/oct1) in the hepatic uptake of organic cations in humans and rats. Xenobiotica 2007, 37, 818–831. [Google Scholar] [CrossRef]
- Segal, E.D.; Yasmeen, A.; Beauchamp, M.C.; Rosenblatt, J.; Pollak, M.; Gotlieb, W.H. Relevance of the oct1 transporter to the antineoplastic effect of biguanides. Biochem. Biophys. Res. Commun. 2011, 414, 694–699. [Google Scholar] [CrossRef]
- Nies, A.T.; Koepsell, H.; Damme, K.; Schwab, M. Organic cation transporters (octs, mates), in vitro and in vivo evidence for the importance in drug therapy. Handb. Exp. Pharmacol. 2011, 201, 105–167. [Google Scholar] [CrossRef]
- Becker, M.L.; Visser, L.E.; van Schaik, R.H.; Hofman, A.; Uitterlinden, A.G.; Stricker, B.H. Genetic variation in the organic cation transporter 1 is associated with metformin response in patients with diabetes mellitus. Pharmacogenomics J. 2009, 9, 242–247. [Google Scholar] [CrossRef]
- Wang, Z.J.; Yin, O.Q.; Tomlinson, B.; Chow, M.S. Oct2 polymorphisms and in vivo renal functional consequence: Studies with metformin and cimetidine. Pharmacogenet. Genomics 2008, 18, 637–645. [Google Scholar] [CrossRef]
- Zhou, K.; Bellenguez, C.; Sutherland, C.; Hardie, G.; Palmer, C.; Donnelly, P.; Pearson, E. The role of atm in response to metformin treatment and activation of ampk. Nat. Genet. 2012, 44, 361–362. [Google Scholar] [CrossRef]
- Brunet, J.; Vazquez-Martin, A.; Colomer, R.; Grana-Suarez, B.; Martin-Castillo, B.; Menendez, J.A. Brca1 and acetyl-coa carboxylase: The metabolic syndrome of breast cancer. Mol. Carcinog. 2008, 47, 157–163. [Google Scholar] [CrossRef]
- Sakamoto, K.; McCarthy, A.; Smith, D.; Green, K.A.; Grahame Hardie, D.; Ashworth, A.; Alessi, D.R. Deficiency of lkb1 in skeletal muscle prevents ampk activation and glucose uptake during contraction. EMBO J. 2005, 24, 1810–1820. [Google Scholar] [CrossRef]
- Hay, N.; Sonenberg, N. Upstream and downstream of mtor. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef]
- Dowling, R.J.; Zakikhani, M.; Fantus, I.G.; Pollak, M.; Sonenberg, N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007, 67, 10804–10812. [Google Scholar] [CrossRef]
- Inoki, K.; Corradetti, M.N.; Guan, K.L. Dysregulation of the tsc-mtor pathway in human disease. Nat. Genet. 2005, 37, 19–24. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.L. Tsc2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. Ampk phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef]
- Woodard, J.; Joshi, S.; Viollet, B.; Hay, N.; Platanias, L.C. Ampk as a therapeutic target in renal cell carcinoma. Cancer Biol. Ther. 2010, 10, 1168–1177. [Google Scholar] [CrossRef]
- Wurth, R.; Pattarozzi, A.; Gatti, M.; Bajetto, A.; Corsaro, A.; Parodi, A.; Sirito, R.; Massollo, M.; Marini, C.; Zona, G.; et al. Metformin selectively affects human glioblastoma tumor-initiating cell viability: A role for metformin-induced inhibition of akt. Cell Cycle 2013, 12, 145–156. [Google Scholar] [CrossRef]
- Na, H.J.; Park, J.S.; Pyo, J.H.; Lee, S.H.; Jeon, H.J.; Kim, Y.S.; Yoo, M.A. Mechanism of metformin: Inhibition of DNA damage and proliferative activity in drosophila midgut stem cell. Mech. Ageing Dev. 2013, 9, 381–390. [Google Scholar]
- Samarajeewa, N.U.; Ham, S.; Yang, F.; Simpson, E.R.; Brown, K.A. Promoter-specific effects of metformin on aromatase transcript expression. Steroids 2011, 76, 768–771. [Google Scholar] [CrossRef]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. Amp-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef]
- Cerezo, M.; Tichet, M.; Abbe, P.; Ohanna, M.; Lehraiki, A.; Rouaud, F.; Allegra, M.; Giacchero, D.; Bahadoran, P.; Bertolotto, C.; et al. Metformin blocks melanoma invasion and metastasis development in ampk/p53-dependent manner. Mol. Cancer Ther. 2013, 12, 1605–1615. [Google Scholar] [CrossRef]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. Ras is regulated by the let-7 microrna family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The emt-activator zeb1 promotes tumorigenicity by repressing stemness-inhibiting micrornas. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef]
- Bao, B.; Wang, Z.; Ali, S.; Ahmad, A.; Azmi, A.S.; Sarkar, S.H.; Banerjee, S.; Kong, D.; Li, Y.; Thakur, S.; et al. Metformin inhibits cell proliferation, migration and invasion by attenuating csc function mediated by deregulating mirnas in pancreatic cancer cells. Cancer Prev. Res. 2012, 5, 355–364. [Google Scholar] [CrossRef]
- Li, W.; Yuan, Y.; Huang, L.; Qiao, M.; Zhang, Y. Metformin alters the expression profiles of micrornas in human pancreatic cancer cells. Diabetes Res. Clin. Pract. 2012, 96, 187–195. [Google Scholar] [CrossRef]
- Kobayashi, M.; Kato, K.; Iwama, H.; Fujihara, S.; Nishiyama, N.; Mimura, S.; Toyota, Y.; Nomura, T.; Nomura, K.; Tani, J.; et al. Antitumor effect of metformin in esophageal cancer: In vitro study. Int. J. Oncol. 2013, 42, 517–524. [Google Scholar]
- Kato, K.; Gong, J.; Iwama, H.; Kitanaka, A.; Tani, J.; Miyoshi, H.; Nomura, K.; Mimura, S.; Kobayashi, M.; Aritomo, Y.; et al. The antidiabetic drug metformin inhibits gastric cancer cell proliferation in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 549–560. [Google Scholar] [CrossRef]
- Avci, C.B.; Harman, E.; Dodurga, Y.; Susluer, S.Y.; Gunduz, C. Therapeutic potential of an anti-diabetic drug, metformin: Alteration of mirna expression in prostate cancer cells. Asian Pac. J. Cancer Prev. 2013, 14, 765–768. [Google Scholar] [CrossRef]
- Cufi, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Quirantes, R.; Segura-Carretero, A.; Micol, V.; Joven, J.; Bosch-Barrera, J.; Del Barco, S.; Martin-Castillo, B.; et al. Metformin lowers the threshold for stress-induced senescence: A role for the microrna-200 family and mir-205. Cell Cycle 2012, 11, 1235–1246. [Google Scholar] [CrossRef]
- Pawelczyk, L.; Spaczynski, R.Z.; Banaszewska, B.; Duleba, A.J. Metformin therapy increases insulin-like growth factor binding protein-1 in hyperinsulinemic women with polycystic ovary syndrome. Eur. J. Obstet. Gynecol. Reprod. Biol. 2004, 113, 209–213. [Google Scholar] [CrossRef]
- Adashi, E.Y. The igf family and folliculogenesis. J. Reprod. Immunol. 1998, 39, 13–19. [Google Scholar] [CrossRef]
- Smith, M.R.; Lee, H.; Nathan, D.M. Insulin sensitivity during combined androgen blockade for prostate cancer. J. Clin. Endocrinol. Metab. 2006, 91, 1305–1308. [Google Scholar] [CrossRef]
- Warshamana-Greene, G.S.; Litz, J.; Buchdunger, E.; Garcia-Echeverria, C.; Hofmann, F.; Krystal, G.W. The insulin-like growth factor-i receptor kinase inhibitor, nvp-adw742, sensitizes small cell lung cancer cell lines to the effects of chemotherapy. Clin. Cancer Res. 2005, 11, 1563–1571. [Google Scholar] [CrossRef]
- Quinn, B.J.; Dallos, M.; Kitagawa, H.; Kunnumakkara, A.B.; Memmott, R.M.; Hollander, M.C.; Gills, J.J.; Dennis, P.A. Inhibition of lung tumorigenesis by metformin is associated with decreased plasma igf-i and diminished receptor tyrosine kinase signaling. Cancer Prev. Res. 2013, 6, 801–810. [Google Scholar] [CrossRef]
- Memmott, R.M.; Mercado, J.R.; Maier, C.R.; Kawabata, S.; Fox, S.D.; Dennis, P.A. Metformin prevents tobacco carcinogen—induced lung tumorigenesis. Cancer Prev. Res. 2010, 3, 1066–1076. [Google Scholar] [CrossRef]
- Arai, M.; Uchiba, M.; Komura, H.; Mizuochi, Y.; Harada, N.; Okajima, K. Metformin, an antidiabetic agent, suppresses the production of tumor necrosis factor and tissue factor by inhibiting early growth response factor-1 expression in human monocytes in vitro. J. Pharmacol. Exp. Ther. 2010, 334, 206–213. [Google Scholar] [CrossRef]
- Algire, C.; Moiseeva, O.; Deschenes-Simard, X.; Amrein, L.; Petruccelli, L.; Birman, E.; Viollet, B.; Ferbeyre, G.; Pollak, M.N. Metformin reduces endogenous reactive oxygen species and associated DNA damage. Cancer Prev. Res. 2012, 5, 536–543. [Google Scholar] [CrossRef]
- Menendez, J.A.; Cufi, S.; Oliveras-Ferraros, C.; Martin-Castillo, B.; Joven, J.; Vellon, L.; Vazquez-Martin, A. Metformin and the atm DNA damage response (ddr): Accelerating the onset of stress-induced senescence to boost protection against cancer. Aging 2011, 3, 1063–1077. [Google Scholar]
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Cufi, S.; Martin-Castillo, B.; Menendez, J.A. Metformin activates an ataxia telangiectasia mutated (atm)/chk2-regulated DNA damage-like response. Cell Cycle 2011, 10, 1499–1501. [Google Scholar] [CrossRef]
- Ma, Y.; Guo, F.C.; Wang, W.; Shi, H.S.; Li, D.; Wang, Y.S. Kras gene mutation as a predictor of cancer cell responsiveness to metformin. Mol. Med. Rep. 2013, 8, 763–768. [Google Scholar]
- Do, M.T.; Kim, H.G.; Khanal, T.; Choi, J.H.; Kim, D.H.; Jeong, T.C.; Jeong, H.G. Metformin inhibits heme oxygenase-1 expression in cancer cells through inactivation of raf-erk-nrf2 signaling and ampk-independent pathways. Toxicol. Appl. Pharmacol. 2013, 271, 229–238. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res. 2011, 71, 3196–3201. [Google Scholar] [CrossRef]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef]
- Ning, X.; Shu, J.; Du, Y.; Ben, Q.; Li, Z. Therapeutic strategies targeting cancer stem cells. Cancer Biol. Ther. 2013, 14, 295–303. [Google Scholar] [CrossRef]
- Sanli, T.; Rashid, A.; Liu, C.; Harding, S.; Bristow, R.G.; Cutz, J.C.; Singh, G.; Wright, J.; Tsakiridis, T. Ionizing radiation activates amp-activated kinase (ampk): A target for radiosensitization of human cancer cells. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 221–229. [Google Scholar] [CrossRef]
- Storozhuk, Y.; Hopmans, S.N.; Sanli, T.; Barron, C.; Tsiani, E.; Cutz, J.C.; Pond, G.; Wright, J.; Singh, G.; Tsakiridis, T. Metformin inhibits growth and enhances radiation response of non-small cell lung cancer (nsclc) through atm and ampk. Br. J. Cancer 2013, 108, 2021–2032. [Google Scholar] [CrossRef]
- Song, C.W.; Lee, H.; Dings, R.P.; Williams, B.; Powers, J.; Santos, T.D.; Choi, B.H.; Park, H.J. Metformin kills and radiosensitizes cancer cells and preferentially kills cancer stem cells. Sci. Rep. 2012, 2. [Google Scholar] [CrossRef]
- Ruiter, R.; Visser, L.E.; van Herk-Sukel, M.P.; Coebergh, J.W.; Haak, H.R.; Geelhoed-Duijvestijn, P.H.; Straus, S.M.; Herings, R.M.; Stricker, B.H. Lower risk of cancer in patients on metformin in comparison with those on sulfonylurea derivatives: Results from a large population-based follow-up study. Diabetes Care 2012, 35, 119–124. [Google Scholar] [CrossRef]
- Currie, C.J.; Poole, C.D.; Jenkins-Jones, S.; Gale, E.A.; Johnson, J.A.; Morgan, C.L. Mortality after incident cancer in people with and without type 2 diabetes: Impact of metformin on survival. Diabetes Care 2012, 35, 299–304. [Google Scholar] [CrossRef]
- Home, P.D.; Kahn, S.E.; Jones, N.P.; Noronha, D.; Beck-Nielsen, H.; Viberti, G.; Group, A.S.; Committee, R.S. Experience of malignancies with oral glucose-lowering drugs in the randomised controlled adopt (a diabetes outcome progression trial) and record (rosiglitazone evaluated for cardiovascular outcomes and regulation of glycaemia in diabetes) clinical trials. Diabetologia 2010, 53, 1838–1845. [Google Scholar] [CrossRef]
- Franciosi, M.; Lucisano, G.; Lapice, E.; Strippoli, G.F.; Pellegrini, F.; Nicolucci, A. Metformin therapy and risk of cancer in patients with type 2 diabetes: Systematic review. PLoS One 2013, 8, e71583. [Google Scholar]
- Thakkar, B.; Aronis, K.N.; Vamvini, M.T.; Shields, K.; Mantzoros, C.S. Metformin and sulfonylureas in relation to cancer risk in type ii diabetes patients: A meta-analysis using primary data of published studies. Metabolism 2013, 62, 922–934. [Google Scholar] [CrossRef]
- Stevens, R.J.; Ali, R.; Bankhead, C.R.; Bethel, M.A.; Cairns, B.J.; Camisasca, R.P.; Crowe, F.L.; Farmer, A.J.; Harrison, S.; Hirst, J.A.; et al. Cancer outcomes and all-cause mortality in adults allocated to metformin: Systematic review and collaborative meta-analysis of randomised clinical trials. Diabetologia 2012, 55, 2593–2603. [Google Scholar] [CrossRef]
- Soranna, D.; Scotti, L.; Zambon, A.; Bosetti, C.; Grassi, G.; Catapano, A.; La Vecchia, C.; Mancia, G.; Corrao, G. Cancer risk associated with use of metformin and sulfonylurea in type 2 diabetes: A meta-analysis. Oncologist 2012, 17, 813–822. [Google Scholar] [CrossRef]
- Noto, H.; Goto, A.; Tsujimoto, T.; Noda, M. Cancer risk in diabetic patients treated with metformin: A systematic review and meta-analysis. PLoS One 2012, 7, e33411. [Google Scholar]
- Suissa, S.; Azoulay, L. Metformin and the risk of cancer: Time-related biases in observational studies. Diabetes Care 2012, 35, 2665–2673. [Google Scholar] [CrossRef]
- Zhang, Z.J.; Zheng, Z.J.; Kan, H.; Song, Y.; Cui, W.; Zhao, G.; Kip, K.E. Reduced risk of colorectal cancer with metformin therapy in patients with type 2 diabetes: A meta-analysis. Diabetes Care 2011, 34, 2323–2328. [Google Scholar] [CrossRef]
- Lai, S.W.; Liao, K.F.; Chen, P.C.; Tsai, P.Y.; Hsieh, D.P.; Chen, C.C. Antidiabetes drugs correlate with decreased risk of lung cancer: A population-based observation in taiwan. Clin. Lung Cancer 2012, 13, 143–148. [Google Scholar] [CrossRef]
- Zhang, Z.J.; Zheng, Z.J.; Shi, R.; Su, Q.; Jiang, Q.; Kip, K.E. Metformin for liver cancer prevention in patients with type 2 diabetes: A systematic review and meta-analysis. J. Clin. Endocrinol. Metab. 2012, 97, 2347–2353. [Google Scholar] [CrossRef]
- Chlebowski, R.T.; McTiernan, A.; Wactawski-Wende, J.; Manson, J.E.; Aragaki, A.K.; Rohan, T.; Ipp, E.; Kaklamani, V.G.; Vitolins, M.; Wallace, R.; et al. Diabetes, metformin, and breast cancer in postmenopausal women. J. Clin. Oncol. 2012, 30, 2844–2852. [Google Scholar] [CrossRef]
- Bosco, J.L.; Antonsen, S.; Sorensen, H.T.; Pedersen, L.; Lash, T.L. Metformin and incident breast cancer among diabetic women: A population-based case-control study in denmark. Cancer Epidemiol. Biomark. Prev. 2011, 20, 101–111. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pulito, C.; Sanli, T.; Rana, P.; Muti, P.; Blandino, G.; Strano, S. Metformin: On Ongoing Journey across Diabetes, Cancer Therapy and Prevention. Metabolites 2013, 3, 1051-1075. https://doi.org/10.3390/metabo3041051
Pulito C, Sanli T, Rana P, Muti P, Blandino G, Strano S. Metformin: On Ongoing Journey across Diabetes, Cancer Therapy and Prevention. Metabolites. 2013; 3(4):1051-1075. https://doi.org/10.3390/metabo3041051
Chicago/Turabian StylePulito, Claudio, Toran Sanli, Punam Rana, Paola Muti, Giovanni Blandino, and Sabrina Strano. 2013. "Metformin: On Ongoing Journey across Diabetes, Cancer Therapy and Prevention" Metabolites 3, no. 4: 1051-1075. https://doi.org/10.3390/metabo3041051