
Metabolic Investigations of the Molecular Mechanisms Associated with Parkinson’s Disease

 ,
,  ,
,  ,
,  and
and
Abstract
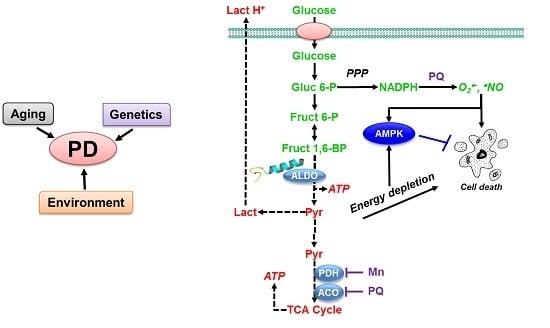
:
1. Introduction
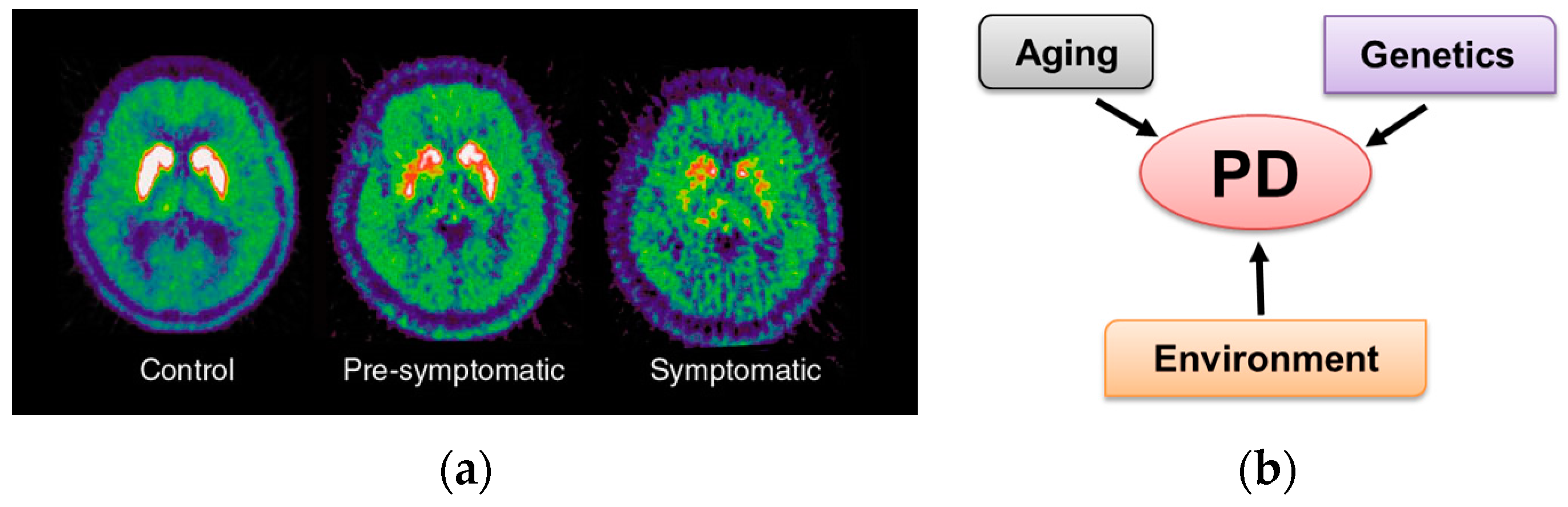
1.1. Parkinson’s Disease Overview
1.2. PD and Environmental Risk Factors
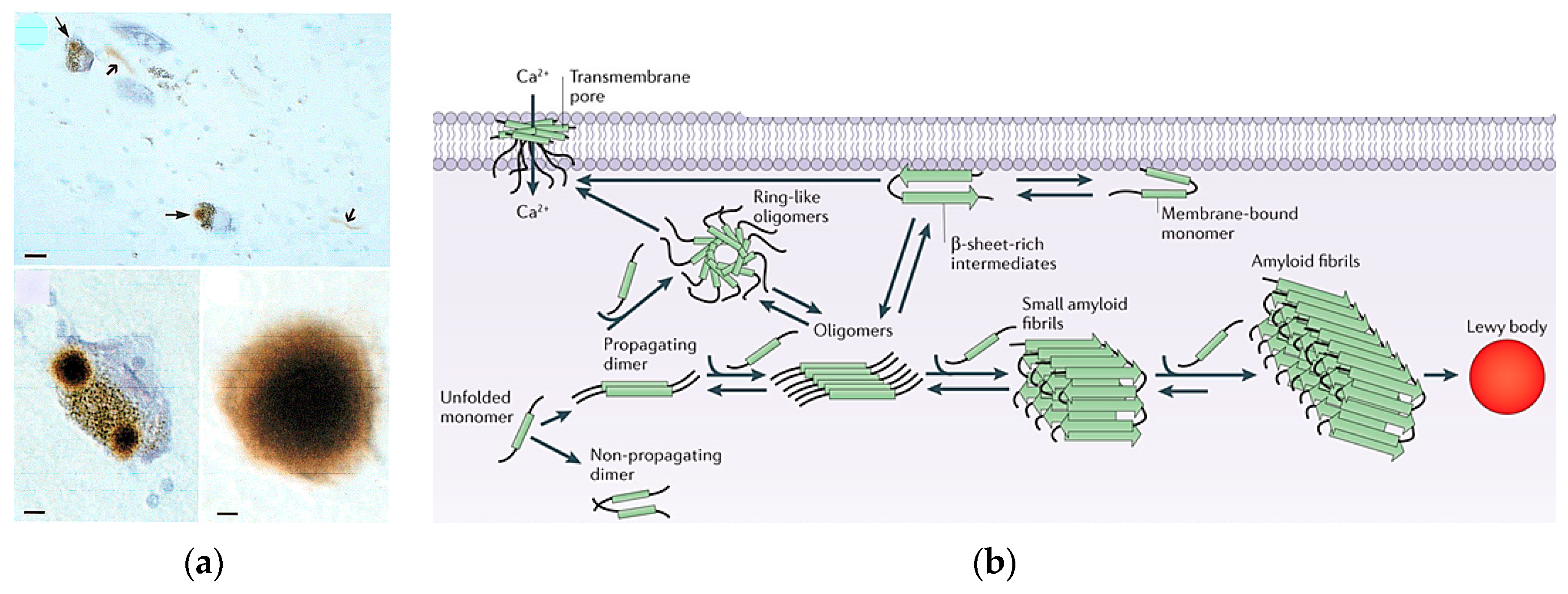
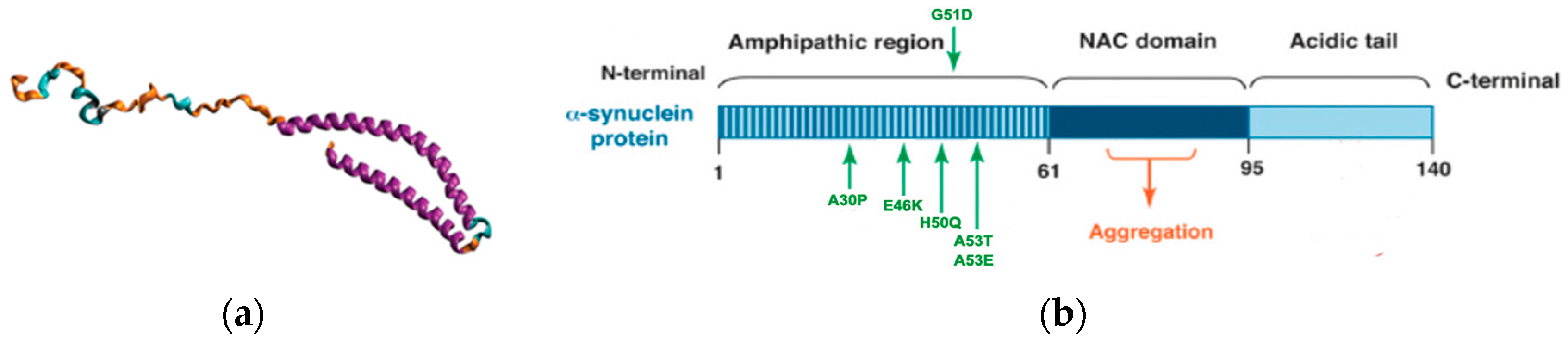
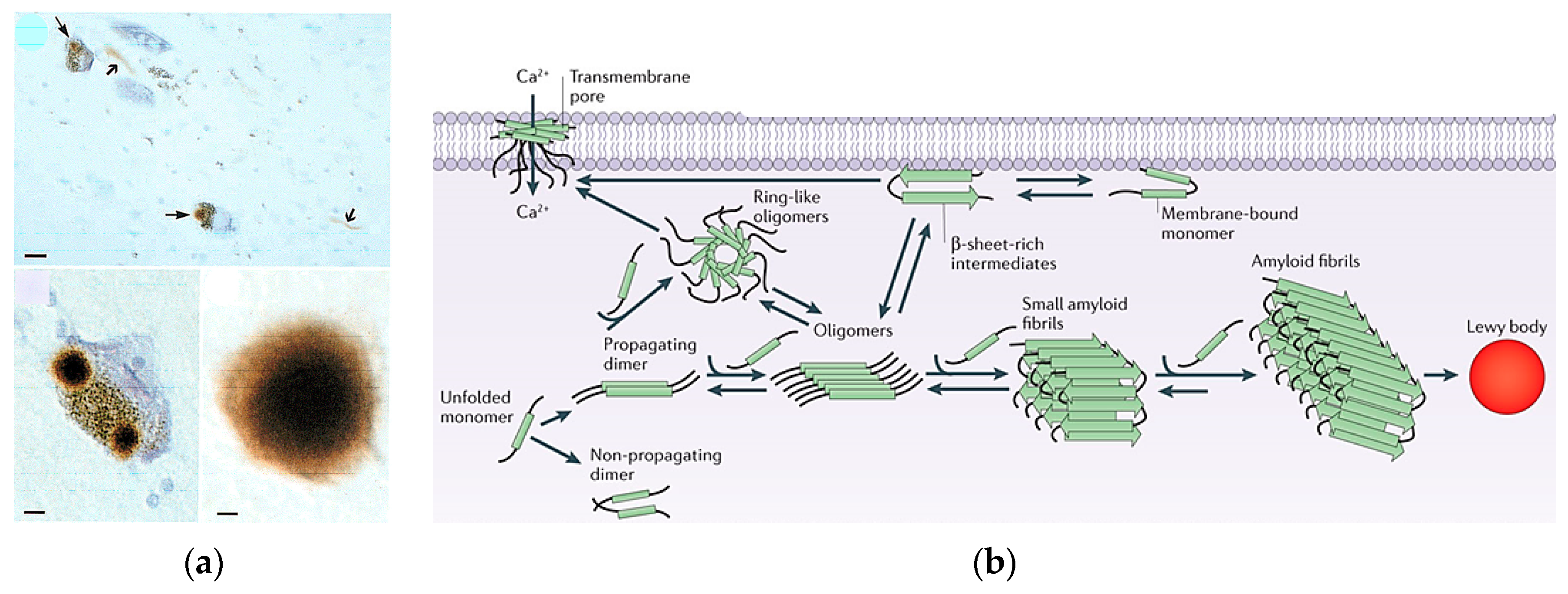
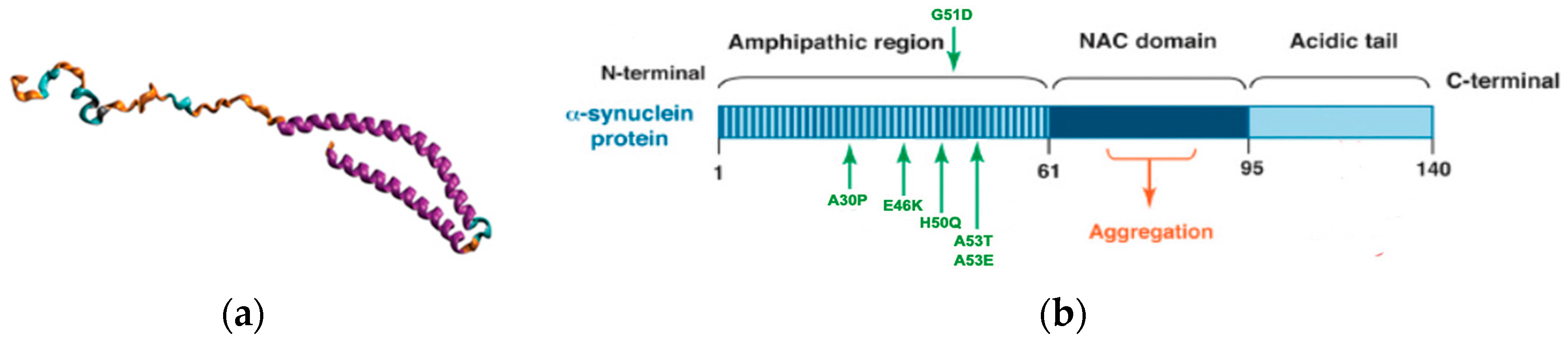
1.3. Lewy Bodies and α-Synuclein
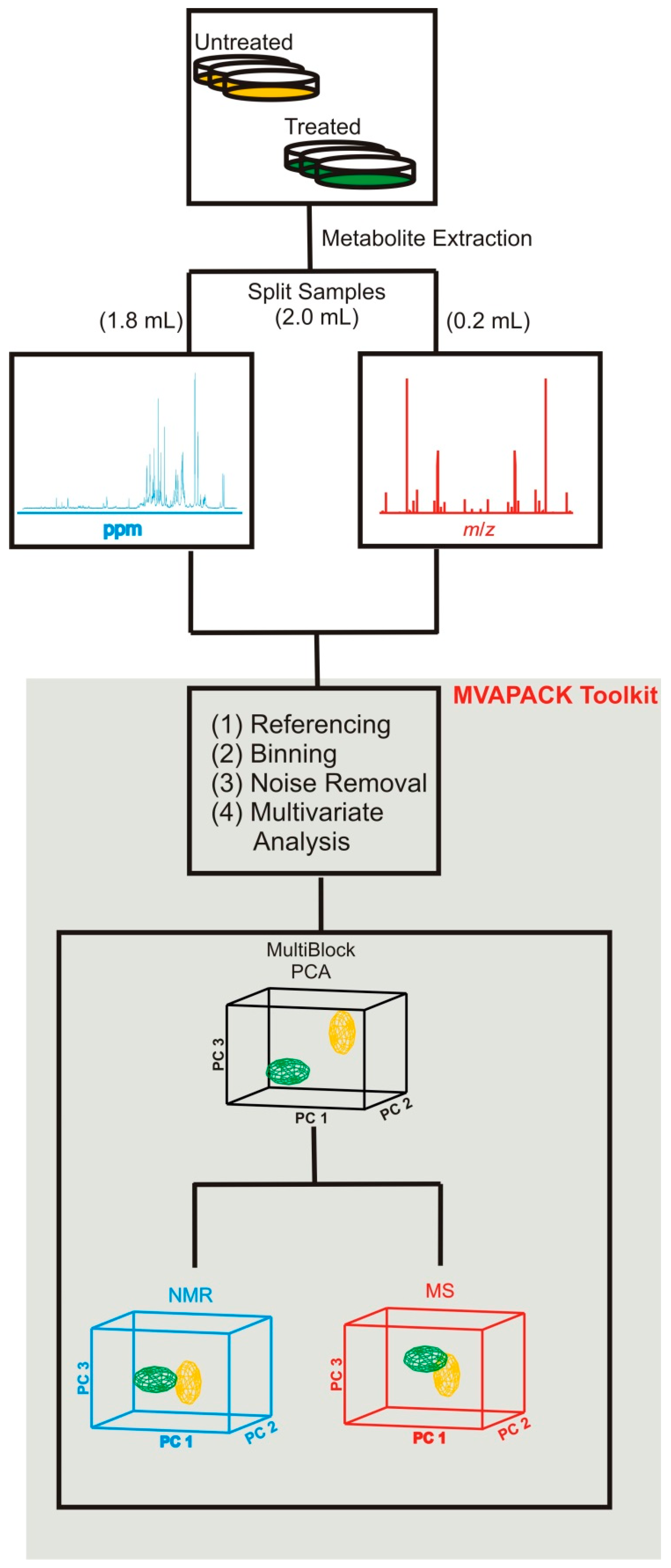
2. NMR and MS Metabolomics Protocol to Investigate PD
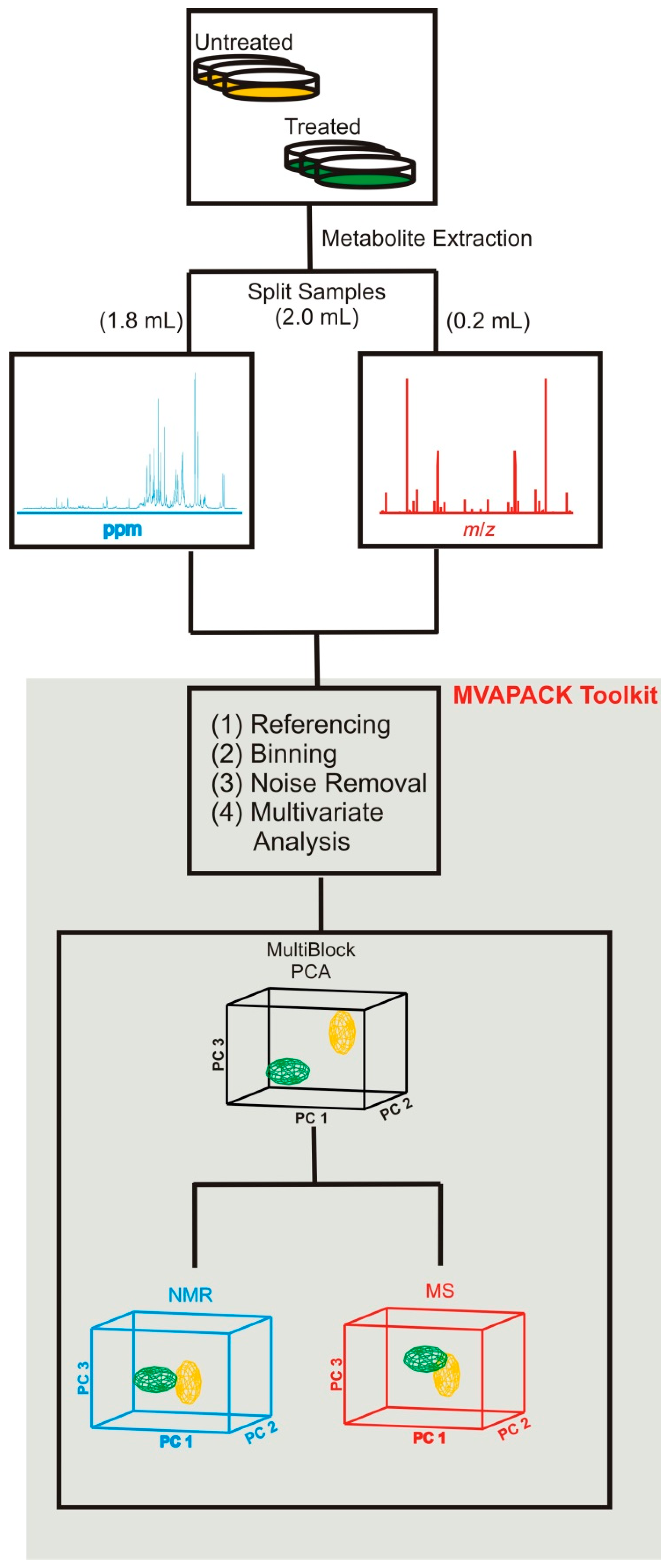
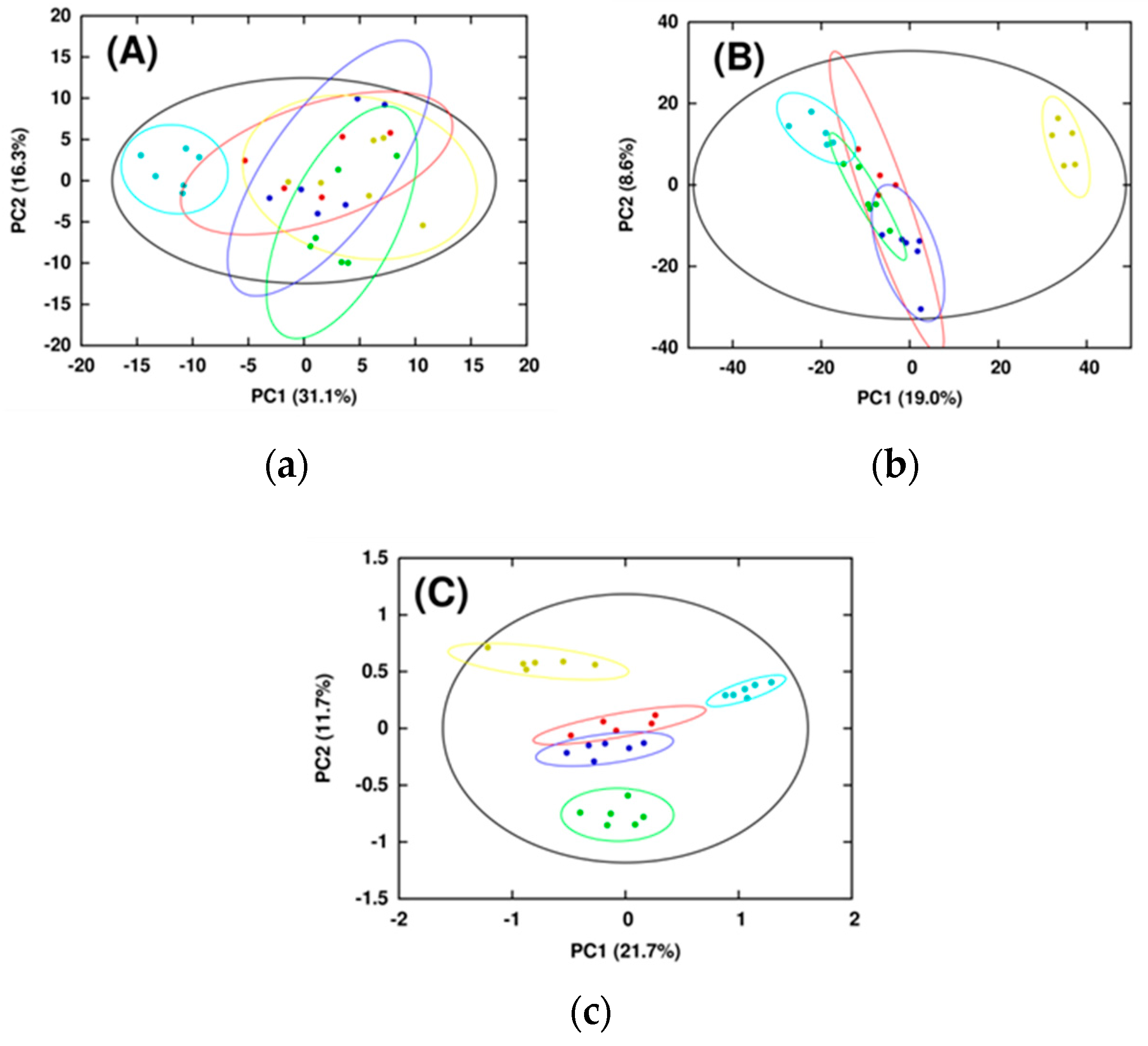
2.1. Combining NMR and MS Improves Coverage of the Metabolome
2.2. Combined NMR and MS Metabolomics Methodology
3. PD and Mitochondrial/Environmental Toxins
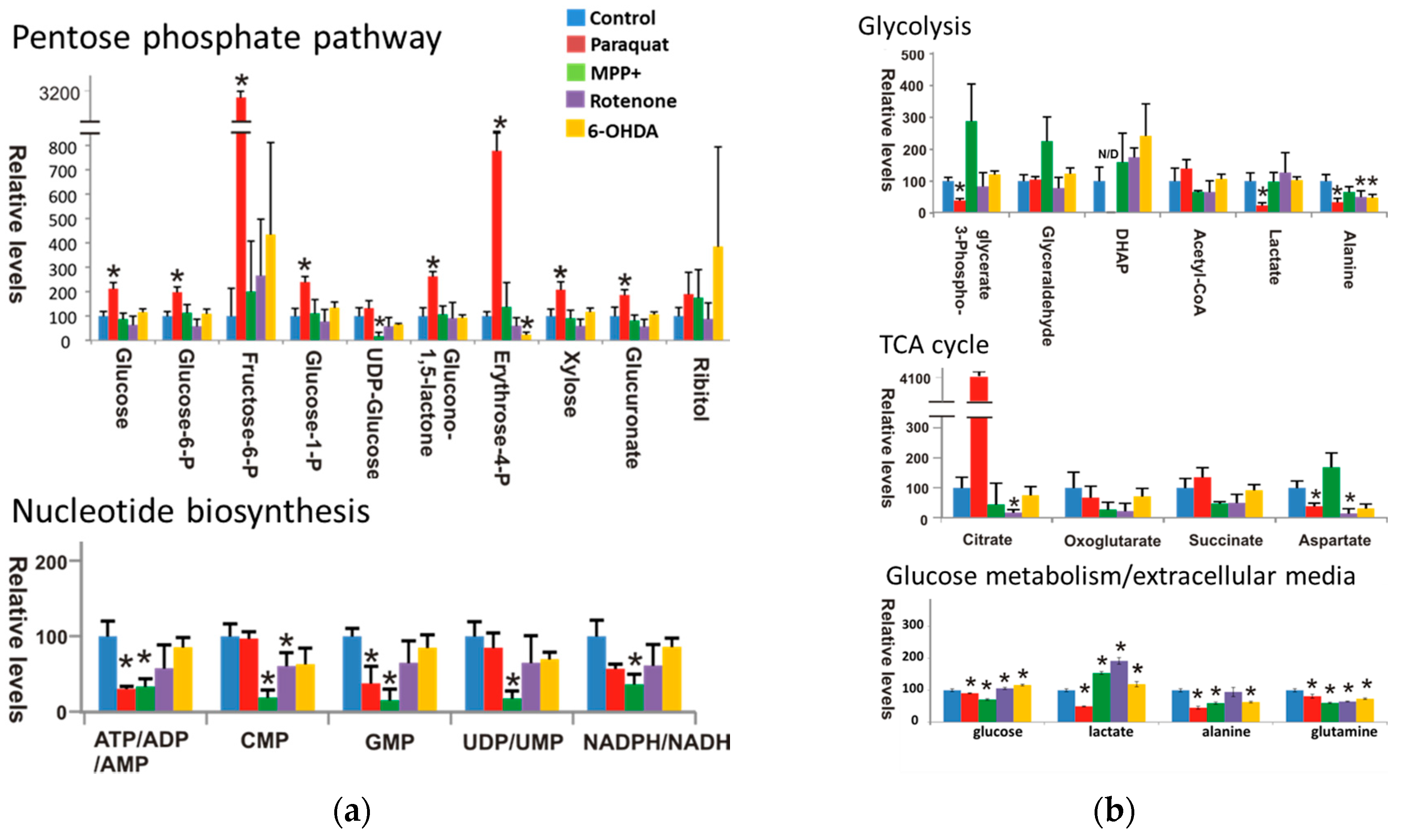
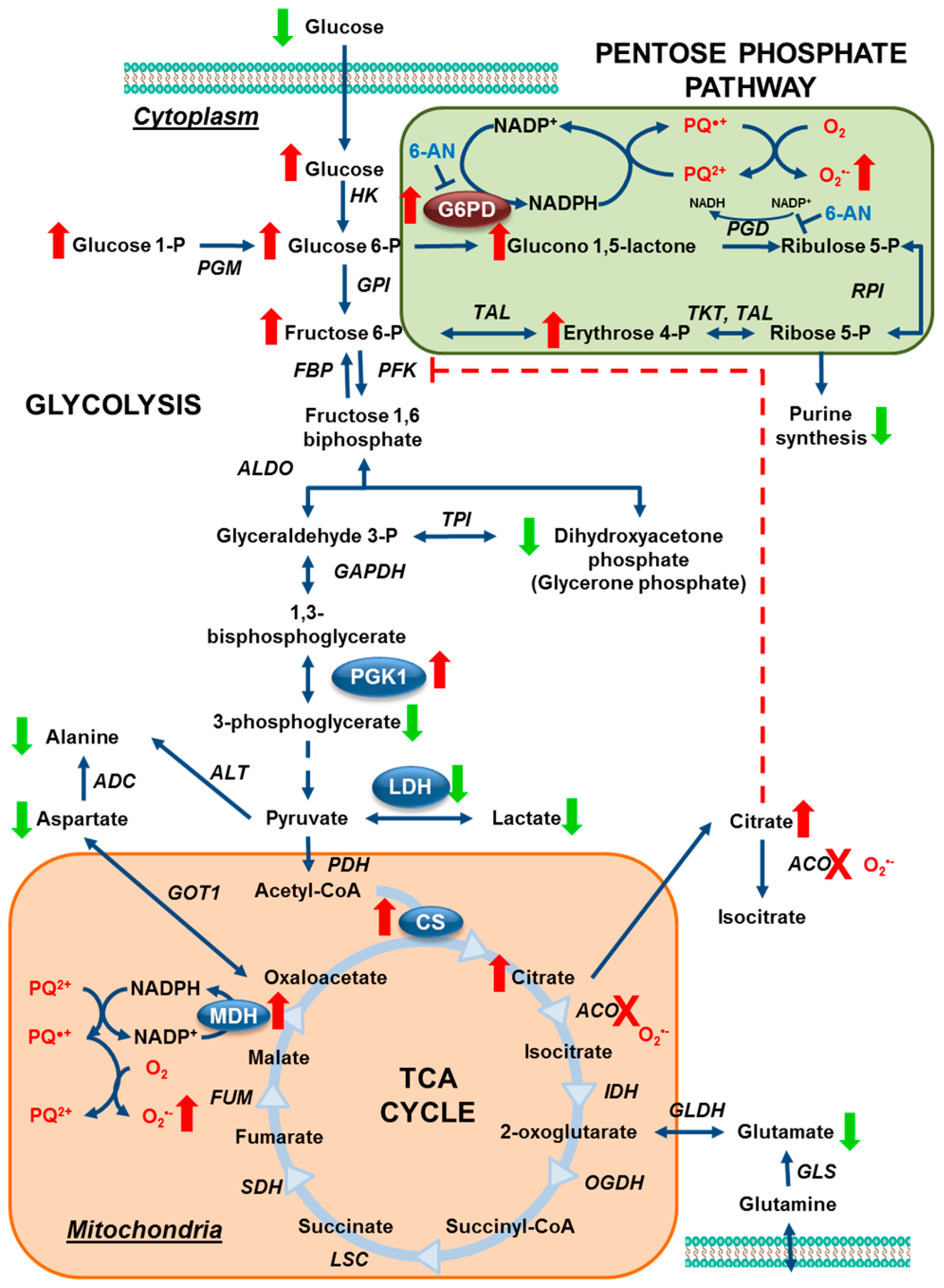
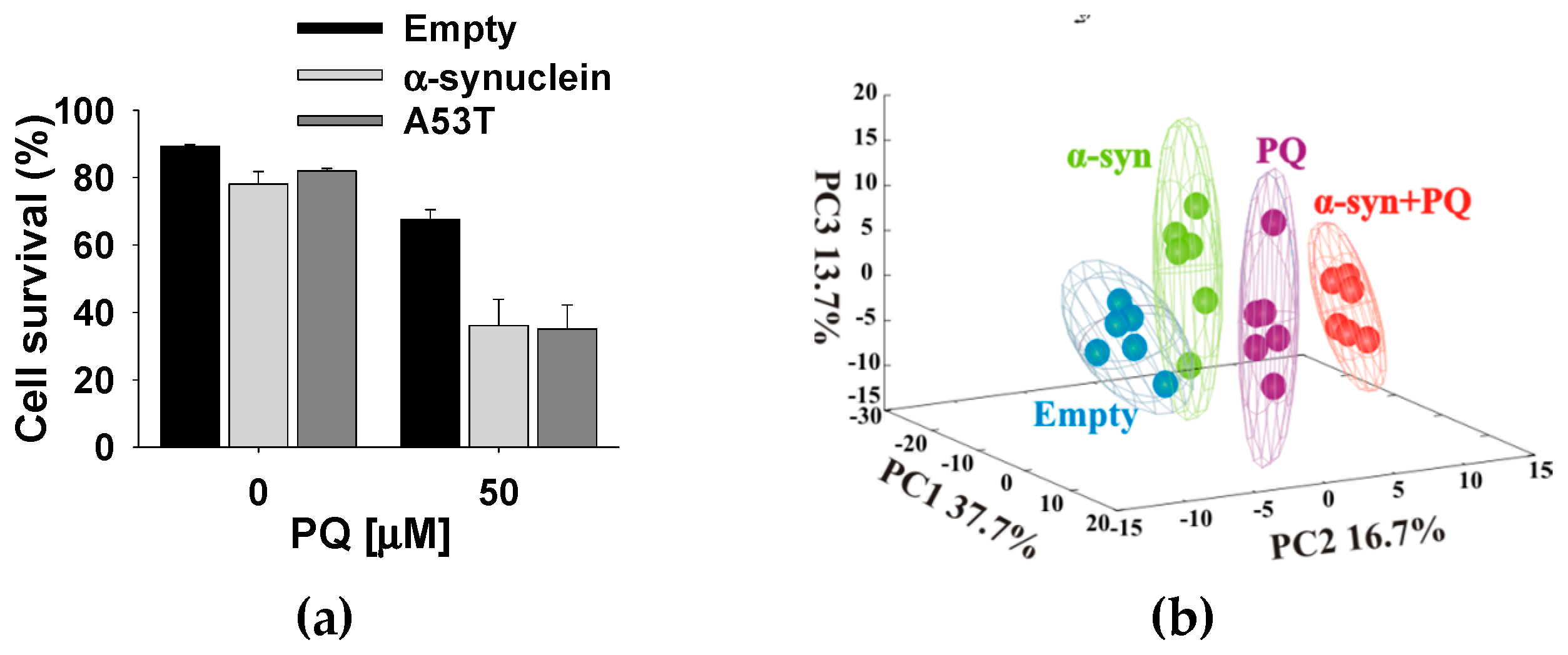
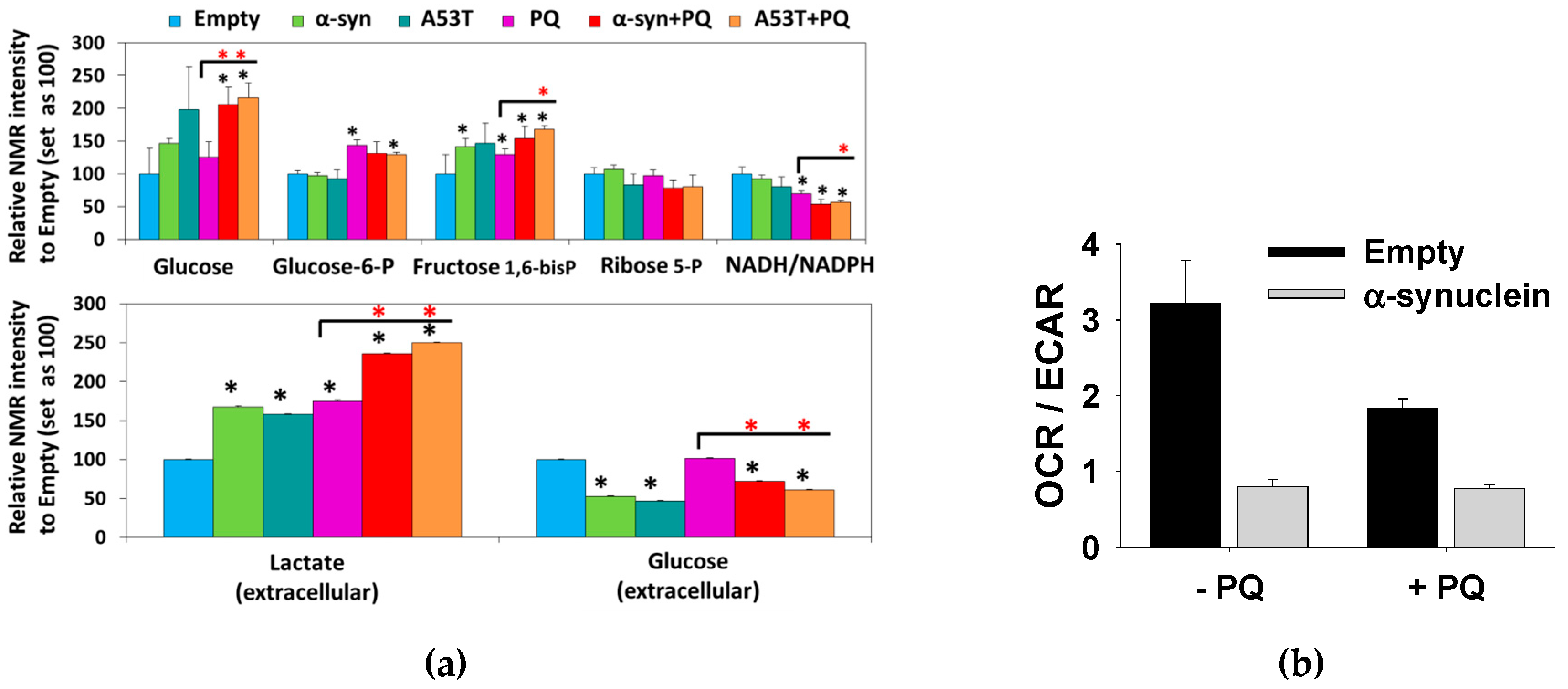
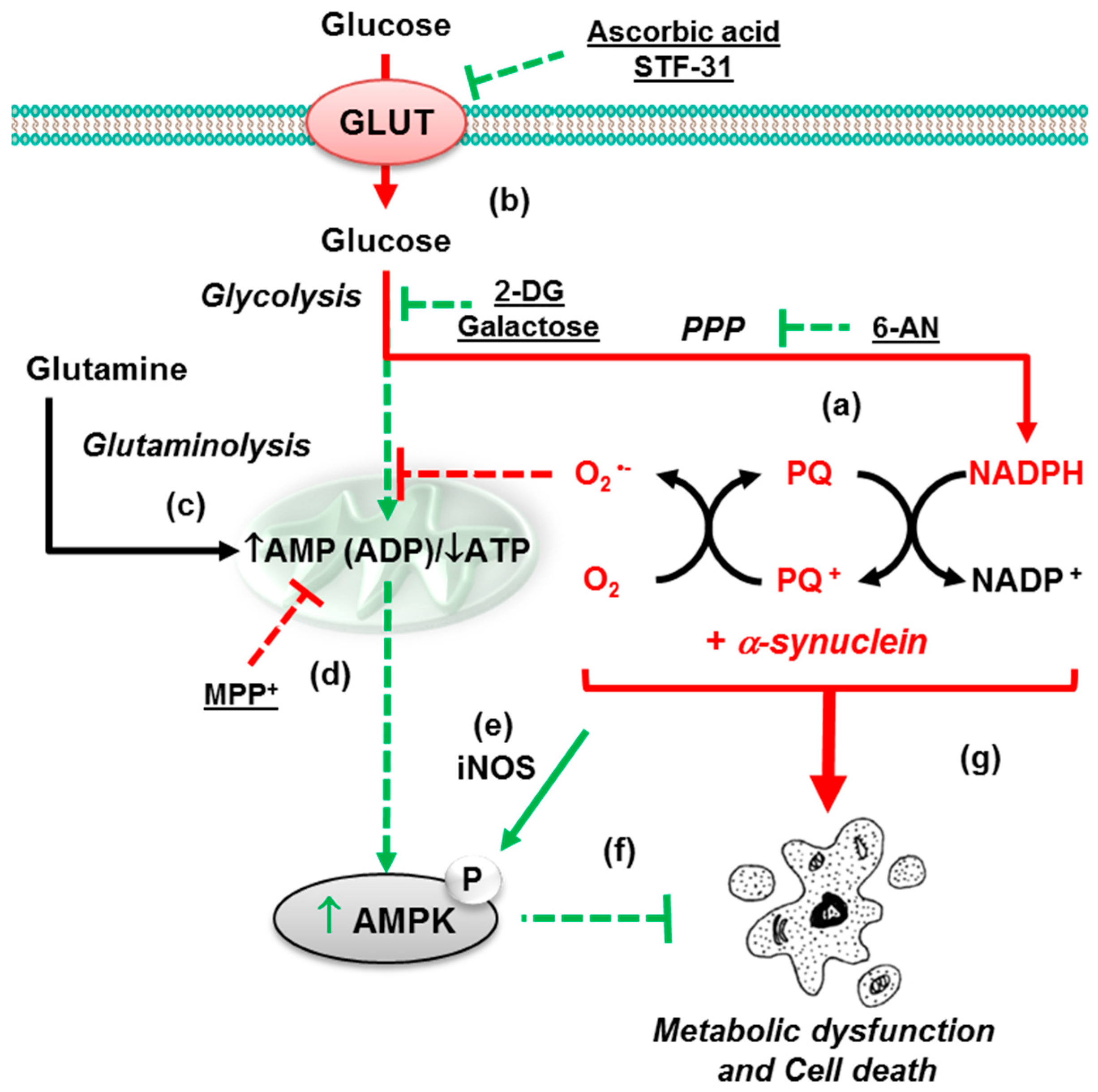
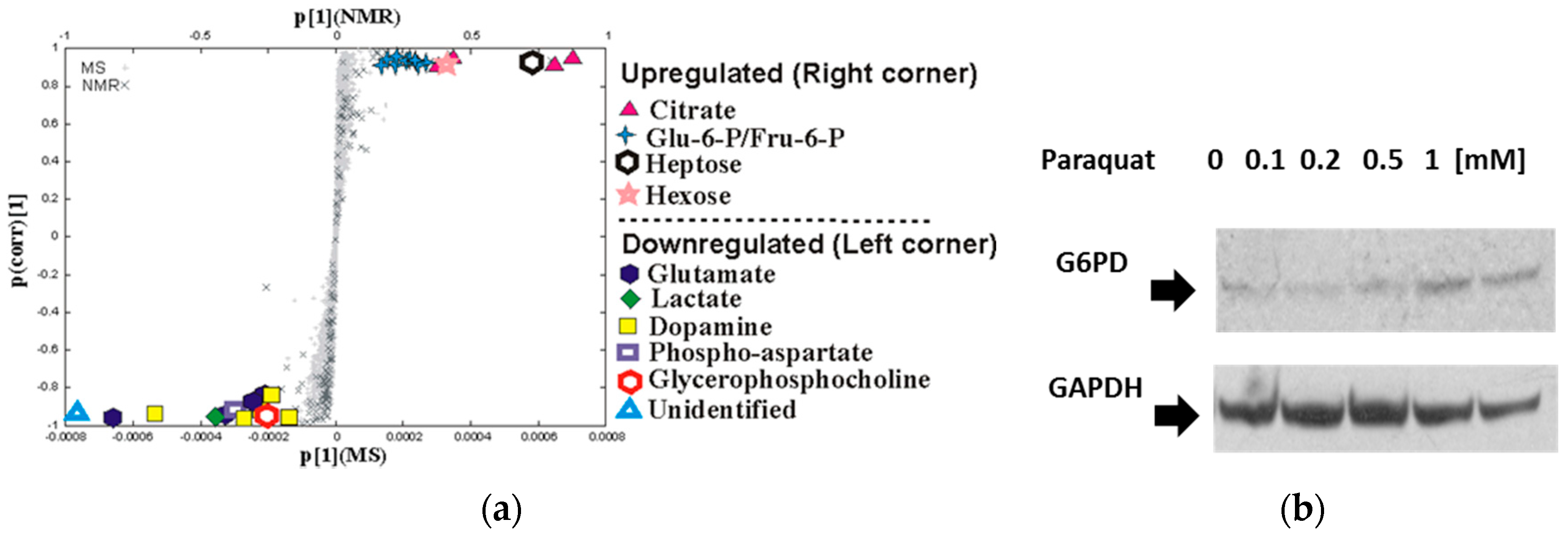
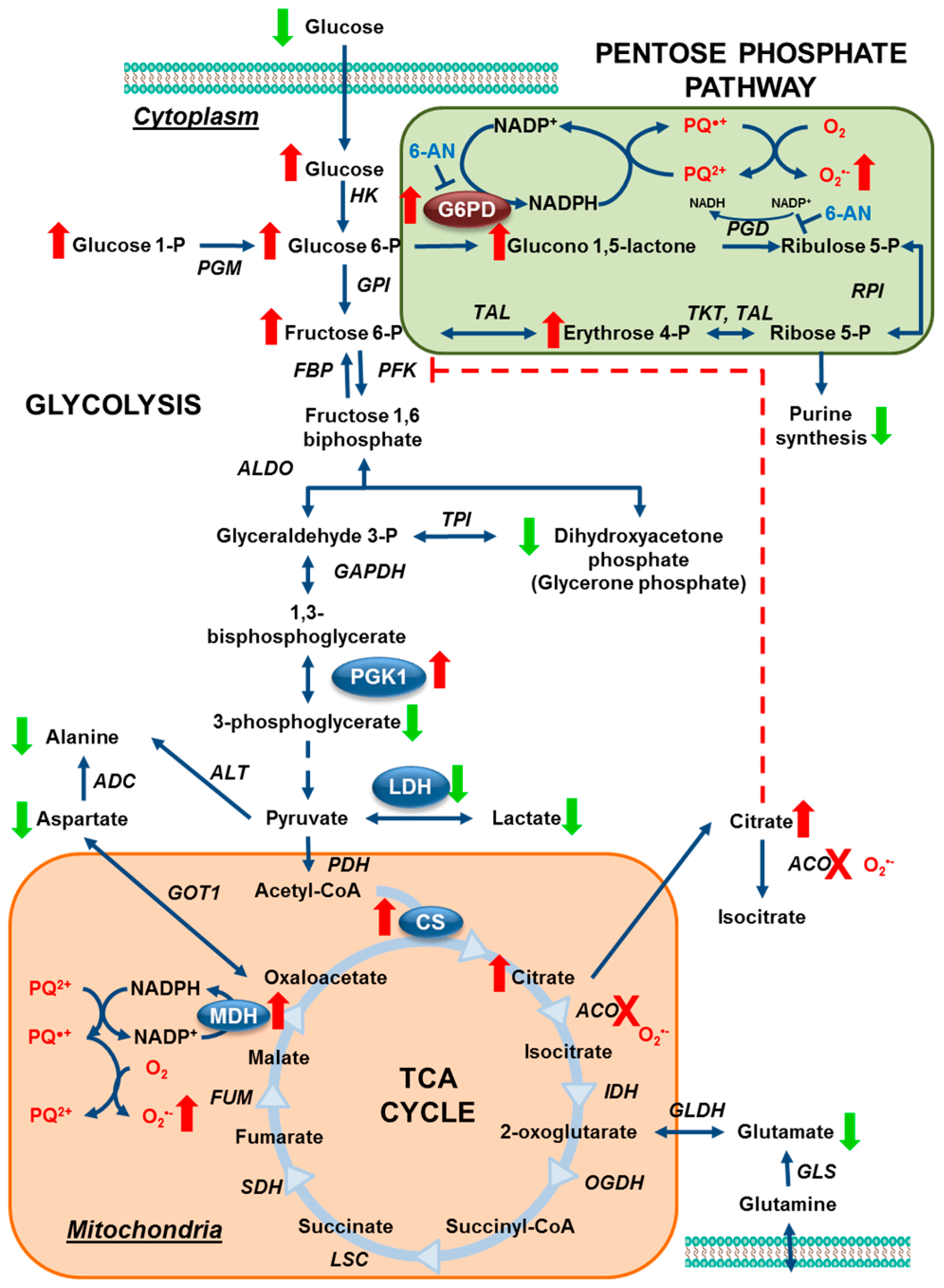
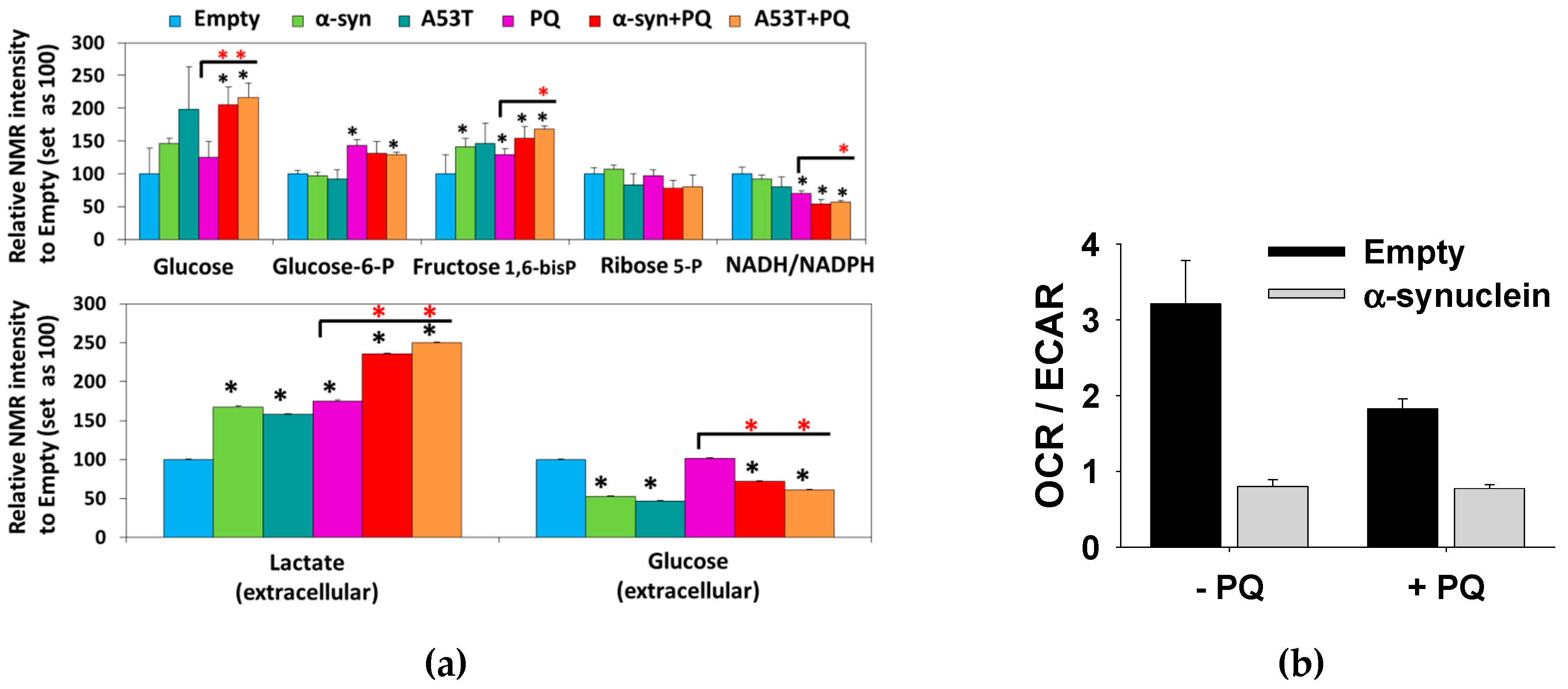
3.1. Paraquat Induces Unique Metabolic Changes
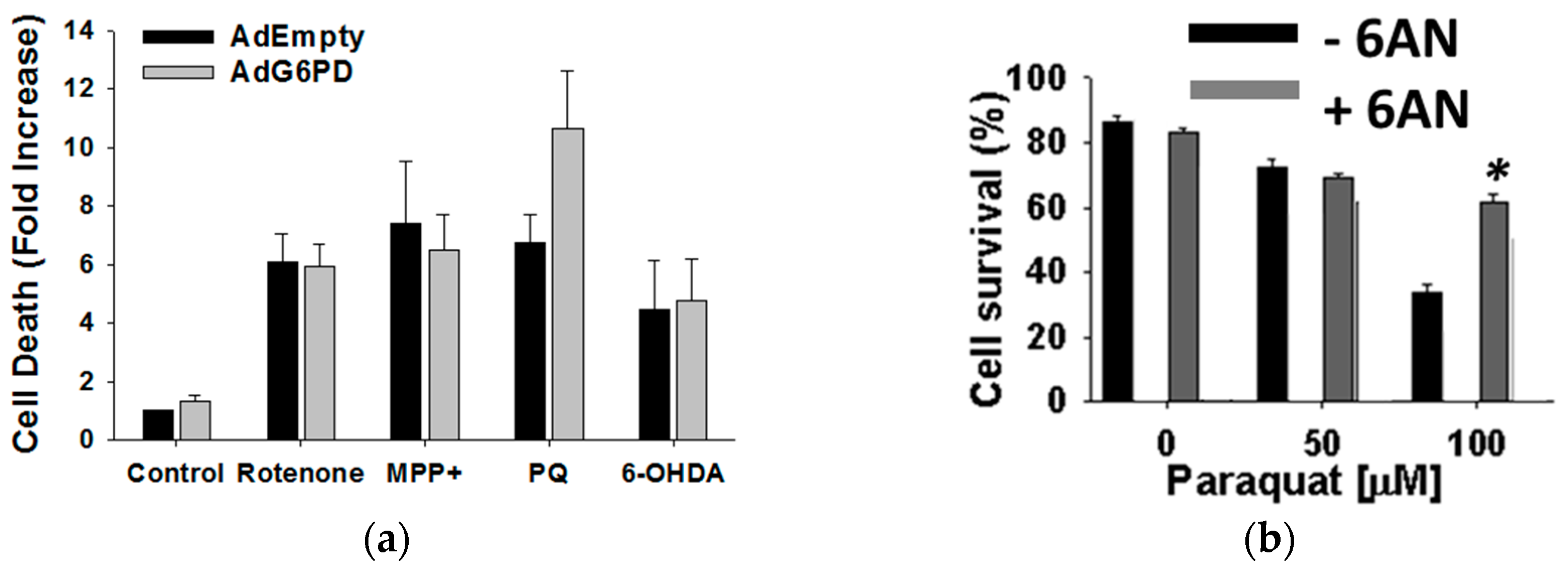
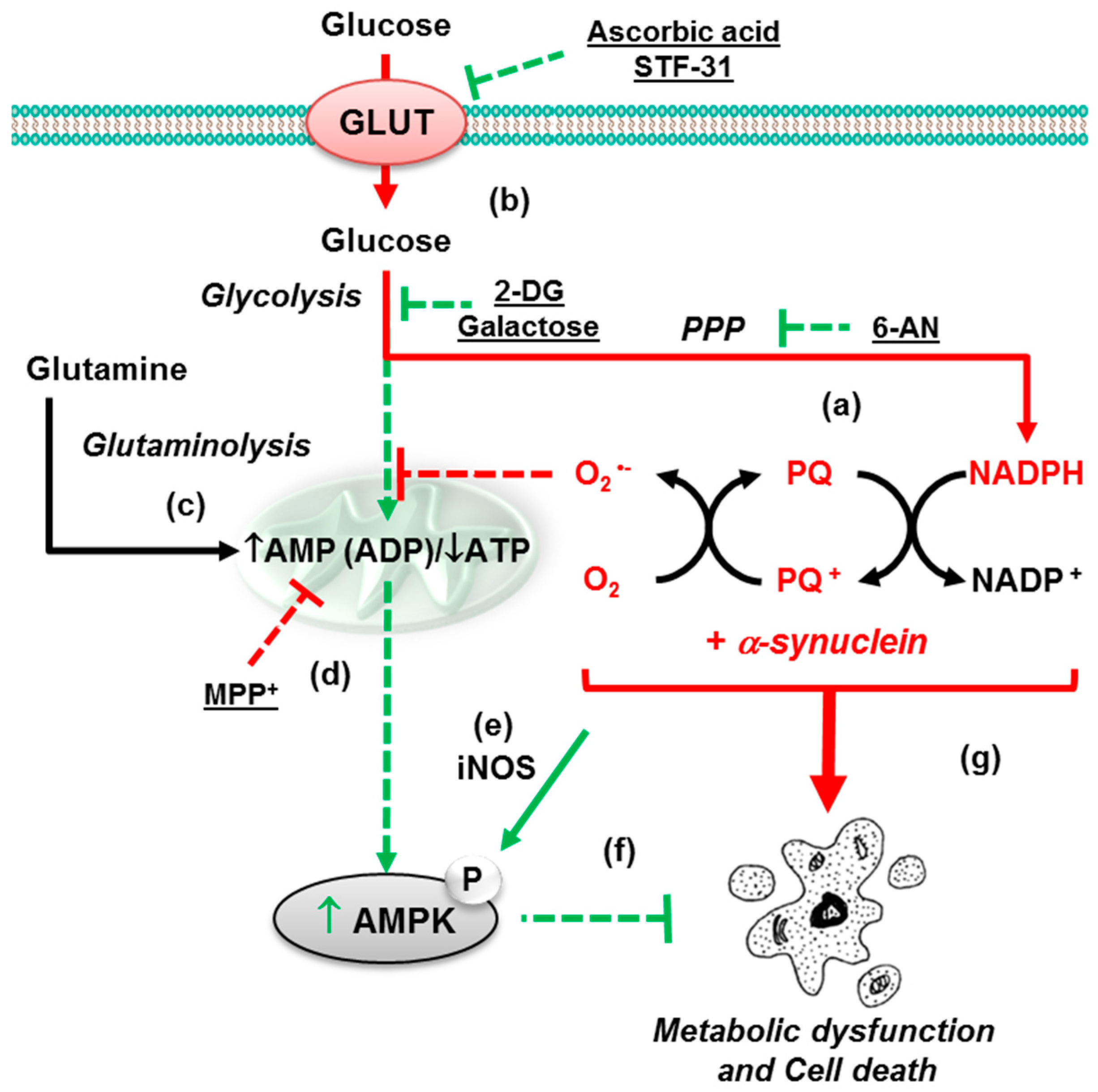
3.2. Glucose 6-Phosphate Dehydrogenase Regulates Paraquat Toxicity
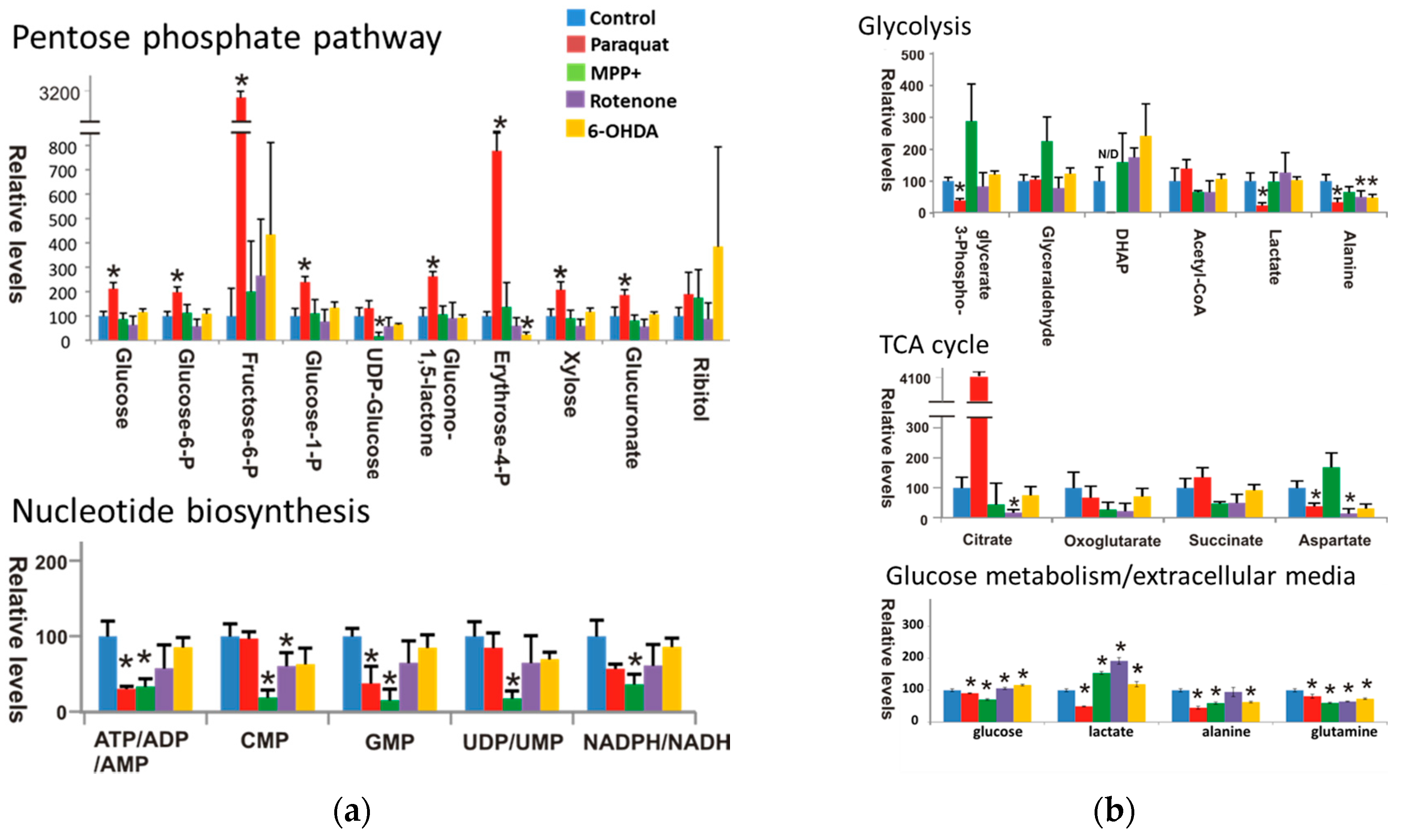
3.3. Paraquat Hijacks the Pentose Phosphate Pathway
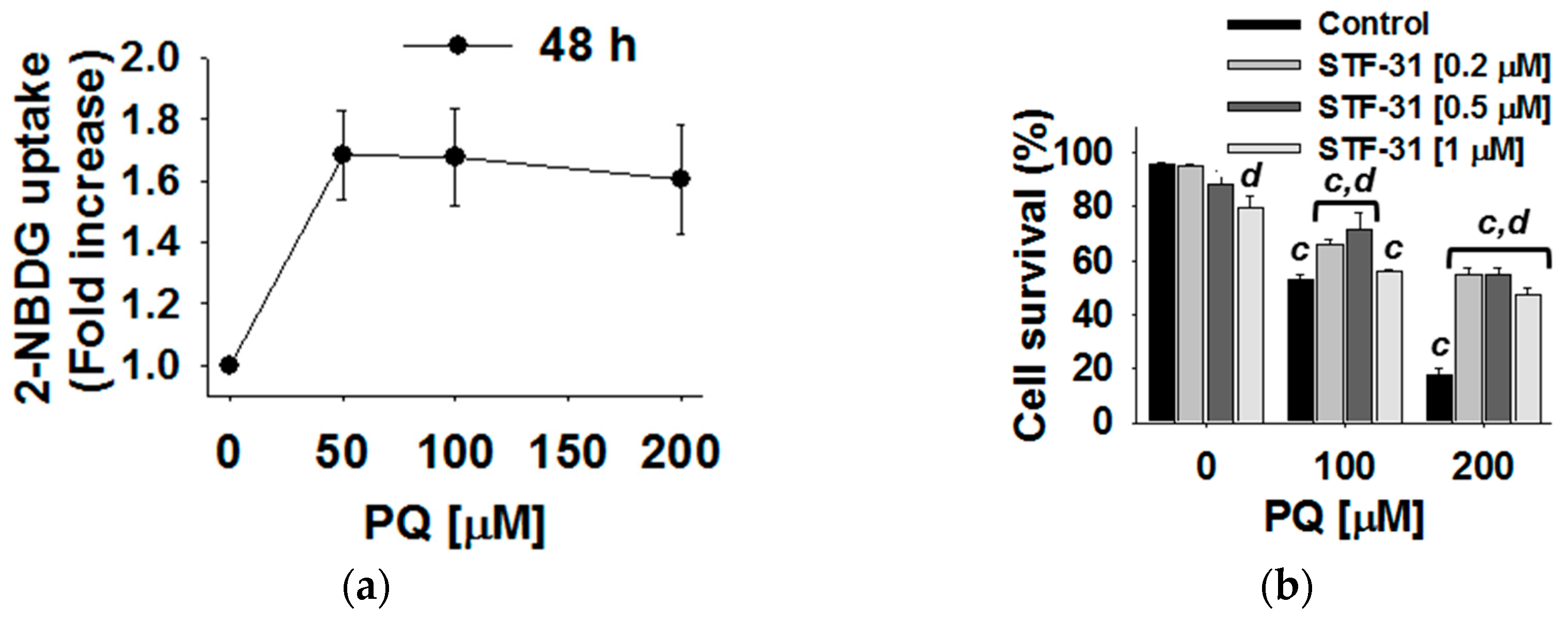
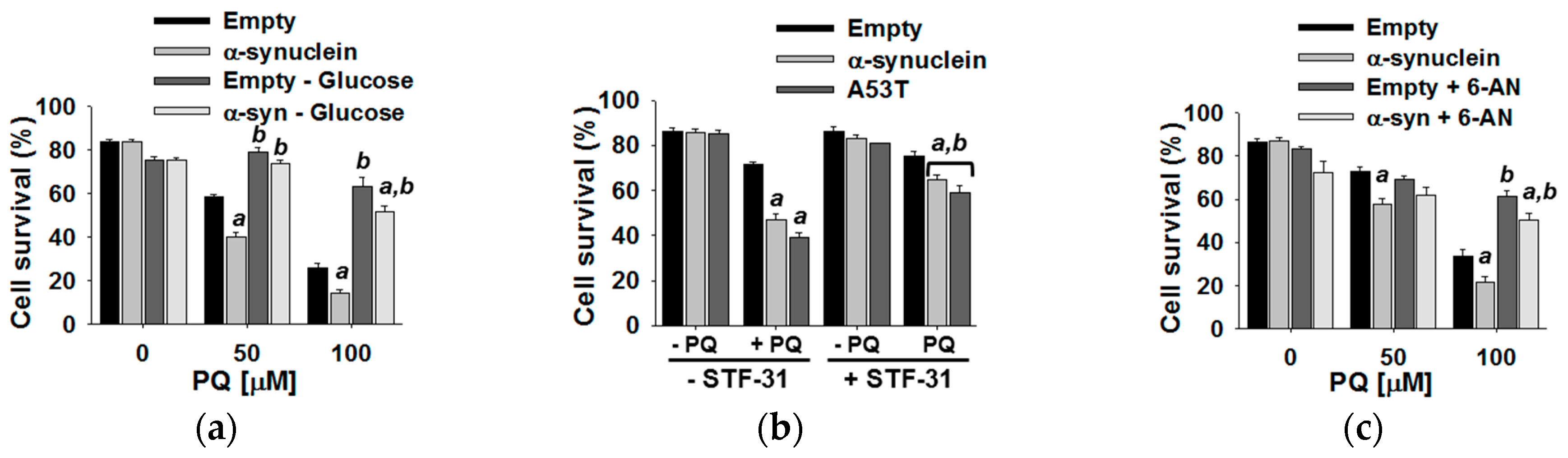
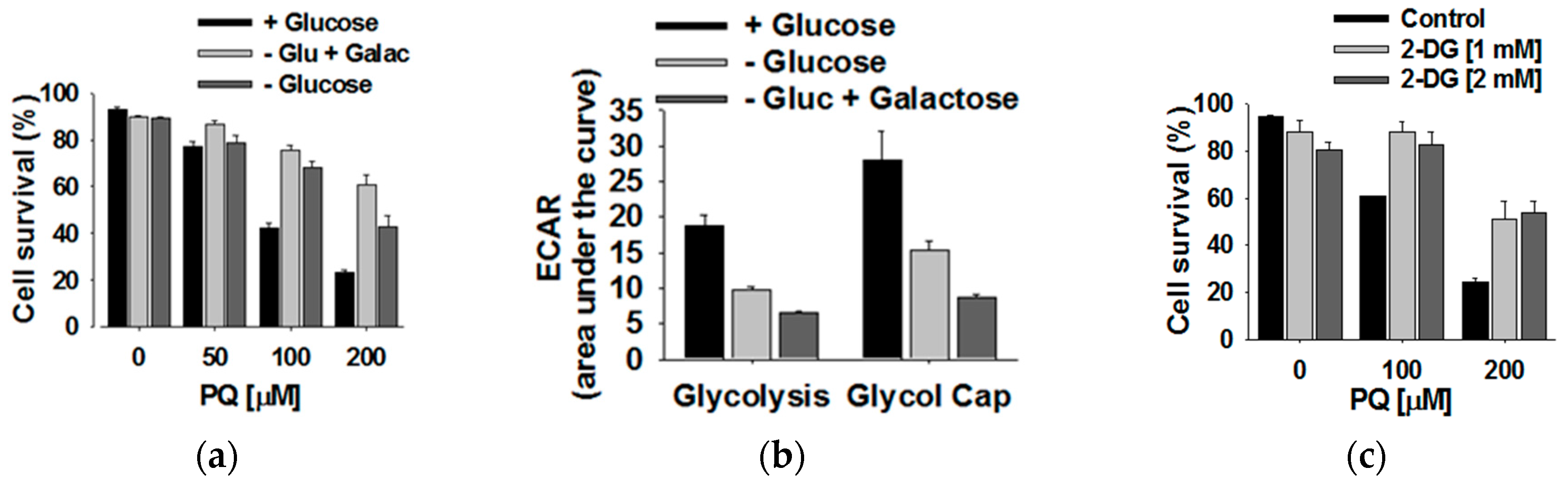
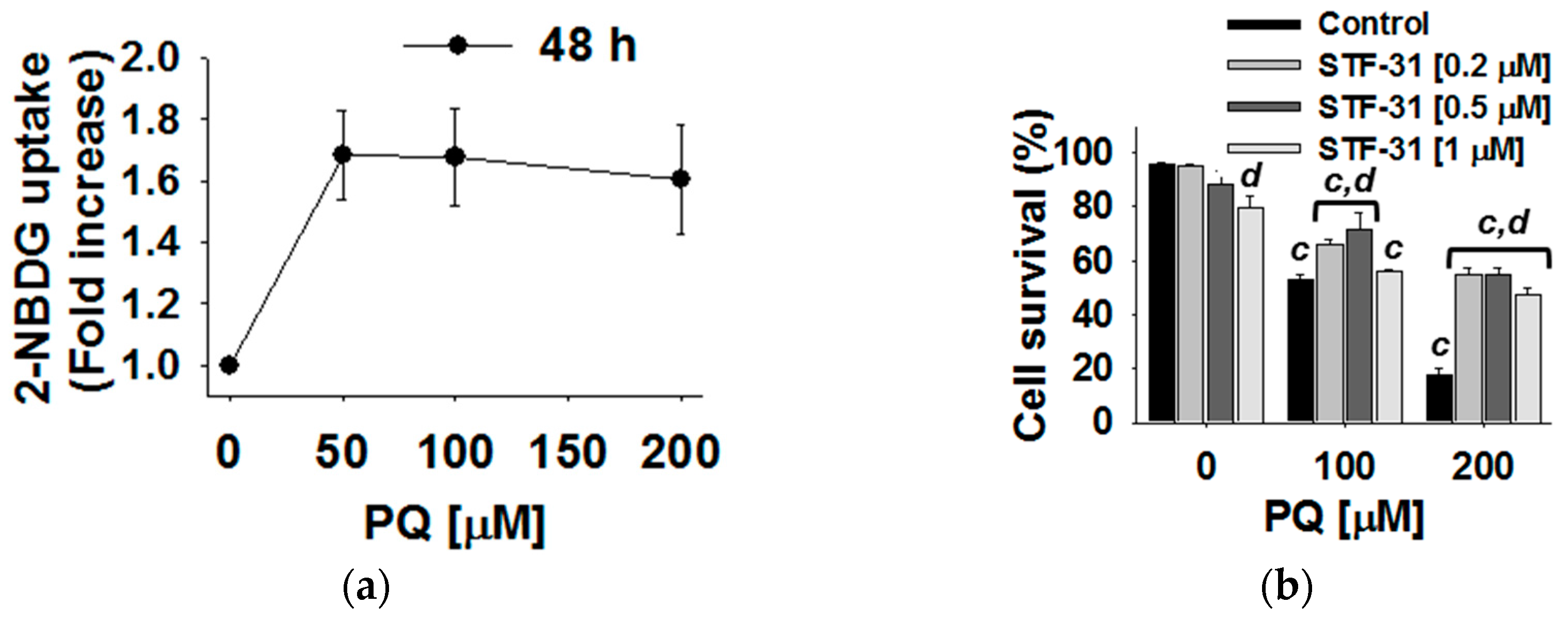
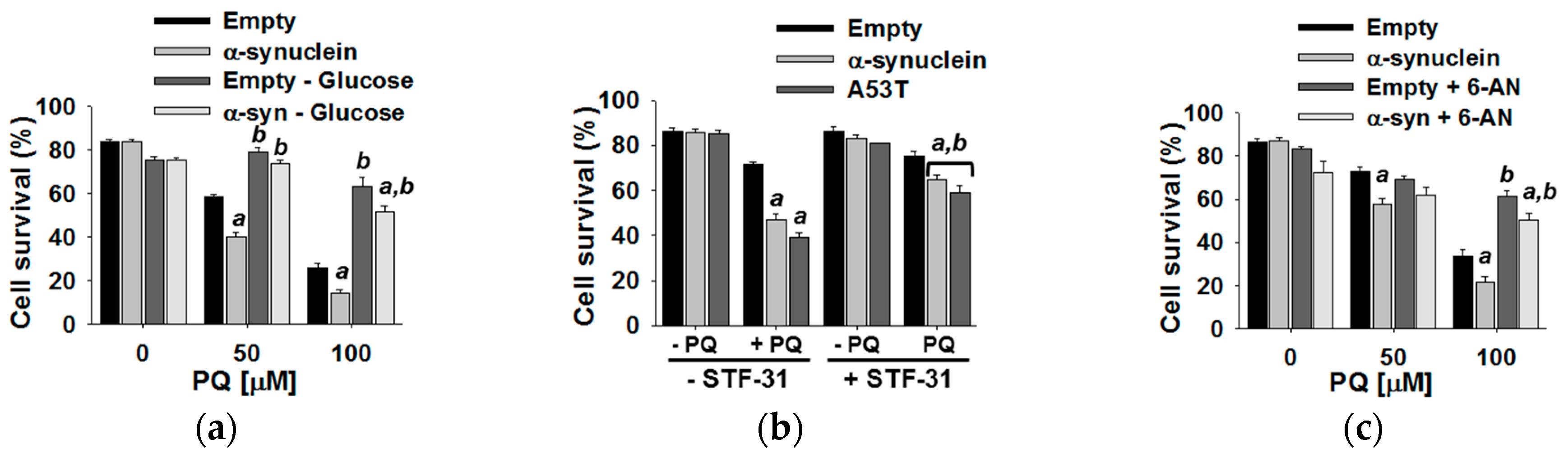
3.4. Glucose Metabolism Regulates Paraquat Toxicity
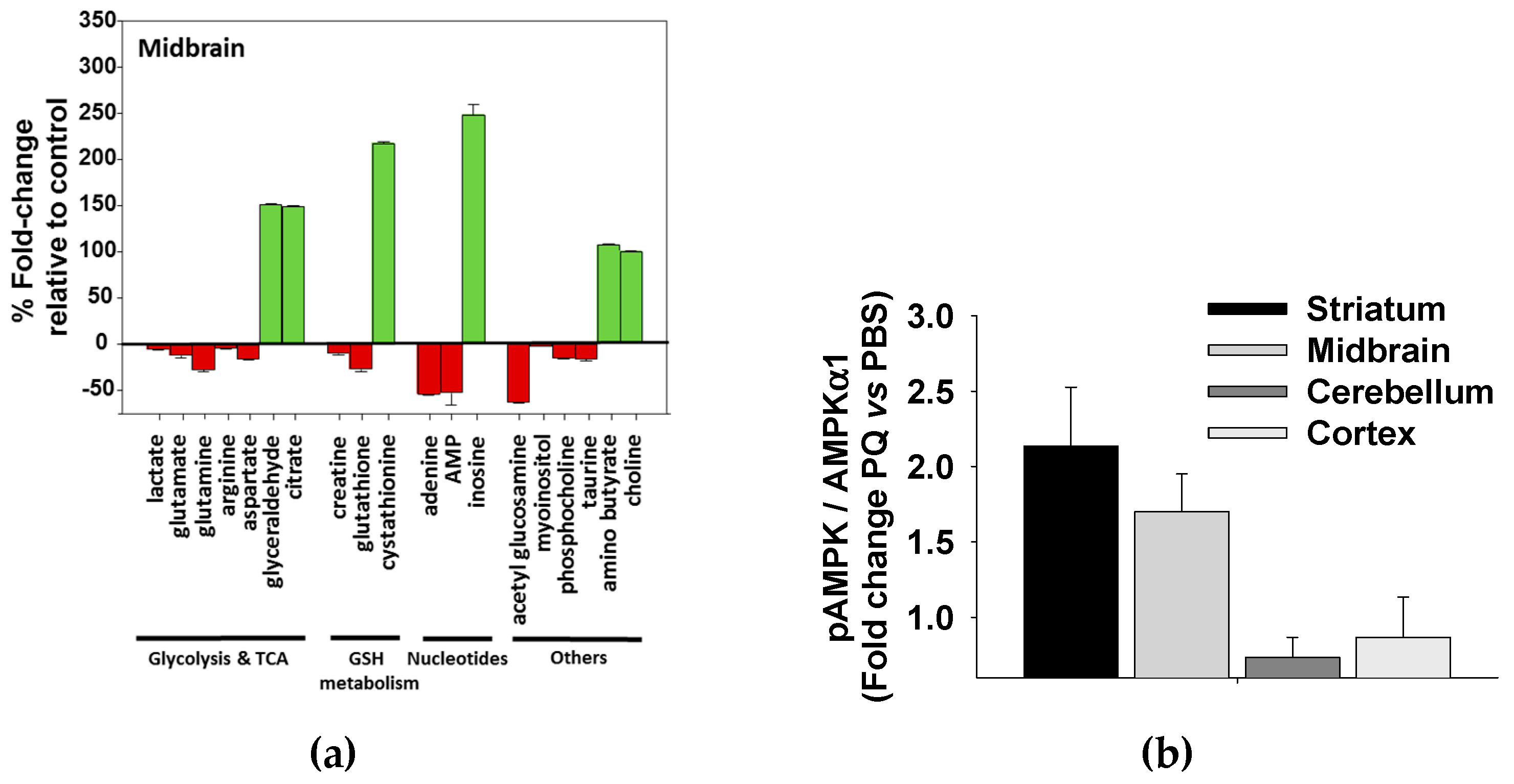
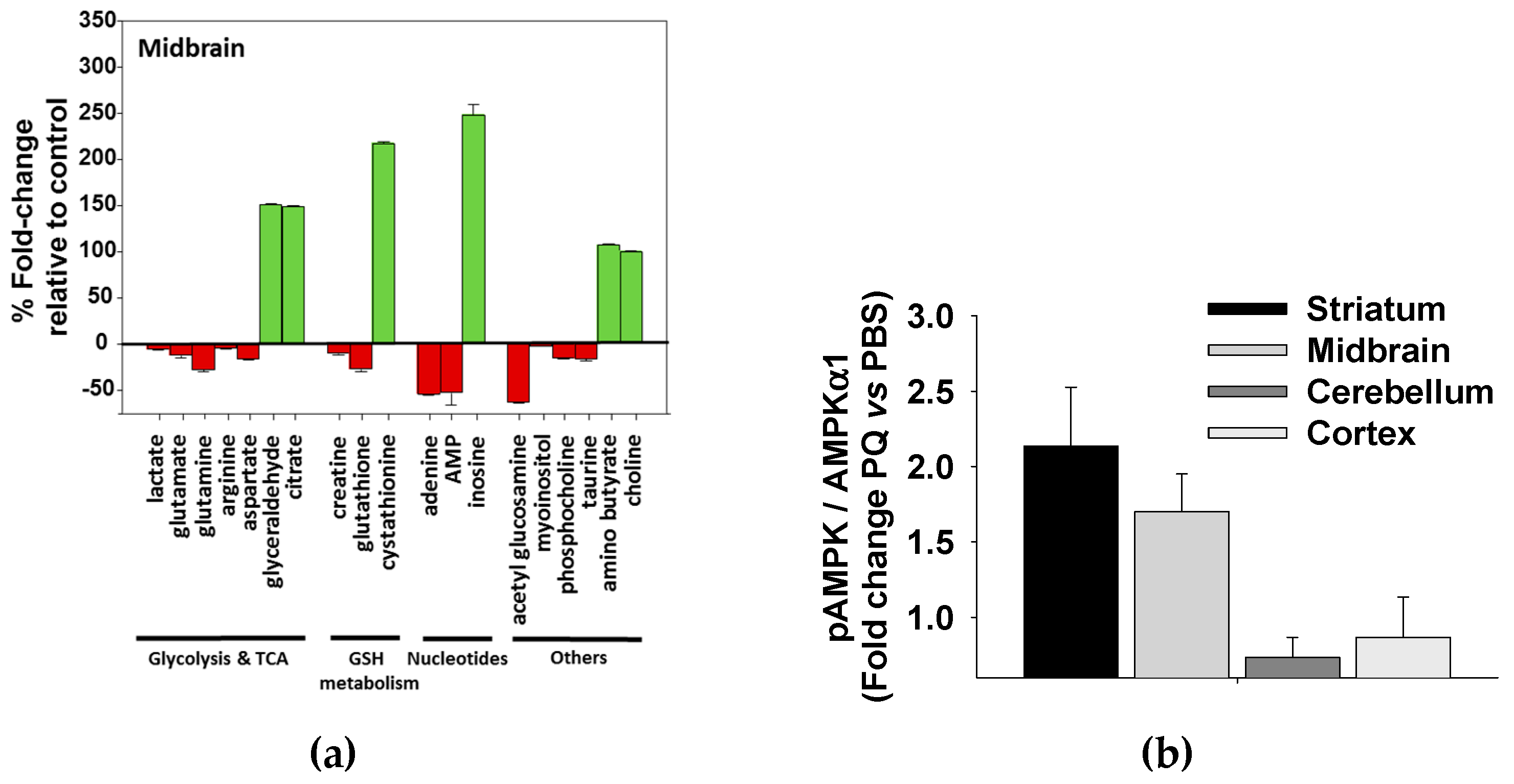
3.5. Paraquat Induced Metabolic Dysfunction in the Mice Midbrain and Striatum
4. Synergy of α-Synuclein Genetic Mutations and Paraquat Toxicity
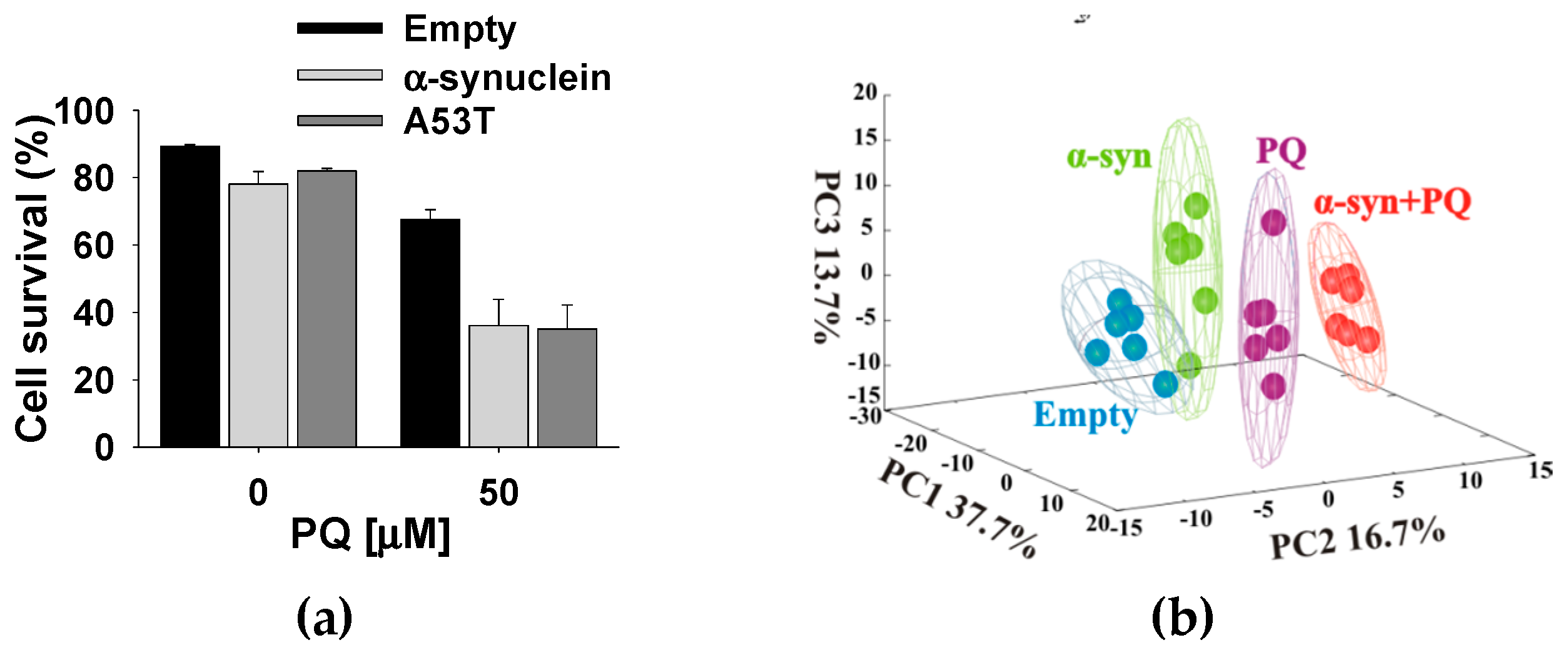
4.1. α-Synuclein Potentiates Paraquat Toxicity and Metabolic Dysfunction
4.2. Glucose Metabolism Contributes to Synergistic Toxicity between Paraquat and α-Synuclein
5. Conclusions
6. Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 1D | one-dimensional |
| 2D | two-dimensional |
| 3D | three-dimensional |
| 2-DG | 2-deoxy-D-glucose |
| 2-NBDG | 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy-D-glucose |
| 6-AN | 6-aminonicotinamide |
| 6-OHDA | 6-hydroxydopamine |
| α-syn | α-synuclein |
| AA | ascorbic acid |
| ACO | aconitase |
| AD | Alzheimer’s disease |
| ADC | aspartate 4-decarboxylase |
| AdEmpty | empty adenovirus |
| AdG6PD | adenovirus encoding for human G6PD |
| ADP | adenosine diphosphate |
| ALDO | fructose-bisphosphate aldolase |
| ALT | Alanine transaminase |
| AMP | adenosine monophosphate |
| AMPK | AMP-activated protein kinase |
| AMPKα1 | AMP-activated protein kinase α1 |
| ANOVA | analysis of variance |
| ATP | adenosine triphosphate |
| BBB | blood brain barrier |
| CS | ATP-citrate synthase |
| CV-ANOVA | ANOVA of the cross-validated residuals |
| DHAP | dihydroxyacetone phosphate |
| DI-ESI-MS | direct infusion-electrospray ionization mass spectrometry |
| DNA | deoxyribonucleic acid |
| ECAR | extracellular acidification rate |
| ECV | extraction voltage |
| FBP | fructose-1,6-bisphosphatase I |
| FUM | fumarate hydratase |
| G6PD | glucose-6-phosphate dehydrogenase |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GLDH | Glutamate dehydrogenase |
| GLS | Glutaminase |
| GLUT | Na+-independent glucose transporters |
| GOT1 | Aspartate aminotransferase, cytoplasmic |
| GPI | Glucose-6-phosphate isomerase |
| GSH | reduced glutathione |
| GSSH | oxidized glutathione |
| HK | hexokinase |
| HSQC | heteronuclear single quantum coherence |
| IDH | isocitrate dehydrogenase |
| iNOS | inducible nitric oxide |
| LDH | L-lactate dehydrogenase |
| LRRK2 | leucine-rich repeat kinase 2 |
| LSC | succinyl-CoA synthetase |
| mBCl | monochlorobimane |
| MB-PCA | multiblock principal component analysis |
| MB-PLS | multiblock partial least squares |
| MDH | malate dehydrogenase |
| Mn | manganese |
| MOI | multiplicity of infection |
| MPP+ | 1-methyl-4-phenylpyridinium |
| MPTP | 1-methyl-4-phenyl-1,2, 3, 6-tetrahydropyridine |
| MS | mass spectrometry |
| NAC | non-amyloid-β component of AD amyloid plaques |
| NADP | nicotinamide adenine dinucleotide phosphate |
| NADPH | nicotinamide adenine dinucleotide phosphate hydrogen |
| NG | glucose free environment |
| NMR | nuclear magnetic resonance spectroscopy |
| OCR | oxygen consumption rate |
| OGDH | 2-oxoglutarate dehydrogenase |
| OPLS | orthogonal projection to latent structures |
| OXPHOS | oxidative phosphorylation |
| pACC | phosphorylated acetyl-CoA carboxylase |
| pAMPK | phosphorylated AMP-activated protein kinase |
| PBS | phosphate buffered saline |
| PC | pyruvate carboxylase |
| PCA | principal component analysis |
| PD | Parkinson’s disease |
| PDH | pyruvate dehydrogenase |
| PET | positron emission tomography |
| PFK | 6-phosphofructokinase 1 |
| PGD | 6-phosphogluconate dehydrogenase |
| PGK1 | phosphoglycerate kinase 1 |
| PGM | phosphoglucomutase |
| PI | propidium iodide |
| PINK1 | phosphatase and tensin homolog (PTEN)-induced kinase 1 |
| PLS | partial least squares |
| PPP | pentose phosphate pathway |
| PQ | paraquat |
| PSC | phase-scatter correction |
| RNA | ribonucleic acid |
| ROS | reactive oxygen species |
| RPI | ribose 5-phosphate isomerase A |
| SCV | sampling cone voltage |
| SDH | succinate dehydrogenase |
| SE | standard error |
| SGLT | Na+-dependent glucose transporters |
| SNARE | soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptor |
| STF-31 | 4-[[[[4-(1,1-dimethylethyl)phenyl]sulfonyl]amino]methyl]-N-3-pyridinyl-benzamide |
| SUS | shared and unique structures |
| TAL | transaldolase |
| TCA | tricarboxylic acid |
| TKT | transketolase |
| TPI | triosephosphate isomerase |
| US | United States |
| UDP | uridine diphosphate |
| UMP | uridine monophosphate |
| WB | Western blot |
| WT | wild-type |
References
- Pringsheim, T.; Jette, N.; Frolkis, A.; Steeves, T.D.L. The prevalence of Parkinson’s disease: A systematic review and meta-analysis. Mov. Disord. 2014, 29, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W. The scientific basis for the current treatment of Parkinson’s disease. Annu. Rev. Med. 2004, 55, 41–60. [Google Scholar] [CrossRef] [PubMed]
- Dunnett, S.B.; Bjorklund, A. Prospects for new restorative and neuroprotective treatments in Parkinson’s disease. Nature 1999, 399, A32–A39. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Olanow, C.W.; Tatton, W.G. Etiology and pathogenesis of Parkinson’s disease. Annu. Rev. Neurosci. 1999, 22, 123–144. [Google Scholar] [CrossRef] [PubMed]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [PubMed]
- Simon-Sanchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. Α-synuclein in Parkinson’s disease. Cold Spring Harb Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.A.; Harper, J.D.; Lansbury, P.T. Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat. Med. 1998, 4, 1318–1320. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in pink1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [PubMed]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.J.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.M. The role of environmental toxins in the etiology of Parkinson’s disease. Trends NeuroSci. 1989, 12, 49–54. [Google Scholar] [CrossRef]
- Wuellner, U.; Kaut, O.; de Boni, L.; Piston, D.; Schmitt, I. DNA methylation in Parkinson’s disease. J. Neurochem. 2016, 139, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Jankovic, J.; Wu, Y.-C. Epigenetic mechanisms in Parkinson’s disease. J. Neurol. Sci. 2015, 349, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Hegarty, S.V.; Sullivan, A.M.; O’Keeffe, G.W. The epigenome as a therapeutic target for Parkinson’s disease. Neural Regen. Res. 2016, 11, 1735–1738. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Kanthasamy, A.; Jin, H.; Anantharam, V.; Kanthasamy, A.G. Paraquat induces epigenetic changes by promoting histone acetylation in cell culture models of dopaminergic degeneration. Neurotoxicology 2011, 32, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Kong, M.; Ba, M.; Liang, H.; Ma, L.; Yu, Q.; Yu, T.; Wang, Y. 5’-aza-dc sensitizes paraquat toxic effects on PC12 cell. Neurosci. Lett. 2012, 524, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Migliore, L.; Coppede, F. Genetics, environmental factors and the emerging role of epigenetics in neurodegenerative diseases. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2009, 667, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Wright, W.A.; Evanoff, B.A.; Lian, M.; Criswell, S.R.; Racette, B.A. Geographic and ethnic variation in Parkinson disease: A population-based study of us medicare beneficiaries. Neuroepidemiology 2010, 34, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Blum, D.; Torch, S.; Lambeng, N.; Nissou, M.F.; Benabid, A.L.; Sadoul, R.; Verna, J.M. Molecular pathways involved in the neurotoxicity of 6-ohda, dopamine and mptp: Contribution to the apoptotic theory in parkinson’s disease. Prog. Neurobiol. 2001, 65, 135–172. [Google Scholar] [CrossRef]
- Smeyne, R.J.; Jackson-Lewis, V. The MPTP model of Parkinson’s disease. Mol. Brain Res. 2005, 134, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, M.; Riederer, P.; Przuntek, H.; Youdim, M.B.H. Mptp mechanisms of neurotoxicity and their implications for Parkinson’s disease. Eur. J. Pharmacol. Mol. Pharmacol. Sect. 1991, 208, 273–286. [Google Scholar] [CrossRef]
- Przedborski, S.; Jackson-Lewis, V.; Djaldetti, R.; Liberatore, G.; Vila, M.; Vukosavic, S.; Almer, G. The parkinsonian toxin mptp: Action and mechanism. Restor. Neurol. Neurosci. 2000, 16, 135–142. [Google Scholar] [PubMed]
- Bartlett, R.M.; Holden, J.E.; Nickles, R.J.; Murali, D.; Barbee, D.L.; Barnhart, T.E.; Christian, B.T.; DeJesus, O.T. Paraquat is excluded by the blood brain barrier in rhesus macaque: An in vivo pet study. Brain Res. 2009, 1259, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, R.M.; Murali, D.; Nickles, R.J.; Barnhart, T.E.; Holden, J.E.; DeJesus, O.T. Assessment of fetal brain uptake of paraquat in utero using in vivo PET/CT imaging. Toxicol. Sci. 2011, 122, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.T.; Woulfe, J.M. Striatal blood-brain barrier permeability in Parkinson’s disease. J. Cereb. Blood Flow Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [PubMed]
- Sherer, T.B.; Betarbet, R.; Testa, C.M.; Seo, B.B.; Richardson, J.R.; Kim, J.H.; Miller, G.W.; Yagi, T.; Matsuno-yagi, A.; Greenamyre, T. Mechanism of toxicity in rotenone models of Parkinson’s disease. J. Neurosci. 2003, 23, 10756–10764. [Google Scholar] [PubMed]
- Glinka, Y.; Gassen, M.; Youdim, M.B. Mechanism of 6-hydroxydopamine neurotoxicity. J Neural Transm. Suppl. 1997, 50, 55–66. [Google Scholar] [PubMed]
- Rodriguez-Pallares, J.; Parga, J.A.; Munoz, A.; Rey, P.; Guerra, M.J.; Labandeira-Garcia, J.L. Mechanism of 6-hydroxydopamine neurotoxicity: The role of nadph oxidase and microglial activation in 6-hydroxydopamine-induced degeneration of dopaminergic neurons. J. Neurochem. 2007, 103, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Strickland, D.; Bertoni, J.M. Parkinson’s prevalence estimated by a state registry. Mov. Disord. 2004, 19, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Tanji, K.; Mori, F.; Takahashi, H. The lewy body in Parkinson’s disease: Molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology 2007, 27, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Kaagedal, K.; Halliday, G.M. Alpha-synuclein biology in lewy body diseases. Alzheimer’s Res. Ther. 2014, 6, 73. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Α-synuclein in lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and dynamics of micelle-bound human α-synuclein. J. Biol. Chem. 2005, 280, 9595–9603. [Google Scholar] [CrossRef] [PubMed]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Suedhof, T.C. Α-synuclein promotes snare-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Kang, S.J.; Lee, K.; Im, H. Human α-synuclein modulates vesicle trafficking through its interaction with prenylated rab acceptor protein 1. Biochem. Biophys. Res. Commun. 2011, 412, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Nemani, V.M.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased expression of α-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 2010, 65, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. Α-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Hartley, D.; Petre, B.M.; Walz, T.; Lansbury, P.T. Neurodegenerative disease: Amyloid pores from pathogenic mutations. Nature 2002, 418, 291. [Google Scholar] [CrossRef] [PubMed]
- Fink, A.L. The aggregation and fibrillation of α-synuclein. Acc. Chem. Res. 2006, 39, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Petre, B.M.; Wall, J.; Simon, M.; Nowak, R.J.; Walz, T.; Lansbury, P.T., Jr. Α-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J. Mol. Biol. 2002, 322, 1089–1102. [Google Scholar] [CrossRef]
- Volles, M.J.; Lansbury, P.T., Jr. Vesicle permeabilization by protofibrillar α-synuclein is sensitive to Parkinson’s disease-linked mutations and occurs by a pore-like mechanism. Biochemistry. 2002, 41, 4595–4602. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Hsu, L.J.; Xia, Y.; Takeda, A.; Sisk, A.; Sundsmo, M.; Masliah, E. Oxidative stress induces amyloid-like aggregate formation of nacp/α-synuclein in vitro. Neuroreport 1999, 10, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Andringa, G.; Lam, K.Y.; Chegary, M.; Wang, X.; Chase, T.N.; Bennett, M.C. Tissue transglutaminase catalyzes the formation of α-synuclein crosslinks in Parkinson’s disease. FASEB J. 2004, 18, 932–934. [Google Scholar] [CrossRef] [PubMed]
- Segers-Nolten, I.M.J.; Wilhelmus, M.M.M.; Veldhuis, G.; Van Rooijen, B.D.; Drukarch, B.; Subramaniam, V. Tissue transglutaminase modulates α-synuclein oligomerization. Protein Sci. 2008, 17, 1395–1402. [Google Scholar] [CrossRef] [PubMed]
- Paleologou, K.E.; Oueslati, A.; Shakked, G.; Rospigliosi, C.C.; Kim, H.-Y.; Lamberto, G.R.; Fernandez, C.O.; Schmid, A.; Chegini, F.; Gai, W.P.; et al. Phosphorylation at s87 is enhanced in synucleinopathies, inhibits α-synuclein oligomerization, and influences synuclein-membrane interactions. J. Neurosci. 2010, 30, 3184–3198. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; West, N.; Colla, E.; Pletnikova, O.; Troncoso, J.C.; Marsh, L.; Dawson, T.M.; Jaekaelae, P.; Hartmann, T.; Price, D.L.; et al. Aggregation promoting c-terminal truncation of α-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc. Natl. Acad. Sci. USA 2005, 102, 2162–2167. [Google Scholar] [CrossRef] [PubMed]
- Dufty, B.M.; Warner, L.R.; Hou, S.T.; Jiang, S.X.; Gomez-Isla, T.; Leenhouts, K.M.; Oxford, J.T.; Feany, M.B.; Masliah, E.; Rohn, T.T. Calpain-cleavage of α-synuclein connecting proteolytic processing to disease-linked aggregation. Am. J. Pathol. 2007, 170, 1725–1738. [Google Scholar] [CrossRef] [PubMed]
- Perrin, R.J.; Woods, W.S.; Clayton, D.F.; George, J.M. Exposure to long chain polyunsaturated fatty acids triggers rapid multimerization of synucleins. J. Biol. Chem. 2001, 276, 41958–41962. [Google Scholar] [CrossRef] [PubMed]
- Sharon, R.; Bar-Joseph, I.; Frosch, M.P.; Walsh, D.M.; Hamilton, J.A.; Selkoe, D.J. The formation of highly soluble oligomers of α-synuclein is regulated by fatty acids and enhanced in Parkinson’s disease. Neuron 2003, 37, 583–595. [Google Scholar] [CrossRef]
- Karube, H.; Sakamoto, M.; Arawaka, S.; Hara, S.; Sato, H.; Ren, C.H.; Goto, S.; Koyama, S.; Wada, M.; Kawanami, T.; et al. N-terminal region of α-synuclein is essential for the fatty acid-induced oligomerization of the molecules. FEBS Lett. 2008, 582, 3693–3700. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.; McLaurin, J.; Yip, C.M.; St. George-Hyslop, P.; Fraser, P.E. Α-synuclein membrane interactions and lipid specificity. J. Biol. Chem. 2000, 275, 34328–34334. [Google Scholar] [CrossRef] [PubMed]
- Binolfi, A.; Rasia, R.M.; Bertoncini, C.W.; Ceolin, M.; Zweckstetter, M.; Griesinger, C.; Jovin, T.M.; Fernandez, C.O. Interaction of α-synuclein with divalent metal ions reveals key differences: A link between structure, binding specificity and fibrillation enhancement. J. Am. Chem. Soc. 2006, 128, 9893–9901. [Google Scholar] [CrossRef] [PubMed]
- Bisaglia, M.; Tessari, I.; Mammi, S.; Bubacco, L. Interaction between α-synuclein and metal ions, still looking for a role in the pathogenesis of Parkinson’s disease. NeuroMol. Med. 2009, 11, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Surguchov, A. Intracellular dynamics of synucleins: “Here, there and everywhere”. Int. Rev. Cell Mol. Biol. 2015, 320, 103–169. [Google Scholar] [PubMed]
- Jethva, P.N.; Kardani, J.R.; Roy, I. Modulation of α-synuclein aggregation by dopamine in the presence of mptp and its metabolite. FEBS J. 2011, 278, 1688–1698. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Chan, P. Interaction between neuromelanin and alpha-synuclein in Parkinson’s disease. Biomolecules 2015, 5, 1122–1142. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Lindon, J.C.; Holmes, E. “Metabonomics”: Understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, K.; Aronov, P.A.; Hammock, B.D. Mass spectrometry-based metabolomics. Mass Spectrom. Rev. 2007, 26, 51–78. [Google Scholar] [CrossRef] [PubMed]
- Marshall, D.D.; Powers, R. Beyond the paradigm: Combining mass spectrometry and nuclear magnetic resonance for metabolomics. Prog. Nucl. Magn. Reson. Spectrosc. 2017, 100, 1–16. [Google Scholar] [CrossRef]
- Markley, J.L.; Bruschweiler, R.; Edison, A.S.; Eghbalnia, H.R.; Powers, R.; Raftery, D.; Wishart, D.S. The future of nmr-based metabolomics. Curr. Opin. Biotechnol. 2017, 43, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Psychogios, N.; Hau, D.D.; Peng, J.; Guo, A.C.; Mandal, R.; Bouatra, S.; Sinelnikov, I.; Krishnamurthy, R.; Eisner, R.; Gautam, B.; et al. The human serum metabolome. PLoS ONE 2011, 6, e16957. [Google Scholar] [CrossRef] [PubMed]
- Moco, S.; Forshed, J.; Vos, R.C.H.; Bino, R.J.; Vervoort, J. Intra- and inter-metabolite correlation spectroscopy of tomato metabolomics data obtained by liquid chromatography-mass spectrometry and nuclear magnetic resonance. Metabolomics 2008, 4, 202–215. [Google Scholar] [CrossRef]
- Lanza, I.R.; Zhang, S.; Ward, L.E.; Karakelides, H.; Raftery, D.; Nair, K.S. Quantitative metabolomics by 1h-nmr and lc-ms/ms confirms altered metabolic pathways in diabetes. PLoS ONE 2010, 5, e10538. [Google Scholar] [CrossRef] [PubMed]
- Kell, D.B. Metabolomics and systems biology: Making sense of the soup. Curr. Opin. Microbiol. 2004, 7, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Canelas, A.B.; ten Pierick, A.; Ras, C.; Seifar, R.M.; van Dam, J.C.; van Gulik, W.M.; Heijnen, J.J. Quantitative evaluation of intracellular metabolite extraction techniques for yeast metabolomics. Analy. Chem. 2009, 81, 7379–7389. [Google Scholar] [CrossRef] [PubMed]
- Kanani, H.; Chrysanthopoulos, P.K.; Klapa, M.I. Standardizing gc-ms metabolomics. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008, 871, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Zou, L.; Ong, C.N. Multiorigination of chromatographic peaks in derivatized GC/MS metabolomics: A confounder that influences metabolic pathway interpretation. J. Proteom. Res. 2009, 8, 5657–5665. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.J. Matrix effects: The achilles heel of quantitative high-performance liquid chromatography-electrospray-tandem mass spectrometry. Clin. Biochem. 2005, 38, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Kopka, J. Current challenges and developments in GC-MS based metabolite profiling technology. J. Biotechnol. 2006, 124, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Liebeke, M.; Sommer, U.; Viant, M.R.; Bundy, J.G.; Ebbels, T.M.D. Statistical correlations between nmr spectroscopy and direct infusion ft-icr mass spectrometry aid annotation of unknowns in metabolomics. Anal. Chem. 2016, 88, 2583–2589. [Google Scholar] [CrossRef] [PubMed]
- Bingol, K.; Brueschweiler, R. Two elephants in the room: New hybrid nuclear magnetic resonance and mass spectrometry approaches for metabolomics. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Bingol, K.; Bruschweiler, R. NMR/MS translator for the enhanced simultaneous analysis of metabolomics mixtures by nmr spectroscopy and mass spectrometry: Application to human urine. J. Proteom. Res. 2015, 14, 2642–2648. [Google Scholar] [CrossRef] [PubMed]
- Bingol, K.; Bruschweiler-Li, L.; Li, D.; Zhang, B.; Xie, M.; Brueschweiler, R. Emerging new strategies for successful metabolite identification in metabolomics. Bioanalysis. 2016, 8, 557–573. [Google Scholar] [CrossRef] [PubMed]
- Bingol, K.; Bruschweiler, L.; Yu, C.; Somogyi, A.; Zhang, F.; Bruschweiler, R. Metabolomics beyond spectroscopic databases: A combined MS/NMR strategy for the rapid identification of new metabolites in complex mixtures. Anal. Chem. 2015, 87, 3864–3870. [Google Scholar] [CrossRef] [PubMed]
- Marshall, D.D.; Lei, S.; Worley, B.; Huang, Y.; Garcia-Garcia, A.; Franco, R.; Dodds, E.D.; Powers, R. Combining DI-ESI–MS and NMR datasets for metabolic profiling. Metabolomics 2015, 11, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Lei, S.; Zavala-Flores, L.; Garcia-Garcia, A.; Nandakumar, R.; Huang, Y.; Madayiputhiya, N.; Stanton, R.C.; Dodds, E.D.; Powers, R.; Franco, R. Alterations in energy/redox metabolism induced by mitochondrial and environmental toxins: A specific role for glucose-6-phosphate-dehydrogenase and the pentose phosphate pathway in paraquat toxicity. ACS Chem. Biol. 2014, 9, 2032–2048. [Google Scholar] [CrossRef] [PubMed]
- Worley, B.; Powers, R. Mvapack: A complete data handling package for nmr metabolomics. ACS Chem. Biol. 2014, 9, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Worley, B.; Powers, R. Simultaneous phase and scatter correction for nmr datasets. Chemom. Intell. Lab. Syst. 2014, 131, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Halouska, S.; Powers, R. Negative impact of noise on the principal component analysis of nmr data. J. Magn. Reson. 2006, 178, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Halouska, S.; Zhang, B.; Gaupp, R.; Lei, S.; Snell, E.; Fenton, R.J.; Barletta, R.G.; Somerville, G.A.; Powers, R. Revisiting protocols for the nmr analysis of bacterial metabolomes. J. Integr. OMICS 2013, 2, 120–137. [Google Scholar]
- De Meyer, T.; Sinnaeve, D.; Van Gasse, B.; Tsiporkova, E.; Rietzschel, E.R.; De Buyzere, M.L.; Gillebert, T.C.; Bekaert, S.; Martins, J.C.; Van Criekinge, W. NMR-based characterization of metabolic alterations in hypertension using an adaptive, intelligent binning algorithm. Anal. Chem. 2008, 80, 3783–3790. [Google Scholar] [CrossRef] [PubMed]
- Wold, S.; Hellbern, S.; Lundstedt, T.; Siostrom, M. PLS Modeling with Latent Variables in Two or More Dimensions; PLS Model Building, Theory and Applications Symposium: Frankfurt, Germany, 1987. [Google Scholar]
- Smilde, A.K.; Westerhuis, J.A.; de Jong, S. A framework for sequential multiblock component methods. J. Chemom. 2003, 17, 323–337. [Google Scholar] [CrossRef]
- Westerhuis, J.A.; Kourti, T.; Macgregor, J.F. Analysis of multiblock and hierarchical PCA and PLS models. J. Chemom. 1998, 12, 301–321. [Google Scholar] [CrossRef]
- Worley, B.; Powers, R. A sequential algorithm for multiblock orthogonal projections to latent structures. Chemometr. Intell. Lab. Syst. 2015, 149, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Martinez, T.N.; Greenamyre, J.T. Toxin models of mitochondrial dysfunction in Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 920–934. [Google Scholar] [CrossRef] [PubMed]
- Cantu, D.; Fulton, R.E.; Drechsel, D.A.; Patel, M. Mitochondrial aconitase knockdown attenuates paraquat-induced dopaminergic cell death via decreased cellular metabolism and release of iron and H2O2. J. Neurochem. 2011, 118, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garcia, A.; Zavala-Flores, L.; Rodriguez-Rocha, H.; Franco, R. Thiol-redox signaling, dopaminergic cell death, and Parkinson’s disease. Antioxid. Redox Signal. 2012, 17, 1764–1784. [Google Scholar] [CrossRef] [PubMed]
- Perry, T.L.; Yong, V.W. Idiopathic Parkinson’s disease, progressive supranuclear palsy and glutathione metabolism in the substantia nigra of patients. Neurosci. Lett. 1986, 67, 269–274. [Google Scholar] [CrossRef]
- Stanton, R.C. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life 2012, 64, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Anandhan, A.; Lei, S.; Levytskyy, R.; Pappa, A.; Panayiotidis, M.I.; Cerny, R.L.; Kalimonchuk, O.; Powers, R.; Franco, R. Glucose metabolism and ampk signaling regulate dopaminergic cell death induced by gene (α-synuclein)-envronment(paraquat) interactions. Mol. Neurobiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Cardaci, S.; Filomeni, G.; Ciriolo, M.R. Redox implications of AMPK-mediated signal transduction beyond energetic clues. J. Cell Sci. 2012, 125, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Gasior, M.; Rogawski, M.A.; Hartman, A.L. Neuroprotective and disease-modifying effects of the ketogenic diet. Behav. Pharmacol. 2006, 17, 431–439. [Google Scholar] [CrossRef] [PubMed]
- VanItallie, T.B.; Nonas, C.; Di Rocco, A.; Boyar, K.; Hyams, K.; Heymsfield, S.B. Treatment of Parkinson disease with diet-induced hyperketonemia: A feasibility study. Neurology 2005, 64, 728–730. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Bourdenx, M.; Gorry, P.; Przedborski, S.; Vila, M.; Hunot, S.; Singleton, A.; Olanow, C.W.; Merchant, K.M.; Bezard, E.; et al. Targeting α-synuclein for treatment of Parkinson’s disease: Mechanistic and therapeutic considerations. Lancet Neurol. 2015, 14, 855–866. [Google Scholar] [CrossRef]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinson’s Dis. 2013, 3, 461–491. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age (Years) | |||
|---|---|---|---|
| Nebraska PD Prevalence | 60–70 | 70–80 | 80+ |
| (Rates per 100,000) | |||
| Men | 406 | 1794 | 4248 |
| Women | 298 | 991 | 2069 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Powers, R.; Lei, S.; Anandhan, A.; Marshall, D.D.; Worley, B.; Cerny, R.L.; Dodds, E.D.; Huang, Y.; Panayiotidis, M.I.; Pappa, A.; et al. Metabolic Investigations of the Molecular Mechanisms Associated with Parkinson’s Disease. Metabolites 2017, 7, 22. https://doi.org/10.3390/metabo7020022
Powers R, Lei S, Anandhan A, Marshall DD, Worley B, Cerny RL, Dodds ED, Huang Y, Panayiotidis MI, Pappa A, et al. Metabolic Investigations of the Molecular Mechanisms Associated with Parkinson’s Disease. Metabolites. 2017; 7(2):22. https://doi.org/10.3390/metabo7020022
Chicago/Turabian StylePowers, Robert, Shulei Lei, Annadurai Anandhan, Darrell D. Marshall, Bradley Worley, Ronald L. Cerny, Eric D. Dodds, Yuting Huang, Mihalis I. Panayiotidis, Aglaia Pappa, and et al. 2017. "Metabolic Investigations of the Molecular Mechanisms Associated with Parkinson’s Disease" Metabolites 7, no. 2: 22. https://doi.org/10.3390/metabo7020022
APA StylePowers, R., Lei, S., Anandhan, A., Marshall, D. D., Worley, B., Cerny, R. L., Dodds, E. D., Huang, Y., Panayiotidis, M. I., Pappa, A., & Franco, R. (2017). Metabolic Investigations of the Molecular Mechanisms Associated with Parkinson’s Disease. Metabolites, 7(2), 22. https://doi.org/10.3390/metabo7020022