The Molten Globule, and Two-State vs. Non-Two-State Folding of Globular Proteins


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
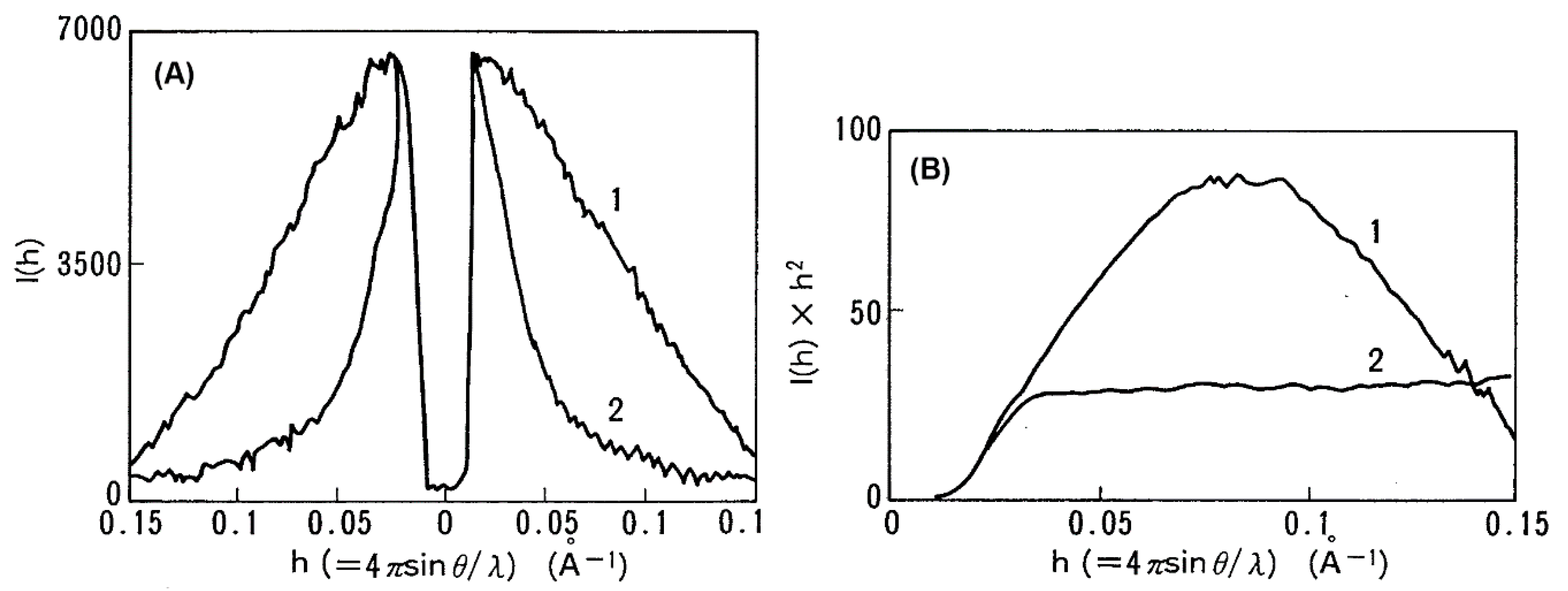
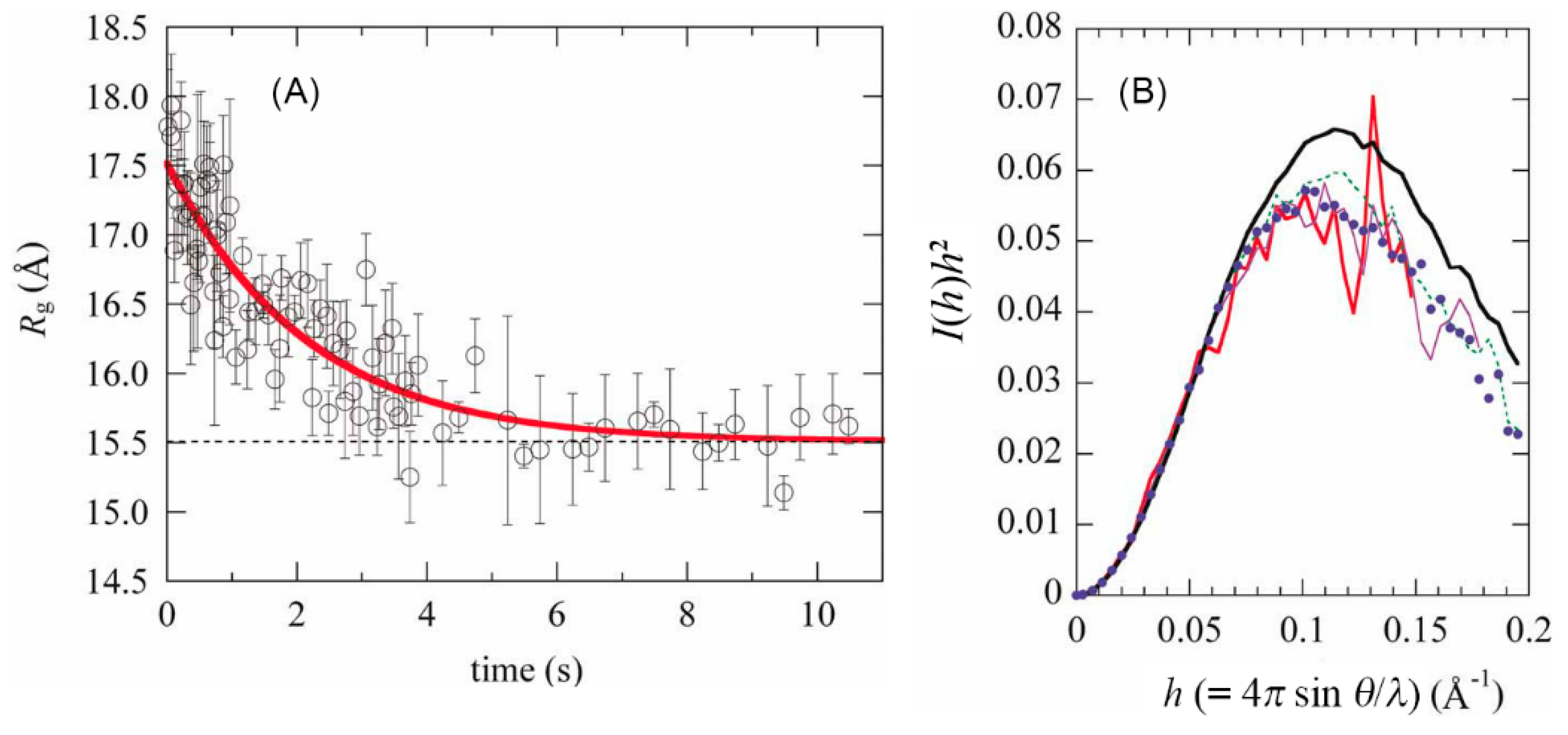
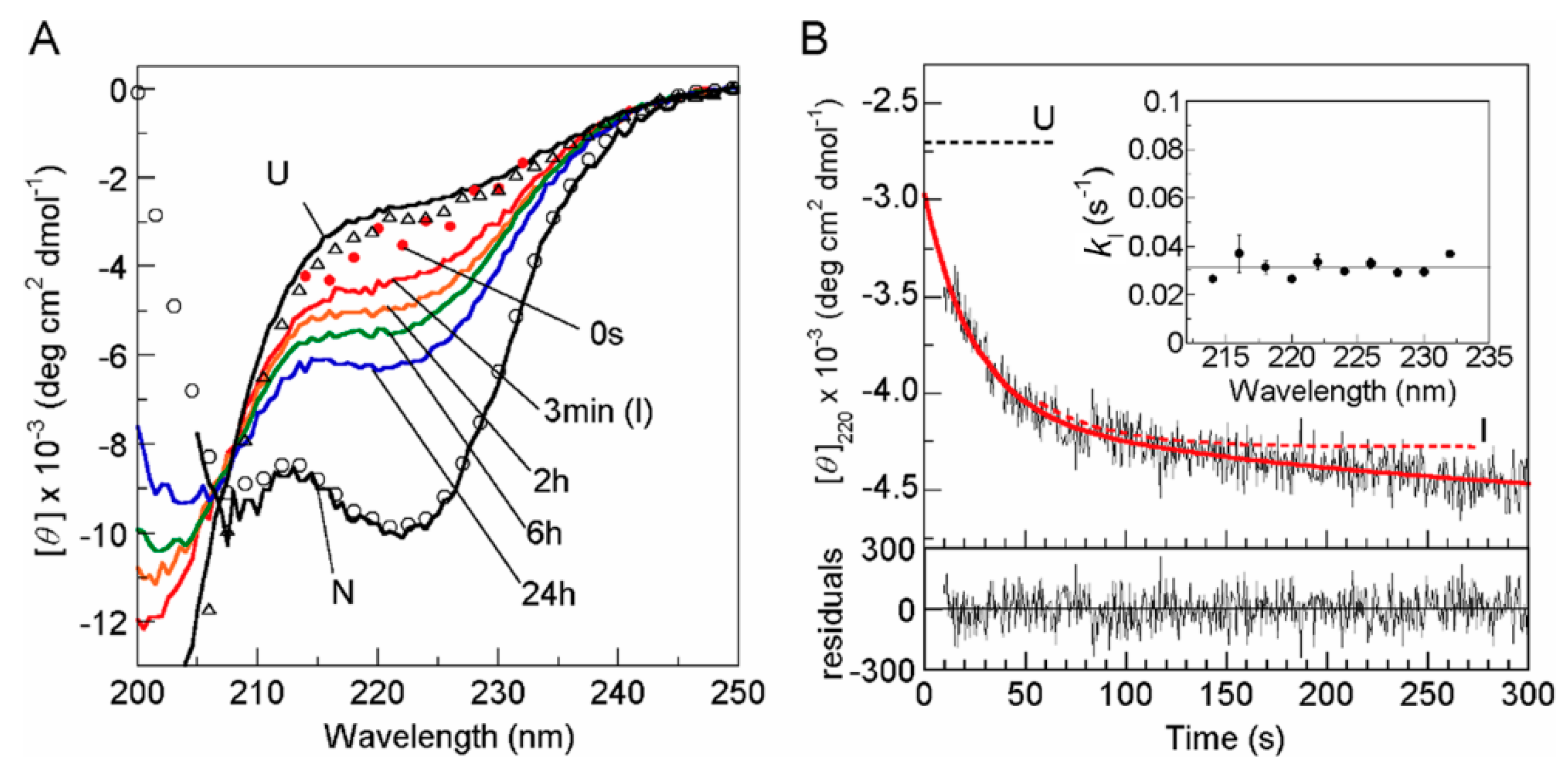
2. Role of The Molten Globule State in Protein Folding
3. Two-State vs. Non-Two-State Folding of Globular Proteins
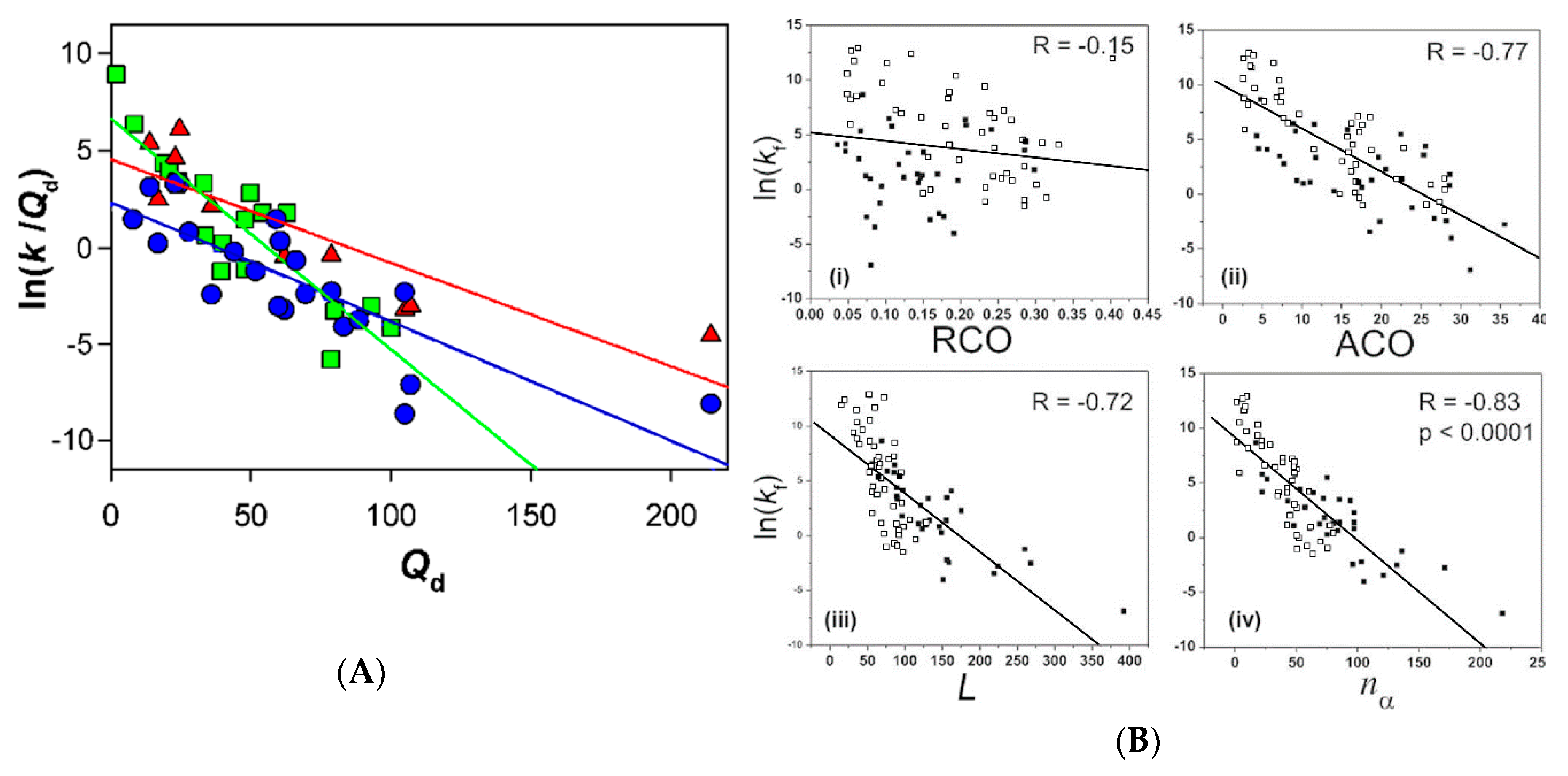
4. The Relationships between the Two-State and Non-Two-State Folding Reactions
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Epstein, C.J.; Goldberger, R.F.; Anfinsen, C.B. The Genetic Control of Tertiary Protein Structure: Studies with Model Systems. Cold Spring Harb. Symp. Quant. Biol. 1963, 28, 439–449. [Google Scholar] [CrossRef]
- Morris, E.; Searle, M. Overview of Protein Folding Mechanisms: Experimental and Theoretical Approaches to Probing Energy Landscapes. Curr. Protoc. Protein Sci. 2012, 68, 28.2.1–28.2.22. [Google Scholar] [CrossRef]
- Sinha, K.K.; Udgaonkar, J.B. Early events in protein folding. Curr. Sci. 2009, 96, 1053–1070. [Google Scholar]
- Bergasa-Caceres, F.; Haas, E.; Rabitz, H. Nature’s Shortcut to Protein Folding. J. Phys. Chem. B 2019, 123, 4463–4476. [Google Scholar] [CrossRef]
- Barrick, D. What have we learned from the studies of two-state folders, and what are the unanswered questions about two-state protein folding? Phys. Biol. 2009, 6, 015001. [Google Scholar] [CrossRef][Green Version]
- Tsytlonok, M.; Itzhaki, L.S. The how’s and why’s of protein folding intermediates. Arch. Biochem. Biophys. 2013, 531, 14–23. [Google Scholar] [CrossRef]
- Levinthal, C. Are there pathways for protein folding? J. Chim. Phys. 1968, 65, 44–45. [Google Scholar] [CrossRef]
- Wetlaufer, D.B. Nucleation, rapid folding, and globular intrachain regions in proteins. Proc. Natl. Acad. Sci. USA 1973, 70, 697–701. [Google Scholar] [CrossRef]
- Ptitsyn, O.; Pain, R.; Semisotnov, G.; Zerovnik, E.; Razgulyaev, O. Evidence for a molten globule state as a general intermediate in protein folding. FEBS Lett. 1990, 262, 20–24. [Google Scholar] [CrossRef]
- Ptitsyn, O. Molten Globule and Protein Folding. Adv. Protein Chem. 1995, 47, 83–229. [Google Scholar]
- Arai, M.; Kuwajima, K. Role of the molten globule state in protein folding. Adv. Protein Chem. 2000, 53, 209–282. [Google Scholar]
- Jackson, S.E.; Fersht, A.R. Folding of chymotrypsin inhibitor 2. 1. Evidence for a two-state transition. Biochemistry 1991, 30, 10428–10435. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.E. How do small single-domain proteins fold? Fold. Des. 1998, 3, R81–R91. [Google Scholar] [CrossRef]
- Onuchic, J.N.; Nymeyer, H.; García, A.E.; Chahine, J.; Socci, N.D. The energy landscape theory of protein folding: Insights into folding mechanisms and scenarios. Adv. Protein Chem. 2000, 53, 87–152. [Google Scholar]
- Roder, H.; Maki, K.; Cheng, H. Early Events in Protein Folding Explored by Rapid Mixing Methods. Chem. Rev. 2006, 106, 1836–1861. [Google Scholar] [CrossRef]
- Okabe, T.; Tsukamoto, S.; Fujiwara, K.; Shibayama, N.; Ikeguchi, M. Delineation of Solution Burst-Phase Protein Folding Events by Encapsulating the Proteins in Silica Gels. Biochemistry 2014, 53, 3858–3866. [Google Scholar] [CrossRef]
- Bychkova, V.E.; Semisotnov, G.V.; Balobanov, V.A.; Finkelstein, A.V. The Molten Globule Concept: 45 Years Later. Biochemistry 2018, 83, S33–S47. [Google Scholar] [CrossRef]
- Semisotnov, G.V.; Rodionova, N.A.; Razgulyaev, O.I.; Uversky, V.N.; Gripas’, A.F.; Gilmanshin, R.I. Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers 1991, 31, 119–128. [Google Scholar] [CrossRef]
- Kamatari, Y. High-pressure NMR spectroscopy for characterizing folding intermediates and denatured states of proteins. Methods 2004, 34, 133–143. [Google Scholar] [CrossRef]
- Kuwajima, K. The molten globule state as a clue for understanding the folding and cooperativity of globular-protein structure. Proteins: Struct. Funct. Bioinform. 1989, 6, 87–103. [Google Scholar] [CrossRef]
- Kuwajima, K.; Hiraoka, Y.; Ikeguchi, M.; Sugai, S. Comparison of the transient folding intermediates in lysozyme and alpha-lactalbumin. Biochemisty 1985, 24, 874–881. [Google Scholar] [CrossRef]
- E Radford, S.; Dobson, C.M.; Evans, P.A. The folding of hen lysozyme involves partially structured intermediates and multiple pathways. Nature 1992, 358, 302–307. [Google Scholar] [CrossRef]
- Roder, H.; Elöve, G.A.; Englander, S.W. Structural characterization of folding intermediates in cytochrome c by H-exchange labelling and proton NMR. Nature 1988, 335, 700–704. [Google Scholar] [CrossRef]
- Udgaonkar, J.B.; Baldwin, R.L. NMR evidence for an early framework intermediate on the folding pathway of ribonuclease A. Nature 1988, 335, 694–699. [Google Scholar] [CrossRef]
- Jennings, P.; Wright, P.E. Formation of a molten globule intermediate early in the kinetic folding pathway of apomyoglobin. Science 1993, 262, 892–896. [Google Scholar] [CrossRef]
- Ikeguchi, M.; Kuwajima, K.; Mitani, M.; Sugai, S. Evidence for identity between the equilibrium unfolding intermediate and a transient folding intermediate: A comparative-study of the folding reactions of α-lactalbumin and lysozyme. Biochemistry 1996, 25, 6965–6972. [Google Scholar] [CrossRef]
- Arai, M.; Kuwajima, K. Rapid formation of a molten globule intermediate in refolding of α-lactalbumin. Fold. Des. 1996, 1, 275–287. [Google Scholar] [CrossRef]
- Semisotnov, G.V.; Kihara, H.; Kotova, N.V.; Kimura, K.; Amemiya, Y.; Wakabayashi, K.; Serdyuk, I.N.; Timchenko, A.A.; Chiba, K.; Nikaido, K.; et al. Protein Globularization During Folding. A Study by Synchrotron Small-angle X-ray Scattering. J. Mol. Biol. 1996, 262, 559–574. [Google Scholar] [CrossRef]
- Glatter, O.; Kratky, O. Small Angle X-ray Scattering; Academic Press: London, UK, 1982. [Google Scholar]
- Arai, M.; Ikura, T.; Semisotnov, G.V.; Kihara, H.; Amemiya, Y.; Kuwajima, K. Kinetic refolding of β-lactoglobulin. Studies by synchrotron X-ray scattering, and circular dichroism, absorption and fluorescence spectroscopy 1 1Edited by P. E. Wright. J. Mol. Biol. 1998, 275, 149–162. [Google Scholar] [CrossRef]
- Arai, M.; Ito, K.; Inobe, T.; Nakao, M.; Maki, K.; Kamagata, K.; Kihara, H.; Amemiya, Y.; Kuwajima, K. Fast Compaction of α-Lactalbumin During Folding Studied by Stopped-flow X-ray Scattering. J. Mol. Biol. 2002, 321, 121–132. [Google Scholar] [CrossRef]
- Kuwajima, K.; Yamaya, H.; Sugai, S. The Burst-phase Intermediate in the Refolding of β-Lactoglobulin Studied by Stopped-flow Circular Dichroism and Absorption Spectroscopy. J. Mol. Biol. 1996, 264, 806–822. [Google Scholar] [CrossRef]
- Plaxco, K.; Millett, I.S.; Segel, D.J.; Doniach, S.; Baker, D. Chain collapse can occur concomitantly with the rate-limiting step in protein folding. Nat. Struct. Biol. 1999, 6, 554–556. [Google Scholar]
- Amemiya, Y.; Ito, K.; Yagi, N.; Asano, Y.; Wakabayashi, K.; Ueki, T.; Endo, T. Large-aperture TV detector with a beryllium-windowed image intensifier for x-ray diffraction. Rev. Sci. Instruments 1995, 66, 2290–2294. [Google Scholar] [CrossRef]
- Ito, K.; Kamikubo, H.; Yagi, N.; Amemiya, Y. Correction Method and Software for Image Distortion and Nonuniform Response in Charge-Coupled Device-Based X-ray Detectors Utilizing X-ray Image intensifier. Jpn. J. Appl. Phys. 2005, 44, 8684–8691. [Google Scholar] [CrossRef]
- Akiyama, S.; Takahashi, S.; Kimura, T.; Ishimori, K.; Morishima, I.; Nishikawa, Y.; Fujisawa, T. Conformational landscape of cytochrome c folding studied by microsecond-resolved small-angle x-ray scattering. Proc. Natl. Acad. Sci. USA 2002, 99, 1329–1334. [Google Scholar] [CrossRef]
- Uzawa, T.; Akiyama, S.; Kimura, T.; Takahashi, S.; Ishimori, K.; Morishima, I.; Fujisawa, T. Collapse and search dynamics of apomyoglobin folding revealed by submillisecond observations of α-helical content and compactness. Proc. Natl. Acad. Sci. USA 2004, 101, 1171–1176. [Google Scholar] [CrossRef]
- Larios, E.; Li, J.; Schulten, K.; Kihara, H.; Gruebele, M. Multiple Probes Reveal a Native-like Intermediate During Low-temperature Refolding of Ubiquitin. J. Mol. Biol. 2004, 340, 115–125. [Google Scholar] [CrossRef]
- Kimura, T.; Akiyama, S.; Uzawa, T.; Ishimori, K.; Morishima, I.; Fujisawa, T.; Takahashi, S. Specifically Collapsed Intermediate in the Early Stage of the Folding of Ribonuclease A. J. Mol. Biol. 2005, 350, 349–362. [Google Scholar] [CrossRef]
- Kimura, T.; Uzawa, T.; Ishimori, K.; Morishima, I.; Takahashi, S.; Konno, T.; Akiyama, S.; Fujisawa, T. Specific collapse followed by slow hydrogen-bond formation of β-sheet in the folding of single-chain monellin. Proc. Natl. Acad. Sci. USA 2005, 102, 2748–2753. [Google Scholar] [CrossRef]
- Arai, M.; Kondrashkina, E.; Kayatekin, C.; Matthews, C.R.; Iwakura, M.; Bilsel, O. Microsecond Hydrophobic Collapse in the Folding of Escherichia coli Dihydrofolate Reductase, an α/β-Type Protein. J. Mol. Biol. 2007, 368, 219–229. [Google Scholar] [CrossRef]
- Konuma, T.; Kimura, T.; Matsumoto, S.; Goto, Y.; Fujisawa, T.; Fersht, A.R.; Takahashi, S. Time-Resolved Small-Angle X-ray Scattering Study of the Folding Dynamics of Barnase. J. Mol. Biol. 2011, 405, 1284–1294. [Google Scholar] [CrossRef] [PubMed]
- Brönnimann, C.; Eikenberry, E.F.; Henrich, B.; Horisberger, R.; Huelsen, G.; Pohl, E.; Schmitt, B.; Schulze-Briese, C.; Suzuki, M.; Tomizaki, T.; et al. The PILATUS 1M detector. J. Synchrotron Radiat. 2006, 13, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Blanchet, C.; Spilotros, A.; Schwemmer, F.; Graewert, M.A.; Kikhney, A.; Jeffries, C.M.; Franke, D.; Mark, D.; Zengerle, R.; Cipriani, F.; et al. Versatile sample environments and automation for biological solution X-ray scattering experiments at the P12 beamline (PETRA III, DESY). J. Appl. Crystallogr. 2015, 48, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Osaka, K.; Matsumoto, T.; Taniguchi, Y.; Inoue, D.; Sato, M.; Sano, N. High-throughput and automated SAXS/USAXS experiment for industrial use at BL19B2 in SPring-8. In AIP Conference Proceedings; AIP Publishing: New York, NY, USA, 2016; Volume 1741, p. 30003. [Google Scholar]
- Brandts, J.F.; Halvorson, H.R.; Brennan, M. Consideration of the possibility that the slow step in protein denaturation reactions is due to cis-trans isomerism of proline residues. Biochemistry 1975, 14, 4953–4963. [Google Scholar] [CrossRef]
- Yoo, T.Y.; Meisburger, S.; Hinshaw, J.; Pollack, L.; Haran, G.; Sosnick, T.R.; Plaxco, K. Small-Angle X-ray Scattering and Single-Molecule FRET Spectroscopy Produce Highly Divergent Views of the Low-Denaturant Unfolded State. J. Mol. Biol. 2012, 418, 226–236. [Google Scholar] [CrossRef]
- Manavalan, B.; Kuwajima, K.; Lee, J. PFDB: A standardized protein folding database with temperature correction. Sci. Rep. 2019, 9, 1588. [Google Scholar] [CrossRef]
- Silow, M.; Oliveberg, M. Transient aggregates in protein folding are easily mistaken for folding intermediates. Proc. Natl. Acad. Sci. USA 1997, 94, 6084–6086. [Google Scholar] [CrossRef]
- Went, H.M.; Benítez-Cardoza, C.G.; Jackson, S.E. Is an intermediate state populated on the folding pathway of ubiquitin? FEBS Lett. 2004, 567, 333–338. [Google Scholar] [CrossRef]
- Colón, W.; Elöve, G.A.; Wakem, L.P.; Sherman, F.; Roder, H. Side Chain Packing of the N- and C-Terminal Helices Plays a Critical Role in the Kinetics of Cytochromec Folding. Biochem. 1996, 35, 5538–5549. [Google Scholar] [CrossRef]
- Houry, W.A.; Rothwarf, D.M.; Scheraga, H.A. Circular Dichroism Evidence for the Presence of Burst-Phase Intermediates on the Conformational Folding Pathway of Ribonuclease A. Biochemistry 1996, 35, 10125–10133. [Google Scholar] [CrossRef]
- Sosnick, T.R.; Mayne, L.; Englander, S.W. Molecular collapse: The rate-limiting step in two-state cytochrome c folding. Proteins: Struct. Funct. Bioinform. 1996, 24, 413–426. [Google Scholar] [CrossRef]
- Sosnick, T.R.; Shtilerman, M.D.; Mayne, L.; Englander, S.W. Ultrafast signals in protein folding and the polypeptide contracted state. Proc. Natl. Acad. Sci. USA 1997, 94, 8545–8550. [Google Scholar] [CrossRef] [PubMed]
- Qi, P.X.; Sosnick, T.R.; Englander, S.W. The burst phase in ribonuclease A folding and solvent dependence of the unfolded state. Nat. Genet. 1998, 5, 882–884. [Google Scholar] [CrossRef] [PubMed]
- Krantz, B.A.; Mayne, L.; Rumbley, J.; Englander, S.W.; Sosnick, T.R. Fast and Slow Intermediate Accumulation and the Initial Barrier Mechanism in Protein Folding. J. Mol. Biol. 2002, 324, 359–371. [Google Scholar] [CrossRef]
- Tanford, C. Protein denaturation. C. Theoretical models for the mechanism of denaturation. Adv. Protein Chem. 1970, 24, 1–95. [Google Scholar]
- Shastry, M.; Roder, H. Evidence for barrier-limited protein folding kinetics on the microsecond time scale. Nat. Struct. Biol. 1998, 5, 385–392. [Google Scholar] [CrossRef]
- Welker, E.; Maki, K.; Shastry, M.C.R.; Juminaga, D.; Bhat, R.; Scheraga, H.A.; Roder, H. Ultrarapid mixing experiments shed new light on the characteristics of the initial conformational ensemble during the folding of ribonuclease A. Proc. Natl. Acad. Sci. USA 2004, 101, 17681–17686. [Google Scholar] [CrossRef]
- Akiyama, S.; Takahashi, S.; Ishimori, K.; Morishima, I. Stepwise formation of α-helices during cytochrome c folding. Nat. Struct Biol. 2000, 7, 514–520. [Google Scholar]
- Qiu, L.; Zachariah, C.; Hagen, S.J. Fast Chain Contraction during Protein Folding: “Foldability” and Collapse Dynamics. Phys. Rev. Lett. 2003, 90, 168103. [Google Scholar] [CrossRef]
- Goldbeck, R.A.; Chen, E.; Kliger, D.S. Early Events, Kinetic Intermediates and the Mechanism of Protein Folding in Cytochrome c. Int. J. Mol. Sci. 2009, 10, 1476–1499. [Google Scholar] [CrossRef]
- Ellerby, L.; Nishida, C.; Nishida, F.; Yamanaka, S.; Dunn, B.; Valentine, J.; Zink, J. Encapsulation of proteins in transparent porous silicate glasses prepared by the sol-gel method. Science 1992, 255, 1113–1115. [Google Scholar] [CrossRef] [PubMed]
- Shibayama, N.; Saigo, S. Fixation of the Quaternary Structures of Human Adult Haemoglobin by Encapsulation in Transparent Porous Silica Gels. J. Mol. Biol. 1995, 251, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Shibayama, N.; Saigo, S. Kinetics of the Allosteric Transition in Hemoglobin within Silicate Sol−Gels. J. Am. Chem. Soc. 1999, 121, 444–445. [Google Scholar] [CrossRef]
- Ronda, L.; Bruno, S.; Campanini, B.; Mozzarelli, A.; Abbruzzetti, S.; Viappiani, C.; Cupane, A.; Levantino, M.; Bettati, S. Immobilization of Proteins in Silica Gel: Biochemical and Biophysical Properties. Curr. Org. Chem. 2015, 19, 1653–1668. [Google Scholar] [CrossRef][Green Version]
- Samuni, U.; Navati, M.S.; Juszczak, L.J.; Dantsker, D.; Yang, M.; Friedman, J.M. Unfolding and Refolding of Sol−Gel Encapsulated Carbonmonoxymyoglobin: An Orchestrated Spectroscopic Study of Intermediates and Kinetics. J. Phys. Chem. B 2000, 104, 10802–10813. [Google Scholar] [CrossRef]
- Peterson, E.S.; Leonard, E.F.; Foulke, J.A.; Oliff, M.C.; Salisbury, R.D.; Kim, D.Y. Folding Myoglobin within a Sol-Gel Glass: Protein Folding Constrained to a Small Volume. Biophys. J. 2008, 95, 322–332. [Google Scholar] [CrossRef]
- Shibayama, N. Slow Motion Analysis of Protein Folding Intermediates within Wet Silica Gels. Biochem. 2008, 47, 5784–5794. [Google Scholar] [CrossRef]
- Shibayama, N. Circular dichroism study on the early folding events of β-lactoglobulin entrapped in wet silica gels. FEBS Lett. 2008, 582, 2668–2672. [Google Scholar] [CrossRef]
- Woody, R.W. Circular Dichroism of Protein-Folding Intermediates. Methods in Enzymology 2004, 380, 242–285. [Google Scholar]
- Ohgushi, M.; Wada, A. ‘Molten-globule state’: A compact form of globular proteins with mobile side-chains. FEBS Lett. 1983, 164, 21–24. [Google Scholar] [CrossRef]
- Nakamura, S.; Seki, Y.; Katoh, E.; Kidokoro, S.-I. Thermodynamic and Structural Properties of the Acid Molten Globule State of Horse Cytochromec. Biochemistry 2011, 50, 3116–3126. [Google Scholar] [CrossRef] [PubMed]
- Colón, W.; Roder, H.; Col, W. Kinetic intermediates in the formation of the cytochrome c molten globule. Nat. Struct. Biol. 1996, 3, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Kamagata, K.; Sawano, Y.; Tanokura, M.; Kuwajima, K. Multiple parallel-pathway folding of proline-free Staphylococcal nuclease. J. Mol. Biol. 2003, 332, 1143–1153. [Google Scholar] [CrossRef] [PubMed]
- Plaxco, K.; Simons, K.T.; Baker, D. Contact order, transition state placement and the refolding rates of single domain proteins. J. Mol. Biol. 1998, 277, 985–994. [Google Scholar] [CrossRef]
- Baker, D. A surprising simplicity to protein folding. Nature 2000, 405, 39–42. [Google Scholar] [CrossRef]
- Plaxco, K.; Simons, K.T.; Ruczinski, I.; Baker, D. Topology, stability, sequence, and length: defining the determinants of two-state protein folding kinetics. Biochemistry 2000, 39, 11177–11183. [Google Scholar] [CrossRef]
- Gromiha, M.M.; Selvaraj, S. Comparison between long-range interactions and contact order in determining the folding rate of two-state proteins: Application of long-range order to folding rate prediction. J. Mol. Biol. 2001, 310, 27–32. [Google Scholar] [CrossRef]
- Harihar, B.; Selvaraj, S. Refinement of the long-range order parameter in predicting folding rates of two-state proteins. Biopolymer 2009, 91, 928–935. [Google Scholar] [CrossRef]
- Makarov, D.E.; Keller, C.A.; Plaxco, K.; Metiu, H. How the folding rate constant of simple, single-domain proteins depends on the number of native contacts. Proc. Natl. Acad. Sci. USA 2002, 99, 3535–3539. [Google Scholar] [CrossRef]
- Makarov, D.E.; Plaxco, K. The topomer search model: A simple, quantitative theory of two-state protein folding kinetics. Protein Sci. 2003, 12, 17–26. [Google Scholar] [CrossRef]
- Nölting, B.; Schälike, W.; Hampel, P.; Grundig, F.; Gantert, S.; Sips, N.; Bandlow, W.; Qi, P.X. Structural determinants of the rate of protein folding. J. Theor. Biol. 2003, 223, 299–307. [Google Scholar] [CrossRef]
- Zhou, H.; Zhou, Y. Folding rate prediction using total contact distance. Biophys. J. 2002, 82, 458–463. [Google Scholar] [CrossRef][Green Version]
- Micheletti, C. Prediction of folding rates and transition-state placement from native-state geometry. Proteins: Struct. Funct. Bioinform. 2003, 51, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Dixit, P.D.; Weikl, T. A simple measure of native-state topology and chain connectivity predicts the folding rates of two-state proteins with and without crosslinks. Proteins: Struct. Funct. Bioinform. 2006, 64, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Galzitskaya, O.V.; Garbuzynskiy, S.O.; Ivankov, D.N.; Finkelstein, A. Chain length is the main determinant of the folding rate for proteins with three-state folding kinetics. Proteins: Struct. Funct. Bioinform. 2003, 51, 162–166. [Google Scholar] [CrossRef]
- Ivankov, D.N.; Garbuzynskiy, S.O.; Alm, E.; Plaxco, K.; Baker, D.; Finkelstein, A. Contact order revisited: Influence of protein size on the folding rate. Protein Sci. 2003, 12, 2057–2062. [Google Scholar] [CrossRef]
- Kamagata, K.; Arai, M.; Kuwajima, K. Unification of the folding mechanisms of non-two-state and two-state proteins. J. Mol. Biol. 2004, 339, 951–965. [Google Scholar] [CrossRef]
- Istomin, A.Y.; Jacobs, N.J.; Livesay, D.R. On the role of structural class of a protein with two-state folding kinetics in determining correlations between its size, topology, and folding rate. Protein Sci. 2007, 16, 2564–2569. [Google Scholar] [CrossRef]
- Harihar, B.; Selvaraj, S. Analysis of rate-limiting long-range contacts in the folding rate of three-state and two-state Proteins. Protein Pept. Lett. 2011, 18, 1042–1052. [Google Scholar] [CrossRef]
- Ivankov, D.N.; Finkelstein, A. Prediction of protein folding rates from the amino acid sequence-predicted secondary structure. Proc. Natl. Acad. Sci. USA 2004, 101, 8942–8944. [Google Scholar] [CrossRef]
- Zhang, L.; Sun, T. Folding rate prediction using n-order contact distance for proteins with two- and three-state folding kinetics. Biophys. Chem. 2005, 113, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Z.; Liang, J. Predicting protein folding rates from geometric contact and amino acid sequence. Protein Sci. 2008, 17, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Wang, J.; Wang, W. Composition-based effective chain length for prediction of protein folding rates. Phys. Rev. E 2010, 82, 12. [Google Scholar] [CrossRef] [PubMed]
- De Sancho, D.; Muñoz, V. Integrated prediction of protein folding and unfolding rates from only size and structural class. Phys. Chem. Chem. Phys. 2011, 13, 17030. [Google Scholar] [CrossRef]
- Huang, S.; Huang, J.T. Inter-residue interaction is a determinant of protein folding kinetics. J. Theor. Biol. 2013, 317, 224–228. [Google Scholar] [CrossRef]
- Baiesi, M.; Orlandini, E.; Seno, F.; Trovato, A. Exploring the correlation between the folding rates of proteins and the entanglement of their native states. J. Phys. A Math. Theor. 2017, 50, 504001. [Google Scholar] [CrossRef]
- Censoni, L.; Martıínez, L. Prediction of kinetics of protein folding with non-redundant contact information. Bioinformatic 2018, 34, 4034–4038. [Google Scholar] [CrossRef]
- Khor, S. Folding with a protein’s native shortcut network. Proteins: Struct. Funct. Bioinform. 2018, 86, 924–934. [Google Scholar] [CrossRef]
- Muñoz, V.; Eaton, W.A. A simple model for calculating the kinetics of protein folding from three-dimensional structures. Proc. Natl. Acad. Sci. USA 1999, 96, 11311–11316. [Google Scholar] [CrossRef]
- Alm, E.; Morozov, A.; Kortemme, T.; Baker, D. Simple physical models connect theory and experiment in protein folding kinetics. J. Mol. Biol. 2002, 322, 463–476. [Google Scholar] [CrossRef]
- Garbuzynskiy, S.O.; Finkelstein, A.; Galzitskaya, O.V. Outlining folding nuclei in globular proteins. J. Mol. Biol. 2004, 336, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Galzitskaya, O.V.; Glyakina, A.V. Nucleation-based prediction of the protein folding rate and its correlation with the folding nucleus size. Proteins: Struct. Funct. Bioinform. 2012, 80, 2711–2727. [Google Scholar] [CrossRef] [PubMed]
- Garbuzynskiy, S.O.; Ivankov, D.N.; Bogatyreva, N.; Finkelstein, A. Golden triangle for folding rates of globular proteins. Proc. Natl. Acad. Sci. USA 2012, 110, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Feng, H.; Zhou, Z. Population and structure determination of hidden folding intermediates by native-state hydrogen exchange-directed protein engineering and nuclear magnetic resonance. Methods Mol. Biol. 2007, 350, 69–81. [Google Scholar]
- Sanchez, I.E.; Kiefhaber, T. Evidence for sequential barriers and obligatory intermediates in apparent two-state protein folding. J. Mol. Biol. 2003, 325, 367–376. [Google Scholar] [CrossRef]
- Dalby, P.A.; Clarke, J.; Johnson, C.M.; Fersht, A.R. Folding intermediates of wild-type and mutants of barnase. II. correlation of changes in equilibrium amide exchange kinetics with the population of the folding intermediate. J. Mol. Biol. 1998, 276, 647–656. [Google Scholar] [CrossRef]
- McCallister, E.L.; Alm, E.; Baker, D. Critical role of β-hairpin formation in protein G folding. Nat. Struct. Biol. 2000, 7, 669–673. [Google Scholar] [CrossRef]
- Spudich, G.M.; Miller, E.J.; Marqusee, S. Destabilization of the Escherichia coli RNase H Kinetic Intermediate: Switching Between a Two-state and Three-state Folding Mechanism. J. Mol. Biol. 2004, 335, 609–618. [Google Scholar] [CrossRef]
- Friel, C.T.; Beddard, G.S.; E Radford, S. Switching Two-state to Three-state Kinetics in the Helical Protein Im9 via the Optimisation of Stabilising Non-native Interactions by Design. J. Mol. Biol. 2004, 342, 261–273. [Google Scholar] [CrossRef]






© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuwajima, K. The Molten Globule, and Two-State vs. Non-Two-State Folding of Globular Proteins. Biomolecules 2020, 10, 407. https://doi.org/10.3390/biom10030407
Kuwajima K. The Molten Globule, and Two-State vs. Non-Two-State Folding of Globular Proteins. Biomolecules. 2020; 10(3):407. https://doi.org/10.3390/biom10030407
Chicago/Turabian StyleKuwajima, Kunihiro. 2020. "The Molten Globule, and Two-State vs. Non-Two-State Folding of Globular Proteins" Biomolecules 10, no. 3: 407. https://doi.org/10.3390/biom10030407
APA StyleKuwajima, K. (2020). The Molten Globule, and Two-State vs. Non-Two-State Folding of Globular Proteins. Biomolecules, 10(3), 407. https://doi.org/10.3390/biom10030407